
Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution

 , , and
, , and
Abstract

1. Introduction
2. Myeloperoxidase
2.1. Endothelial Dysfunction
2.2. Plaque Destabilization
2.3. Lipoprotein Oxidation
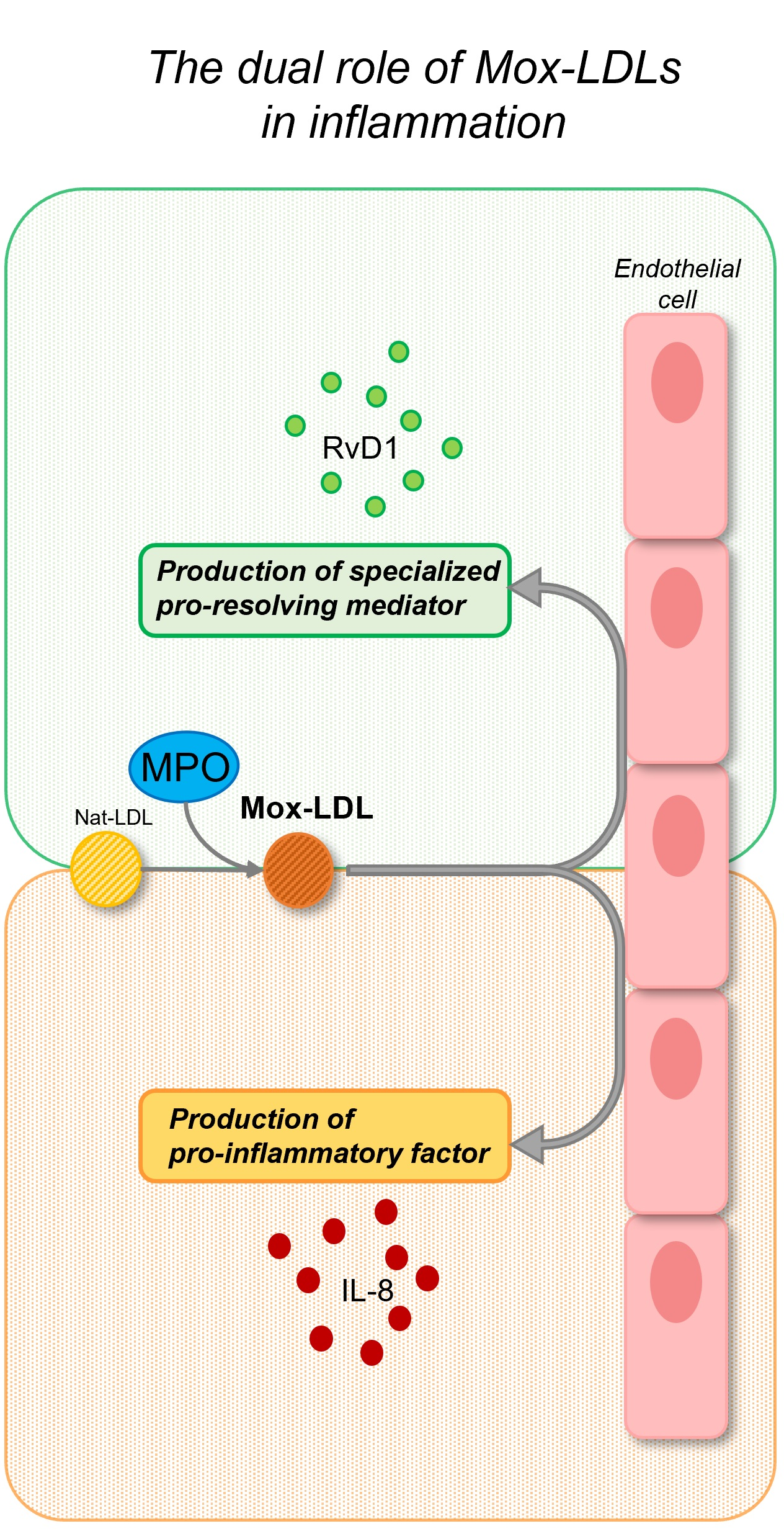
3. Mox-LDL-Cell Interactions
3.1. Mox-LDL-Macrophages
3.2. Mox-LDL-Endothelial Cells
4. Bioactive Lipid Mediators: Implications in Atherosclerosis

{kind=link}
{kind=link}
| SPM | Cell Type | Effect | Reference |
|---|---|---|---|
| RvD1 | Macrophage | Stimulation of phagocytosis | Krishnamoorthy et al. [78] |
| Macrophage | Enhanced efferocytosis | Rymut et al. [84] | |
| Macrophage | Polarization of primary macrophages and repolarization of previously polarized M1-macrophages to a pro-resolution phenotype | Schmid et al. [88] | |
| Macrophage | Switch from a M1 phenotype to a pro-resolution M2-like phenotype | Dalli et al. [92] | |
| Neutrophil | Decreased actin polymerization which is essential to neutrophil migration. Limited infiltration and transendothelial migration | Krishnamoorthy et al. [78] | |
| Cardiac fibroblast | Decreased expression of adhesion molecules ICAM-1 and VCAM-1 | Salas-Hernández et al. [81] | |
| Macrophage | Decreased production of pro-inflammatory cytokines. Increased expression of pro-resolving markers. Polarization of macrophages toward a pro-resolving phenotype | Kain et al. [82] | |
| Endothelial cell | Protection of endothelial cell adherens junction. Maintenance of endothelial barrier integrity and permeability | Chattopadhyay et al. [83] | |
| Macrophage | Translocation of 5-LOX from the nucleus to the cytoplasm, inducing increased synthesis of LXA4 and decreased synthesis of LTB4 | Fredman et al. [87] | |
| 17-HDHA/RvD1 | Endothelial cell and VSMC | Translocation of 5-LOX from the nucleus to the cytoplasm | Chatterjee et al. [86] |
5. Monocyte-Derived Macrophages and SPMs: Interplay in Atherosclerosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Falk, E. Pathogenesis of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and Its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An Imbalance between Specialized Pro-Resolving Lipid Mediators and pro-Inflammatory Leukotrienes Promotes Instability of Atherosclerotic Plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef]
- Fredman, G. Can Inflammation-Resolution Provide Clues to Treat Patients According to Their Plaque Phenotype? Front. Pharmacol. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Mayyas, F.A.; Al-Jarrah, M.I.; Ibrahim, K.S.; Alzoubi, K.H. Level and Significance of Plasma Myeloperoxidase and the Neutrophil to Lymphocyte Ratio in Patients with Coronary Artery Disease. Exp. Ther. Med. 2014, 8, 1951–1957. [Google Scholar] [CrossRef]
- Khan, A.; Alsahli, M.; Rahmani, A. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef]
- Zhang, R.; Brennan, M.-L.; Fu, X.; Aviles, R.J.; Pearce, G.L.; Penn, M.S.; Topol, E.J.; Sprecher, D.L.; Hazen, S.L. Association Between Myeloperoxidase Levels and Risk of Coronary Artery Disease. JAMA 2001, 286, 2136. [Google Scholar] [CrossRef]
- Yoshida, H.; Kisugi, R. Mechanisms of LDL Oxidation. Clin. Chim. Acta 2010, 411, 1875–1882. [Google Scholar] [CrossRef]
- Shao, B.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Myeloperoxidase: An Oxidative Pathway for Generating Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2010, 23, 447–454. [Google Scholar] [CrossRef]
- Ndrepepa, G. Myeloperoxidase—A Bridge Linking Inflammation and Oxidative Stress with Cardiovascular Disease. Clin. Chim. Acta 2019, 493, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Boudjeltia, K.Z.; Legssyer, I.; Van Antwerpen, P.; Kisoka, R.L.; Babar, S.; Moguilevsky, N.; Delree, P.; Ducobu, J.; Remacle, C.; Vanhaeverbeek, M.; et al. Triggering of Inflammatory Response by Myeloperoxidase-Oxidized LDL. Biochem. Cell Biol. 2006, 84, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Aratani, Y. Myeloperoxidase: Its Role for Host Defense, Inflammation, and Neutrophil Function. Arch. Biochem. Biophys. 2018, 640, 47–52. [Google Scholar] [CrossRef]
- Rocca, G.; Stefano, A.; Eleuteri, E.; Anzalone, R.; Magno, F.; Corrao, S.; Loria, T.; Martorana, A.; Gangi, C.; Colombo, M.; et al. Oxidative Stress Induces Myeloperoxidase Expression in Endocardial Endothelial Cells from Patients with Chronic Heart Failure. Basic Res. Cardiol. 2009, 104, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.E.; Schultz, J. Studies on the Chlorinating Activity of Myeloperoxidase. J. Biol. Chem. 1976, 251, 1371–1374. [Google Scholar] [CrossRef]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A Key Regulator of Neutrophil Oxidant Production. Redox Rep. 1997, 3, 3–15. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J.; Hawkins, C.L. Reactions and Reactivity of Myeloperoxidase-Derived Oxidants: Differential Biological Effects of Hypochlorous and Hypothiocyanous Acids. Free Radic. Res. 2012, 46, 975–995. [Google Scholar] [CrossRef]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.-L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes. Characterization, Purification, Molecular Cloning, and Functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a Leukocyte-Derived Vascular NO Oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef]
- Xu, J.; Xie, Z.; Reece, R.; Pimental, D.; Zou, M.-H. Uncoupling of Endothelial Nitric Oxidase Synthase by Hypochlorous Acid: Role of NAD(P)H Oxidase–Derived Superoxide and Peroxynitrite. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2688–2695. [Google Scholar] [CrossRef]
- Zhang, C.; Reiter, C.; Eiserich, J.P.; Boersma, B.; Parks, D.A.; Beckman, J.S.; Barnes, S.; Kirk, M.; Baldus, S.; Darley-Usmar, V.M.; et al. L-Arginine Chlorination Products Inhibit Endothelial Nitric Oxide Production. J. Biol. Chem. 2001, 276, 27159–27165. [Google Scholar] [CrossRef]
- Etwebi, Z.; Landesberg, G.; Preston, K.; Eguchi, S.; Scalia, R. Mechanistic Role of the Calcium-Dependent Protease Calpain in the Endothelial Dysfunction Induced by MPO (Myeloperoxidase). Hypertension 2018, 71, 761–770. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 20. [Google Scholar]
- Wang, Y.; Rosen, H.; Madtes, D.K.; Shao, B.; Martin, T.R.; Heinecke, J.W.; Fu, X. Myeloperoxidase Inactivates TIMP-1 by Oxidizing Its N-Terminal Cysteine Residue. J. Biol. Chem. 2007, 282, 31826–31834. [Google Scholar] [CrossRef]
- Weiss, S.J.; Peppin, G.J.; Ortiz, X.; Ragsdale, C.; Test, S. Oxidative Autoactivation of Latent Collagenase by Human Neutrophils. Science 1985, 227, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Peppin, G.J.; Weiss, S.J. Activation of the Endogenous Metalloproteinase, Gelatinase, by Triggered Human Neutrophils. Proc. Natl. Acad. Sci. USA 1986, 83, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous Acid Oxygenates the Cysteine Switch Domain of Pro-Matrilysin (MMP-7). J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, S.; Kugiyama, K.; Aikawa, M.; Nakamura, S.; Ogawa, H.; Libby, P. Hypochlorous Acid, a Macrophage Product, Induces Endothelial Apoptosis and Tissue Factor Expression: Involvement of Myeloperoxidase-Mediated Oxidant in Plaque Erosion and Thrombogenesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1309–1314. [Google Scholar] [CrossRef]
- Rashid, I.; Maghzal, G.J.; Chen, Y.-C.; Cheng, D.; Talib, J.; Newington, D.; Ren, M.; Vajandar, S.K.; Searle, A.; Maluenda, A.; et al. Myeloperoxidase Is a Potential Molecular Imaging and Therapeutic Target for the Identification and Stabilization of High-Risk Atherosclerotic Plaque. Eur. Heart J. 2018, 39, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Shi, R. Mammalian Peroxidasin (PXDN): From Physiology to Pathology. Free Radic. Biol. Med. 2022, 182, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ero-Tolliver, I.A.; Hudson, B.G.; Bhave, G. The Ancient Immunoglobulin Domains of Peroxidasin Are Required to Form Sulfilimine Cross-Links in Collagen IV. J. Biol. Chem. 2015, 290, 21741–21748. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Wang, L.; Zhang, J.-J.; Xiong, X.-M.; Zhang, D.; Tang, X.-M.; Luo, X.-J.; Ma, Q.-L.; Peng, J. Vascular Peroxide 1 Promotes Ox-LDL-Induced Programmed Necrosis in Endothelial Cells through a Mechanism Involving β-Catenin Signaling. Atherosclerosis 2018, 274, 128–138. [Google Scholar] [CrossRef]
- Medfai, H.; Khalil, A.; Rousseau, A.; Nuyens, V.; Paumann-Page, M.; Sevcnikar, B.; Furtmüller, P.G.; Obinger, C.; Moguilevsky, N.; Peulen, O.; et al. Human Peroxidasin 1 Promotes Angiogenesis through ERK1/2, Akt, and FAK Pathways. Cardiovasc. Res. 2019, 115, 463–475. [Google Scholar] [CrossRef]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a Catalyst for Lipoprotein Oxidation, Is Expressed in Human Atherosclerotic Lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Hazell, L.J.; Arnold, L.; Flowers, D.; Waeg, G.; Malle, E.; Stocker, R. Presence of Hypochlorite-Modified Proteins in Human Atherosclerotic Lesions. J. Clin. Investig. 1996, 97, 1535–1544. [Google Scholar] [CrossRef]
- Carr, A.C.; McCall, M.R.; Frei, B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species: Reaction Pathways and Antioxidant Protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Kostevich, V.A.; Runova, O.L.; Gorudko, I.V.; Vasilyev, V.B.; Cherenkevich, S.N.; Panasenko, O.M. Proatherogenic Modification of LDL by Surface-Bound Myeloperoxidase. Chem. Phys. Lipids 2014, 180, 72–80. [Google Scholar] [CrossRef]
- Malle, E.; Waeg, G.; Schreiber, R.; Gröne, E.F.; Sattler, W.; Gröne, H.-J. Immunohistochemical Evidence for the Myeloperoxidase/H2O2/Halide System in Human Atherosclerotic Lesions: Colocalization of Myeloperoxidase and Hypochlorite-Modified Proteins. Eur. J. Biochem. 2000, 267, 4495–4503. [Google Scholar] [CrossRef]
- Maiolino, G.; Rossitto, G.; Caielli, P.; Bisogni, V.; Rossi, G.P.; Calò, L.A. The Role of Oxidized Low-Density Lipoproteins in Atherosclerosis: The Myths and the Facts. Mediat. Inflamm. 2013, 2013, 714653. [Google Scholar] [CrossRef] [PubMed]
- Moguilevsky, N.; Zouaoui Boudjeltia, K.; Babar, S.; Delrée, P.; Legssyer, I.; Carpentier, Y.; Vanhaeverbeek, M.; Ducobu, J. Monoclonal Antibodies against LDL Progressively Oxidized by Myeloperoxidase React with ApoB-100 Protein Moiety and Human Atherosclerotic Lesions. Biochem. Biophys. Res. Commun. 2004, 323, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Myzak, M.C.; Stocker, R.; McCall, M.R.; Frei, B. Myeloperoxidase Binds to Low-Density Lipoprotein: Potential Implications for Atherosclerosis. FEBS Lett. 2000, 487, 176–180. [Google Scholar] [CrossRef]
- Delporte, C.; Boudjeltia, K.Z.; Noyon, C.; Furtmüller, P.G.; Nuyens, V.; Slomianny, M.-C.; Madhoun, P.; Desmet, J.-M.; Raynal, P.; Dufour, D.; et al. Impact of Myeloperoxidase-LDL Interactions on Enzyme Activity and Subsequent Posttranslational Oxidative Modifications of ApoB-100. J. Lipid Res. 2014, 55, 747–757. [Google Scholar] [CrossRef]
- Yang, C.; Gu, Z.-W.; Yang, M.; Lin, S.-N.; Garcia-Prats, A.J.; Rogers, L.K.; Welty, S.E.; Smith, C.V. Selective Modification of ApoB-100 in the Oxidation of Low Density Lipoproteins by Myeloperoxidase In Vitro. J. Lipid Res. 1999, 40, 686–698. [Google Scholar] [CrossRef]
- Zouaoui Boudjeltia, K.; Moguilevsky, N.; Legssyer, I.; Babar, S.; Guillaume, M.; Delree, P.; Vanhaeverbeek, M.; Brohee, D.; Ducobu, J.; Remacle, C. Oxidation of Low Density Lipoproteins by Myeloperoxidase at the Surface of Endothelial Cells: An Additional Mechanism to Subendothelium Oxidation. Biochem. Biophys. Res. Commun. 2004, 325, 434–438. [Google Scholar] [CrossRef]
- Malle, E.; Marsche, G.; Arnhold, J.; Davies, M.J. Modification of Low-Density Lipoprotein by Myeloperoxidase-Derived Oxidants and Reagent Hypochlorous Acid. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2006, 1761, 392–415. [Google Scholar] [CrossRef]
- Kodama, T.; Reddy, P.; Kishimoto, C.; Krieger, M. Purification and Characterization of a Bovine Acetyl Low Density Lipoprotein Receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 9238–9242. [Google Scholar] [CrossRef]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 Is a Receptor for Oxidized Low Density Lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An Endothelial Receptor for Oxidized Low-Density Lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Delporte, C.; Van Antwerpen, P.; Vanhamme, L.; Roumeguère, T.; Zouaoui Boudjeltia, K. Low-Density Lipoprotein Modified by Myeloperoxidase in Inflammatory Pathways and Clinical Studies. Mediat. Inflamm. 2013, 2013, 971579. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Zimmermann, R.; Horiuchi, S.; Tandon, N.N.; Sattler, W.; Malle, E. Class B Scavenger Receptors CD36 and SR-BI Are Receptors for Hypochlorite-Modified Low Density Lipoprotein. J. Biol. Chem. 2003, 278, 47562–47570. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a Novel Family of Oxidized Phospholipids That Serve as Ligands for the Macrophage Scavenger Receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.-L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I Is a Selective Target for Myeloperoxidase-Catalyzed Oxidation and Functional Impairment in Subjects with Cardiovascular Disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Q.; Zhang, Y.-Z.; Wu, Y.; Zhang, J.-J.; Li, T.-B.; Jiang, T.; Xiong, X.-M.; Luo, X.-J.; Ma, Q.-L.; Peng, J. Myeloperoxidase-Derived Hypochlorous Acid Promotes Ox-LDL-Induced Senescence of Endothelial Cells through a Mechanism Involving β-Catenin Signaling in Hyperlipidemia. Biochem. Biophys. Res. Commun. 2015, 467, 859–865. [Google Scholar] [CrossRef]
- Stadler, N.; Lindner, R.A.; Davies, M.J. Direct Detection and Quantification of Transition Metal Ions in Human Atherosclerotic Plaques: Evidence for the Presence of Elevated Levels of Iron and Copper. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef]
- Daher, J. Other Forms of Oxidized LDL: Emerging Functions (Review). World Acad. Sci. J. 2020, 2, 4. [Google Scholar] [CrossRef]
- Calay, D.; Rousseau, A.; Mattart, L.; Nuyens, V.; Delporte, C.; Van Antwerpen, P.; Moguilevsky, N.; Arnould, T.; Boudjeltia, K.Z.; Raes, M. Copper and Myeloperoxidase-Modified LDLs Activate Nrf2 through Different Pathways of ROS Production in Macrophages. Antioxid. Redox Signal. 2010, 13, 1491–1502. [Google Scholar] [CrossRef]
- Pireaux, V.; Sauvage, A.; Bihin, B.; Van Steenbrugge, M.; Rousseau, A.; Van Antwerpen, P.; Zouaoui Boudjeltia, K.; Raes, M. Myeloperoxidase-Oxidized LDLs Enhance an Anti-Inflammatory M2 and Antioxidant Phenotype in Murine Macrophages. Mediat. Inflamm. 2016, 2016, 8249476. [Google Scholar] [CrossRef]
- Zouaoui Boudjeltia, K.; Daher, J.; Van Antwerpen, P.; Moguilevsky, N.; Delree, P.; Ducobu, J.; Raes, M.; Badran, B.; Vanhaeverbeek, M.; Brohee, D.; et al. Exposure of Endothelial Cells to Physiological Levels of Myeloperoxidase-Modified LDL Delays Pericellular Fibrinolysis. PLoS ONE 2012, 7, e38810. [Google Scholar] [CrossRef] [PubMed]
- Heinloth, A.; Heermeier, K.; Raff, U.; Wanner, C.; Galle, J. Stimulation of NADPH Oxidase by Oxidized Low-Density Lipoprotein Induces Proliferation of Human Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2000, 11, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- El Samad, G.; Bazzi, S.; Karam, M.; Zouaoui Boudjeltia, K.; Vanhamme, L.; Daher, J. Effect of Myeloperoxidase Modified LDL on Bovine and Human Aortic Endothelial Cells. Exp. Ther. Med. 2019, 18, 4567–4574. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.; Martin, M.; Rousseau, A.; Nuyens, V.; Fayyad-Kazan, H.; Van Antwerpen, P.; Courbebaisse, G.; Martiat, P.; Badran, B.; Dequiedt, F.; et al. Myeloperoxidase Oxidized LDL Interferes with Endothelial Cell Motility through MiR-22 and Heme Oxygenase 1 Induction: Possible Involvement in Reendothelialization of Vascular Injuries. Mediat. Inflamm. 2014, 2014, 134635. [Google Scholar] [CrossRef]
- Dufour, D.; Khalil, A.; Nuyens, V.; Rousseau, A.; Delporte, C.; Noyon, C.; Cortese, M.; Reyé, F.; Pireaux, V.; Nève, J.; et al. Native and Myeloperoxidase-Oxidized Low-Density Lipoproteins Act in Synergy to Induce Release of Resolvin-D1 from Endothelial Cells. Atherosclerosis 2018, 272, 108–117. [Google Scholar] [CrossRef]
- El-Hajjar, L.; Hindieh, J.; Andraos, R.; El-Sabban, M.; Daher, J. Myeloperoxidase-Oxidized LDL Activates Human Aortic Endothelial Cells through the LOX-1 Scavenger Receptor. Int. J. Mol. Sci. 2022, 23, 2837. [Google Scholar] [CrossRef]
- Dufour, D.; Khalil, A.; Nuyens, V.; Rousseau, A.; Delporte, C.; Noyon, C.; Cortese, M.; Reyé, F.; Pireaux, V.; Nève, J.; et al. Data on Myeloperoxidase-Oxidized Low-Density Lipoproteins Stimulation of Cells to Induce Release of Resolvin-D1. Data Brief 2018, 18, 1160–1171. [Google Scholar] [CrossRef]
- Brennan, M.-L.; Anderson, M.M.; Shih, D.M.; Qu, X.-D.; Wang, X.; Mehta, A.C.; Lim, L.L.; Shi, W.; Hazen, S.L.; Jacob, J.S.; et al. Increased Atherosclerosis in Myeloperoxidase-Deficient Mice. J. Clin. Investig. 2001, 107, 419–430. [Google Scholar] [CrossRef]
- Rehring, J.F.; Bui, T.M.; Galán-Enríquez, C.S.; Urbanczyk, J.M.; Ren, X.; Wiesolek, H.L.; Sullivan, D.P.; Sumagin, R. Released Myeloperoxidase Attenuates Neutrophil Migration and Accumulation in Inflamed Tissue. Front. Immunol. 2021, 12, 654259. [Google Scholar] [CrossRef]
- Klinke, A.; Nussbaum, C.; Kubala, L.; Friedrichs, K.; Rudolph, T.K.; Rudolph, V.; Paust, H.-J.; Schröder, C.; Benten, D.; Lau, D.; et al. Myeloperoxidase Attracts Neutrophils by Physical Forces. Blood 2011, 117, 1350–1358. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving Inflammation: Dual Anti-Inflammatory and pro-Resolution Lipid Mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Specialized Pro-Resolving Mediator Network: An Update on Production and Actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Das, U. Bioactive Lipids in Age-Related Disorders. In Reviews on New Drug Targets in Age-Related Disorders; Advances in Experimental Medicine and Biology; Guest, P.C., Ed.; Springer International Publishing: Cham, Switzerland, 2020; Volume 1260, pp. 33–84. ISBN 978-3-030-42666-8. [Google Scholar]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2–Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in Inflammation: Emergence of the pro-Resolving Superfamily of Mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, A.T.; Fokin, V.V.; Petasis, N.A.; Serhan, C.N.; Madara, J.L. LXA4, Aspirin-Triggered 15-Epi-LXA4, and Their Analogs Selectively Downregulate PMN Azurophilic Degranulation. Am. J. Physiol.-Cell Physiol. 1999, 276, C988–C994. [Google Scholar] [CrossRef]
- Sun, Y.-P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and Its Aspirin-Triggered 17R Epimer. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 Binds Human Phagocytes with Evidence for Proresolving Receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef]
- Norling, L.V.; Dalli, J.; Flower, R.J.; Serhan, C.N.; Perretti, M. Resolvin D1 Limits Polymorphonuclear Leukocyte Recruitment to Inflammatory Loci: Receptor-Dependent Actions. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1970–1978. [Google Scholar] [CrossRef]
- Thul, S.; Labat, C.; Temmar, M.; Benetos, A.; Bäck, M. Low Salivary Resolvin D1 to Leukotriene B4 Ratio Predicts Carotid Intima Media Thickness: A Novel Biomarker of Non-Resolving Vascular Inflammation. Eur. J. Prev. Cardiol. 2017, 24, 903–906. [Google Scholar] [CrossRef]
- Salas-Hernández, A.; Espinoza-Pérez, C.; Vivar, R.; Espitia-Corredor, J.; Lillo, J.; Parra-Flores, P.; Sánchez-Ferrer, C.F.; Peiró, C.; Díaz-Araya, G. Resolvin D1 and E1 Promote Resolution of Inflammation in Rat Cardiac Fibroblast In Vitro. Mol. Biol. Rep. 2021, 48, 57–66. [Google Scholar] [CrossRef]
- Kain, V.; Halade, G.V. Immune Responsive Resolvin D1 Programs Peritoneal Macrophages and Cardiac Fibroblast Phenotypes in Diversified Metabolic Microenvironment. J. Cell. Physiol. 2019, 234, 3910–3920. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, R.; Raghavan, S.; Rao, G.N. Resolvin D1 via Prevention of ROS-Mediated SHP2 Inactivation Protects Endothelial Adherens Junction Integrity and Barrier Function. Redox Biol. 2017, 12, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 Promotes Efferocytosis in Aging by Limiting Senescent Cell-induced MerTK Cleavage. FASEB J. 2020, 34, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent Insights into the Cellular Biology of Atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Komshian, S.; Sansbury, B.E.; Wu, B.; Mottola, G.; Chen, M.; Spite, M.; Conte, M.S. Biosynthesis of Proresolving Lipid Mediators by Vascular Cells and Tissues. FASEB J. 2017, 31, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin D1 Limits 5-Lipoxygenase Nuclear Localization and Leukotriene B4 Synthesis by Inhibiting a Calcium-Activated Kinase Pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 14530–14535. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Gemperle, C.; Rimann, N.; Hersberger, M. Resolvin D1 Polarizes Primary Human Macrophages toward a Proresolution Phenotype through GPR32. J. Immunol. 2016, 196, 3429–3437. [Google Scholar] [CrossRef]
- Wu, B.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Chen, M.; Siguenza, I.O.; Desai, T.A.; Conte, M.S. Perivascular Delivery of Resolvin D1 Inhibits Neointimal Hyperplasia in a Rat Model of Arterial Injury. J. Vasc. Surg. 2017, 65, 207–217.e3. [Google Scholar] [CrossRef]
- Poreba, M.; Mostowik, M.; Siniarski, A.; Golebiowska-Wiatrak, R.; Malinowski, K.P.; Haberka, M.; Konduracka, E.; Nessler, J.; Undas, A.; Gajos, G. Treatment with High-Dose n-3 PUFAs Has No Effect on Platelet Function, Coagulation, Metabolic Status or Inflammation in Patients with Atherosclerosis and Type 2 Diabetes. Cardiovasc. Diabetol. 2017, 16, 50. [Google Scholar] [CrossRef]
- Barden, A.; Shinde, S.; Burke, V.; Puddey, I.B.; Beilin, L.J.; Irish, A.; Watts, G.; Mori, T.A. The Effect of N-3 Fatty Acids and Coenzyme Q10 Supplementation on Neutrophil Leukotrienes, Mediators of Inflammation Resolution and Myeloperoxidase in Chronic Kidney Disease. Prostaglandins Lipid Mediat. 2018, 136, 1–8. [Google Scholar] [CrossRef]
- Dalli, J.; Zhu, M.; Vlasenko, N.A.; Deng, B.; Haeggström, J.Z.; Petasis, N.A.; Serhan, C.N. The Novel 13S, 14S-epoxy-maresin Is Converted by Human Macrophages to Maresin 1 (MaR1), Inhibits Leukotriene A4 Hydrolase (LTA4H), and Shifts Macrophage Phenotype. FASEB J. 2013, 27, 2573–2583. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of Macrophage Phenotypes and Responses in Atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Waldo, S.W.; Li, Y.; Buono, C.; Zhao, B.; Billings, E.M.; Chang, J.; Kruth, H.S. Heterogeneity of Human Macrophages in Culture and in Atherosclerotic Plaques. Am. J. Pathol. 2008, 172, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.Y.; Miyoshi, H.; Kuroda, S.; Yasuda, H.; Kamiyama, K.; Nakagawara, J.; Takigami, M.; Kondo, T.; Atsumi, T. The Phenotype of Infiltrating Macrophages Influences Arteriosclerotic Plaque Vulnerability in the Carotid Artery. J. Stroke Cerebrovasc. Dis. 2013, 22, 910–918. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C.; Locati, M. Macrophage Diversity and Polarization in Atherosclerosis: A Question of Balance. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1419–1423. [Google Scholar] [CrossRef]
- Pireaux, V.; Delporte, C.; Rousseau, A.; Desmet, J.-M.; Van Antwerpen, P.; Raes, M.; Zouaoui Boudjeltia, K. M2 Monocyte Polarization in Dialyzed Patients Is Associated with Increased Levels of M-CSF and Myeloperoxidase-Associated Oxidative Stress: Preliminary Results. Biomedicines 2021, 9, 84. [Google Scholar] [CrossRef]
- Chiu, C.-Y.; Gomolka, B.; Dierkes, C.; Huang, N.R.; Schroeder, M.; Purschke, M.; Manstein, D.; Dangi, B.; Weylandt, K.H. Omega-6 Docosapentaenoic Acid-Derived Resolvins and 17-Hydroxydocosahexaenoic Acid Modulate Macrophage Function and Alleviate Experimental Colitis. Inflamm. Res. 2012, 61, 967–976. [Google Scholar] [CrossRef]
- Adamson, S.; Leitinger, N. Phenotypic Modulation of Macrophages in Response to Plaque Lipids. Curr. Opin. Lipidol. 2011, 22, 335–342. [Google Scholar] [CrossRef]
- Yao, Q.; Liu, J.; Zhang, Z.; Li, F.; Zhang, C.; Lai, B.; Xiao, L.; Wang, N. Peroxisome Proliferator–Activated Receptor γ (PPARγ) Induces the Gene Expression of Integrin AVβ5 to Promote Macrophage M2 Polarization. J. Biol. Chem. 2018, 293, 16572–16582. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a Novel Macrophage Phenotype That Develops in Response to Atherogenic Phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Specific Lipid Mediator Signatures of Human Phagocytes: Microparticles Stimulate Macrophage Efferocytosis and pro-Resolving Mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.-S.; Spite, M.; Fredman, G.; Tabas, I. MerTK Cleavage Limits Proresolving Mediator Biosynthesis and Exacerbates Tissue Inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W.; Newson, J.; Sawmynaden, P.; Willoughby, D.A.; Croxtall, J.D. A Novel Role for Phospholipase A2 Isoforms in the Checkpoint Control of Acute Inflammation. FASEB J. 2004, 18, 489–498. [Google Scholar] [CrossRef]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a Key Enzyme for Leukotriene Biosynthesis in Health and Disease. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-Associated Phospholipase A2 Protein Expression in the Natural Progression of Human Coronary Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar] [CrossRef]
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of Macrophages toward M2 Phenotype Is Favored by Reduction in IPLA2β (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangeten, C.; Zouaoui Boudjeltia, K.; Delporte, C.; Van Antwerpen, P.; Korpak, K. Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution. Antioxidants 2022, 11, 874. https://doi.org/10.3390/antiox11050874
Tangeten C, Zouaoui Boudjeltia K, Delporte C, Van Antwerpen P, Korpak K. Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution. Antioxidants. 2022; 11(5):874. https://doi.org/10.3390/antiox11050874
Chicago/Turabian StyleTangeten, Cecilia, Karim Zouaoui Boudjeltia, Cedric Delporte, Pierre Van Antwerpen, and Keziah Korpak. 2022. "Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution" Antioxidants 11, no. 5: 874. https://doi.org/10.3390/antiox11050874
APA StyleTangeten, C., Zouaoui Boudjeltia, K., Delporte, C., Van Antwerpen, P., & Korpak, K. (2022). Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution. Antioxidants, 11(5), 874. https://doi.org/10.3390/antiox11050874