
Substance P Hinders Bile Acid-Induced Hepatocellular Injury by Modulating Oxidative Stress and Inflammation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Western Blot
2.5. SP Administration
2.6. Cytokine Measurement of Cytokines Using Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. ROS Measurement
2.8. Alanine Aminotransferase (ALT)/Alkaline Phosphatase (ALP) Assay
2.9. Tube Formation Assay
2.10. Statistical Analysis
3. Results
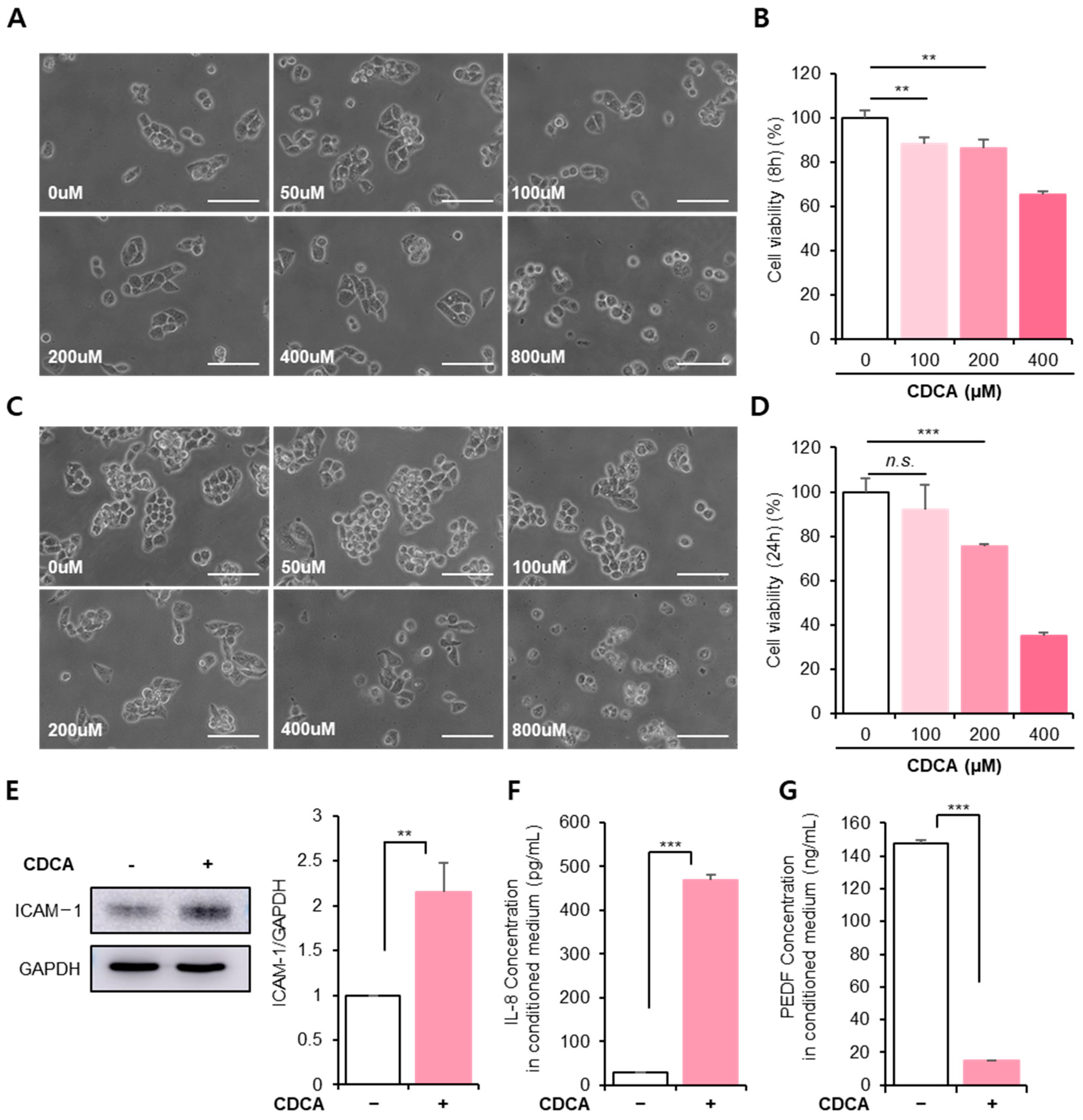
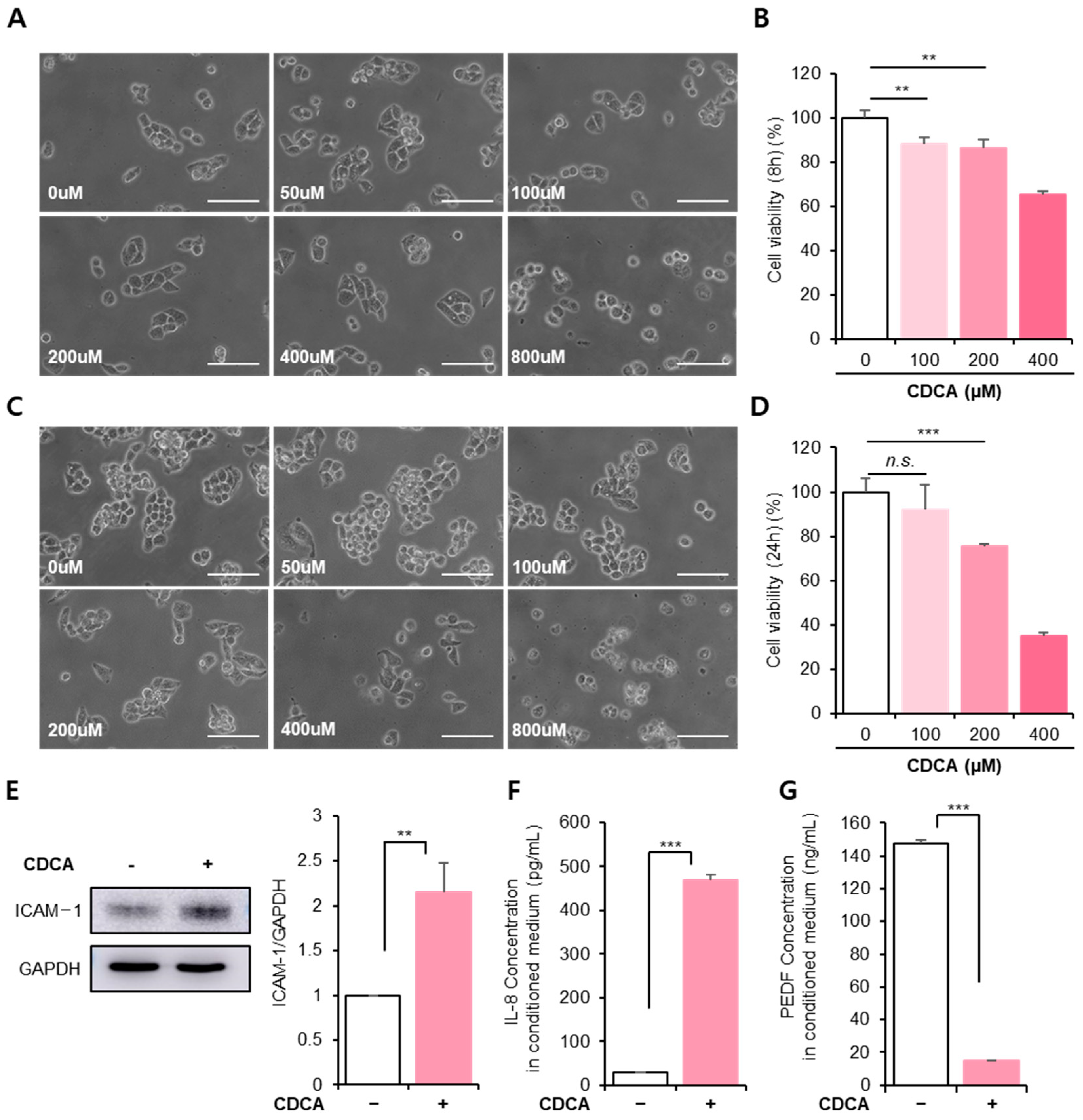
3.1. BA Reduced Hepatocyte Cellular Activity, Accompanied by Inflammation
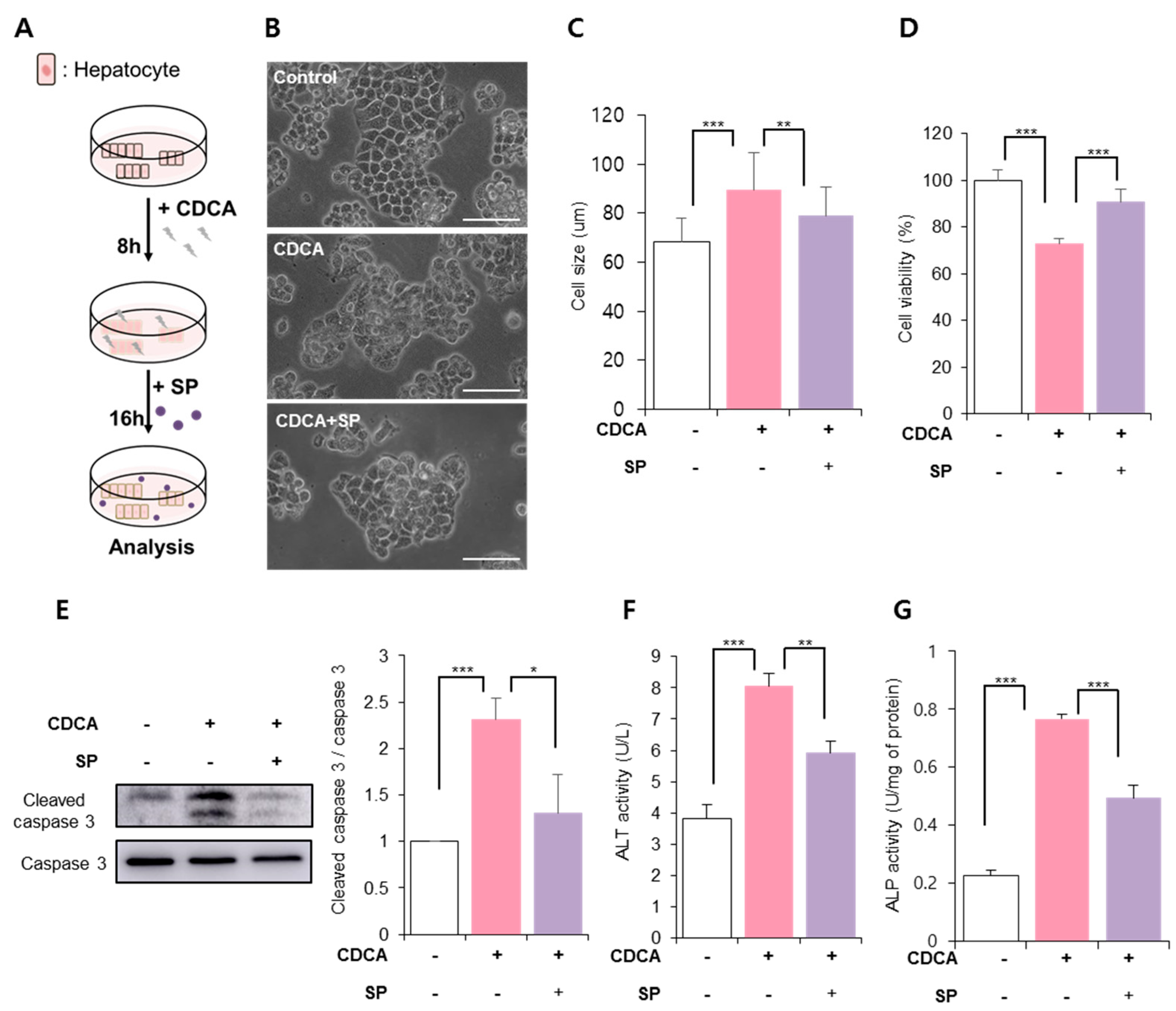
3.2. SP Prevented CDCA-Induced Hepatic Cell Death
3.3. SP Ameliorated BA-Induced Hepatic Inflammation and Oxidative Stress
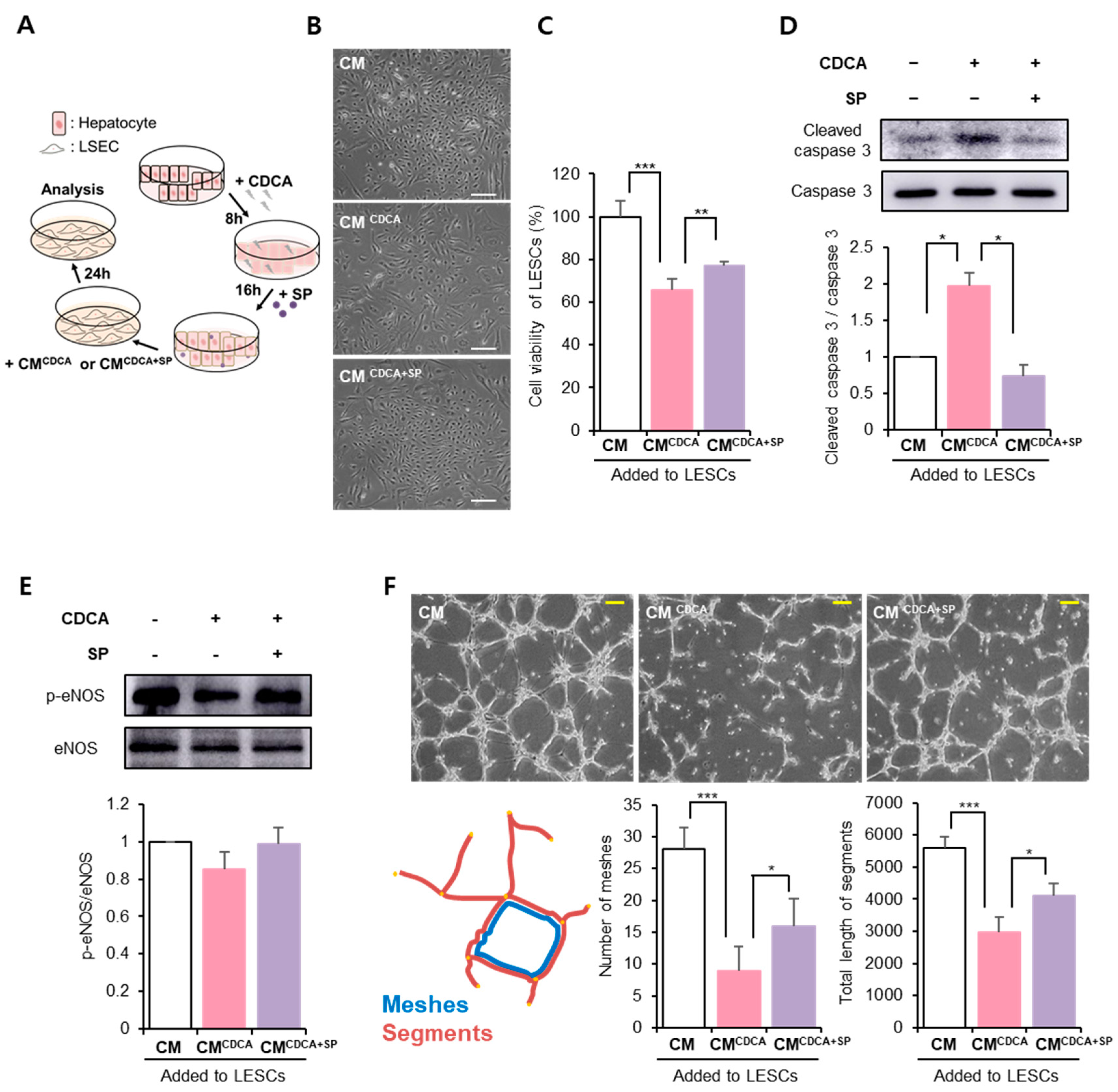
3.4. SP Protected the Hepatic Endothelium against BA-Damaged Hepatocyte Paracrine Action
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michalopoulos, G.K. Liver regeneration. Liver Biol. Pathobiol. 2020, 45, 566–584. [Google Scholar]
- Zhang, Y.; Hong, J.Y.; Rockwell, C.E.; Copple, B.L.; Jaeschke, H.; Klaassen, C.D. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int. 2012, 32, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Ma, X.; Zhao, Y. Mechanism of hydrophobic bile acid-induced hepatocyte injury and drug discovery. Front. Pharmacol. 2020, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Li, M.K.; Crawford, J.M. The pathology of cholestasis. Semin. Liver Dis. 2004, 24, 21–42. [Google Scholar] [PubMed]
- Martinez-Diez, M.C.; Serrano, M.A.; Monte, M.J.; Marin, J.J. Comparison of the effects of bile acids on cell viability and DNA synthesis by rat hepatocytes in primary culture. Biochim. Et Biophys. Acta 2000, 1500, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.Y.; Yu, D.; Soroka, C.J.; Wang, J.; Boyer, J.L. Hepatic NFAT signaling regulates the expression of inflammatory cytokines in cholestasis. J. Hepatol. 2021, 74, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kaminaga, T.; Yasuda, H.; Kamiya, T.; Hara, H. The involvement of endoplasmic reticulum stress in bile acid-induced hepatocellular injury. J. Clin. Biochem. Nutr. 2014, 54, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrasa, J.I.; Olmo, N.; Pérez-Ramos, P.; Santiago-Gómez, A.; Lecona, E.; Turnay, J.; Antonia Lizarbe, M. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis 2011, 16, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.Y.; Ouyang, X.; Chen, Y.; Soroka, C.J.; Wang, J.; Mennone, A.; Boyer, J.L. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017, 2, e90780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujral, J.S.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile ductligated Mice. Hepatology 2003, 38, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [PubMed] [Green Version]
- Wang, Z.; Lv, Q.; Liu, H.; Wu, Y.; Bai, Y.; Cheng, Y.; Su, Y.; Cai, Y.; Yu, J.; Ma, J.; et al. Caveolae depletion contributes to vasorelaxant effects of chenodeoxycholic acid. Cell Physiol. Biochem. 2017, 42, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.D.; MEaretti-Mira, A.C. Liver sinusoidal endothelial cell: An update. In Seminars in Liver Disease; Thieme Medical Publishers: Stuttgart, Germany, 2017; pp. 377–387. [Google Scholar]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Manatsathit, W.; Dies, D.; Shokouh-Amiri, H.; Zibari, G.; Boktor, M.; Alexander, J.S. Cholestasis liver disease: An era of emerging therapies. World J. Clin. Cases 2019, 7, 1571–1581. [Google Scholar] [CrossRef]
- Fang, S. Bile acid receptor farnesoid X receptor: A novel therapeutic target for metabolic disease. J. Lipid Atheroscler. 2017, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Purohit, T. Primary biliary cirrhosis: Pathophysiology, clinical presentation and therapy. World J. Hepatol. 2015, 7, 926. [Google Scholar] [CrossRef]
- Sharanek, A.; Burban, A.; Humbert, L.; Guguen-Guillouzo, C.; Rainteau, D.; Guillouzo, A. Progressive and preferential cellular accumulation of hydrophobic bile acids induced by cholestatic drugs is associated with inhibition of their amidation and sulfation. Drug Metab. Dispos. 2017, 45, 1292–1303. [Google Scholar] [CrossRef]
- Kumar, D.; Tandon, R.K. Use of ursodeoxycholic acid in liver diseases. J. Gastroenterol. Hepatol. 2001, 16, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive analysis of serum and fecal bile acid profiles and interaction with gut microbiota in primary biliary cholangitis. J. Allergy Clin. Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Yerushalmi, B.; Dahl, R.; Devereaux, M.W.; Gumpricht, E.; Sokol, R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001, 33, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Piao, J.; Park, J.S.; Lee, D.; Hong, H.S. Substance P ameliorates TNF-α-mediated impairment of human aortic vascular cells in vitro. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1288–1297. [Google Scholar] [CrossRef]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell. Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Piao, J.; Park, G.; Hong, H.S. Substance-P Restores Cellular Activity of ADSC Impaired by Oxidative Stress. Antioxidants 2020, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Kim, S.; Yoo, K.; Hong, H.S. Substance P blocks ethanol-induced hepatotoxicity. Life Sci. 2018, 203, 268–275. [Google Scholar] [CrossRef]
- Kim, S.; Hong, H.S. Substance-P prevents the cholestatic liver injury by regulating inflammatory responses. Peptides 2021, 137, 170494. [Google Scholar] [CrossRef]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes; a novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef]
- Baek, S.M.; Yu, S.Y.; Son, Y.; Hong, H.S. Substance P promotes the recovery of oxidative stress-damaged retinal pigmented epithelial cells by modulating Akt/GSK-3β signaling. Mol. Vis. 2016, 22, 1015. [Google Scholar]
- Kim, D.Y.; Piao, J.; Hong, H.S. Substance-P Inhibits Cardiac Microvascular Endothelial Dysfunction Caused by High Glucose-Induced Oxidative Stress. Antioxidants 2021, 10, 1084. [Google Scholar] [CrossRef]
- Pan, W.C.; Wu, C.D.; Chen, M.J.; Huang, Y.T.; Chen, C.J.; Su, H.J.; Yang, H.I. Fine particle pollution, alanine transaminase, and liver cancer: A Taiwanese prospective cohort study (REVEAL-HBV). J. Natl. Cancer Inst. 2016, 108, djv341. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Guo, X.; Hamada, T.; Yokoyama, S.; Nakamura, Y.; Zheng, J.; Kurose, N.; Ishigaki, Y.; Uramoto, H.; Tanimoto, A.; et al. Protective effects of peroxiredoxin 4 (PRDX4) on cholestatic liver injury. Int. J. Mol. Sci. 2018, 19, 2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Qing, W.; Sun, M.; Lv, L.; Guo, D.; Jiang, Y. Melatonin protects hepatocytes against bile acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radic. Res. 2015, 49, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. Nitric oxide: From good to bad. Ann. Vasc. Dis. 2018, 11, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Saha, M.R.; Sen, A. An overview on drug-induced hepatotoxicity. Asian J. Pharm. Clin. Res. 2013, 6, 1–4. [Google Scholar]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of hepatotoxicity. Toxicol. Sci. 2002, 65, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: Differential activation and binding of the transcription factors AP-1 and NF-kappaB. Int. J. Mol. Med. 1999, 4, 223–253. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Park, J.S.; Kim, D.; Hong, H.S. Substance P Hinders Bile Acid-Induced Hepatocellular Injury by Modulating Oxidative Stress and Inflammation. Antioxidants 2022, 11, 920. https://doi.org/10.3390/antiox11050920
Lee D, Park JS, Kim D, Hong HS. Substance P Hinders Bile Acid-Induced Hepatocellular Injury by Modulating Oxidative Stress and Inflammation. Antioxidants. 2022; 11(5):920. https://doi.org/10.3390/antiox11050920
Chicago/Turabian StyleLee, Dahyeon, Jeong Seop Park, Doyoung Kim, and Hyun Sook Hong. 2022. "Substance P Hinders Bile Acid-Induced Hepatocellular Injury by Modulating Oxidative Stress and Inflammation" Antioxidants 11, no. 5: 920. https://doi.org/10.3390/antiox11050920
APA StyleLee, D., Park, J. S., Kim, D., & Hong, H. S. (2022). Substance P Hinders Bile Acid-Induced Hepatocellular Injury by Modulating Oxidative Stress and Inflammation. Antioxidants, 11(5), 920. https://doi.org/10.3390/antiox11050920