
Hepatic Encephalopathy and Melatonin



Abstract
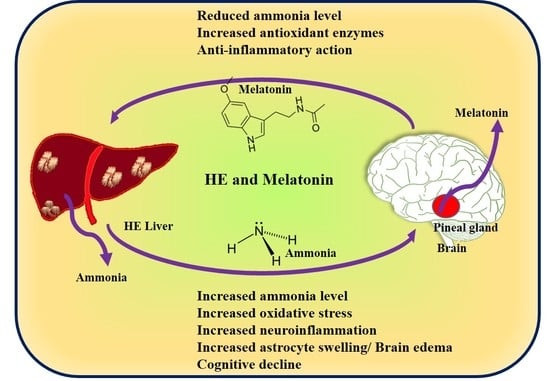
1. Introduction
2. Melatonin in the CNS
3. Hepatic Encephalopathy (HE) and Melatonin (Hyperammonemia)
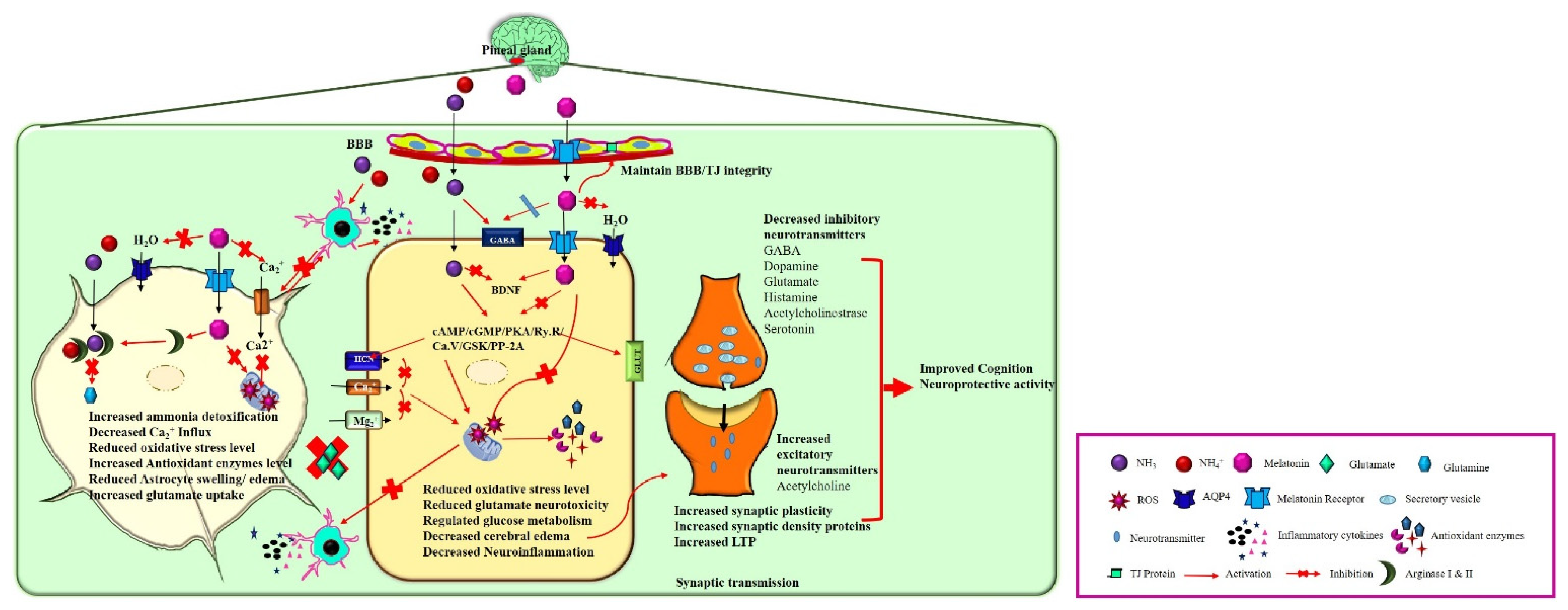
4. HE and Melatonin (Neuroinflammation and BBB Disruption)
5. HE and Melatonin (Neurotransmitters)
6. HE and Melatonin (Insulin Resistance)
7. HE and Melatonin (Cognitive Function)
8. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Zhang, J.J.; Meng, X.; Li, Y.; Zhou, Y.; Xu, D.P.; Li, S.; Li, H.B. Effects of Melatonin on Liver Injuries and Diseases. Int. J. Mol. Sci. 2017, 18, 673. [Google Scholar] [CrossRef] [PubMed]
- Ciecko-Michalska, I.; Szczepanek, M.; Slowik, A.; Mach, T. Pathogenesis of hepatic encephalopathy. Gastroenterol. Res. Pract. 2012, 2012, 642108. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.Y.; Song, J. The Association between Hepatic Encephalopathy and Diabetic Encephalopathy: The Brain-Liver Axis. Int. J. Mol. Sci. 2021, 22, 463. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Norenberg, M.D.; Felipo, V.; Ferenci, P.; Albrecht, J.; Blei, A.T.; ISHEN Commission on Experimental Models of HE. Experimental models of hepatic encephalopathy: ISHEN guidelines. Liver Int. 2009, 29, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Hepatic encephalopathy. Gastroenterol. Rep. 2017, 5, 138–147. [Google Scholar] [CrossRef]
- Stinton, L.M.; Jayakumar, S. Minimal hepatic encephalopathy. Can J. Gastroenterol. 2013, 27, 572–574. [Google Scholar] [CrossRef]
- Khungar, V.; Poordad, F. Management of overt hepatic encephalopathy. Clin. Liver Dis. 2012, 16, 73–89. [Google Scholar] [CrossRef]
- Garcia-Garcia, R.; Cruz-Gomez, A.J.; Urios, A.; Mangas-Losada, A.; Forn, C.; Escudero-Garcia, D.; Kosenko, E.; Torregrosa, I.; Tosca, J.; Giner-Duran, R.; et al. Learning and Memory Impairments in Patients with Minimal Hepatic Encephalopathy are Associated with Structural and Functional Connectivity Alterations in Hippocampus. Sci. Rep. 2018, 8, 9664. [Google Scholar] [CrossRef]
- Felipo, V.; Butterworth, R.F. Neurobiology of ammonia. Prog. Neurobiol. 2002, 67, 259–279. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. A model of blood-ammonia homeostasis based on a quantitative analysis of nitrogen metabolism in the multiple organs involved in the production, catabolism, and excretion of ammonia in humans. Clin. Exp. Gastroenterol. 2018, 11, 193–215. [Google Scholar] [CrossRef]
- Olde Damink, S.W.; Deutz, N.E.; Dejong, C.H.; Soeters, P.B.; Jalan, R. Interorgan ammonia metabolism in liver failure. Neurochem. Int. 2002, 41, 177–188. [Google Scholar] [CrossRef]
- Sorensen, M. Update on cerebral uptake of blood ammonia. Metab. Brain Dis. 2013, 28, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, A.; Fernandez, M.A. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann. Hepatol. 2009, 8, 95–102. [Google Scholar] [CrossRef]
- Montoliu, C.; Piedrafita, B.; Serra, M.A.; del Olmo, J.A.; Urios, A.; Rodrigo, J.M.; Felipo, V. IL-6 and IL-18 in blood may discriminate cirrhotic patients with and without minimal hepatic encephalopathy. J. Clin. Gastroenterol. 2009, 43, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Palomero-Gallagher, N.; Bidmon, H.J.; Cremer, M.; Schleicher, A.; Kircheis, G.; Reifenberger, G.; Kostopoulos, G.; Haussinger, D.; Zilles, K. Neurotransmitter receptor imbalances in motor cortex and basal ganglia in hepatic encephalopathy. Cell Physiol. Biochem. 2009, 24, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Norenberg, M.D. Brain energy metabolism and mitochondrial dysfunction in acute and chronic hepatic encephalopathy. Neurochem. Int. 2012, 60, 697–706. [Google Scholar] [CrossRef]
- Gorg, B.; Karababa, A.; Haussinger, D. Hepatic Encephalopathy and Astrocyte Senescence. J. Clin. Exp. Hepatol. 2018, 8, 294–300. [Google Scholar] [CrossRef]
- Bosoi, C.R.; Zwingmann, C.; Marin, H.; Parent-Robitaille, C.; Huynh, J.; Tremblay, M.; Rose, C.F. Increased brain lactate is central to the development of brain edema in rats with chronic liver disease. J. Hepatol. 2014, 60, 554–560. [Google Scholar] [CrossRef]
- Patidar, K.R.; Bajaj, J.S. Antibiotics for the treatment of hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 307–312. [Google Scholar] [CrossRef]
- Bemeur, C.; Desjardins, P.; Butterworth, R.F. Role of nutrition in the management of hepatic encephalopathy in end-stage liver failure. J. Nutr. Metab. 2010, 2010, 489823. [Google Scholar] [CrossRef]
- Zoratti, C.; Moretti, R.; Rebuzzi, L.; Albergati, I.V.; Di Somma, A.; Decorti, G.; Di Bella, S.; Croce, L.S.; Giuffre, M. Antibiotics and Liver Cirrhosis: What the Physicians Need to Know. Antibiotics 2021, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Neurosteroids in hepatic encephalopathy: Novel insights and new therapeutic opportunities. J. Steroid Biochem. Mol. Biol. 2016, 160, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, S.; Summerskill, W.H.; White, L.P.; Phear, E.A. Portal-systemic encephalopathy; neurological complications of liver disease. Lancet 1954, 267, 454–457. [Google Scholar] [CrossRef]
- Velissaris, D.; Karamouzos, V.; Polychronopoulos, P.; Karanikolas, M. Chronotypology and melatonin alterations in minimal hepatic encephalopathy. J. Circadian Rhythm. 2009, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.D.F.; Mellanby, R.J.; Gow, A.G. Serum melatonin in dogs with congenital portosystemic shunting, with and without hepatic encephalopathy. Vet. Rec. 2020, 187, e23. [Google Scholar] [CrossRef]
- Amaral, F.G.D.; Cipolla-Neto, J. A brief review about melatonin, a pineal hormone. Arch. Endocrinol. Metab. 2018, 62, 472–479. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Role and Therapeutic Potential of Melatonin in the Central Nervous System and Cancers. Cancers 2020, 12, 1567. [Google Scholar] [CrossRef]
- Claustrat, B.; Brun, J.; Chazot, G. The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 2005, 9, 11–24. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Zheng, H.; Ho, J.; Chan, M.T.; Wu, W.K. Protective roles of melatonin in central nervous system diseases by regulation of neural stem cells. Cell Prolif. 2017, 50, e12323. [Google Scholar] [CrossRef]
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.H.; Chen, Y.C.; Sheen, J.M.; Li, S.W.; Huang, L.T. Melatonin prevented spatial deficits and increases in brain asymmetric dimethylarginine in young bile duct ligation rats. Neuroreport 2018, 29, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Tiao, M.M.; Tain, Y.L.; Chen, C.C.; Hsieh, C.S. Melatonin ameliorates bile duct ligation-induced systemic oxidative stress and spatial memory deficits in developing rats. Pediatr. Res. 2009, 65, 176–180. [Google Scholar] [CrossRef]
- Zhao, L.; An, R.; Yang, Y.; Yang, X.; Liu, H.; Yue, L.; Li, X.; Lin, Y.; Reiter, R.J.; Qu, Y. Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: The role of SIRT1 signaling. J. Pineal. Res. 2015, 59, 230–239. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, H.; Yue, L.; Zhang, J.; Li, X.; Wang, B.; Lin, Y.; Qu, Y. Melatonin Attenuates Early Brain Injury via the Melatonin Receptor/Sirt1/NF-kappaB Signaling Pathway Following Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2017, 54, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Babaee, A.; Eftekhar-Vaghefi, S.H.; Asadi-Shekaari, M.; Shahrokhi, N.; Soltani, S.D.; Malekpour-Afshar, R.; Basiri, M. Melatonin treatment reduces astrogliosis and apoptosis in rats with traumatic brain injury. Iran J. Basic Med. Sci. 2015, 18, 867–872. [Google Scholar] [PubMed]
- Sinha, B.; Wu, Q.; Li, W.; Tu, Y.; Sirianni, A.C.; Chen, Y.; Jiang, J.; Zhang, X.; Chen, W.; Zhou, S.; et al. Protection of melatonin in experimental models of newborn hypoxic-ischemic brain injury through MT1 receptor. J. Pineal. Res. 2018, 64, e12443. [Google Scholar] [CrossRef]
- Das, A.; Belagodu, A.; Reiter, R.J.; Ray, S.K.; Banik, N.L. Cytoprotective effects of melatonin on C6 astroglial cells exposed to glutamate excitotoxicity and oxidative stress. J. Pineal. Res. 2008, 45, 117–124. [Google Scholar] [CrossRef]
- Khan, A.; Ayub, M.; Khan, W.M. Hyperammonemia Is Associated with Increasing Severity of Both Liver Cirrhosis and Hepatic Encephalopathy. Int. J. Hepatol. 2016, 2016, 6741754. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Norenberg, M.D. Hyperammonemia in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2018, 8, 272–280. [Google Scholar] [CrossRef]
- Mohiuddin, S.S.; Khattar, D. Biochemistry, Ammonia. In StatPearls; StatPearls Publisher: Treasure Island, FL, USA, 2022. [Google Scholar]
- Choi, J.M.; Kim, Y.H.; Roh, S.Y. Acute hepatic encephalopathy presenting as cortical laminar necrosis: Case report. Korean J. Radiol. 2013, 14, 324–328. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olde Damink, S.W.; Jalan, R.; Redhead, D.N.; Hayes, P.C.; Deutz, N.E.; Soeters, P.B. Interorgan ammonia and amino acid metabolism in metabolically stable patients with cirrhosis and a TIPSS. Hepatology 2002, 36, 1163–1171. [Google Scholar] [CrossRef]
- Matsuda, I.; Matsuura, T.; Hoshide, R.; Uchino, T.; Matsubasa, T. Molecular basis of urea cycle disorders. Nihon Rinsho. 1993, 51, 520–524. [Google Scholar]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A Multifaceted Enzyme Important in Health and Disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Aydogdu, N.; Erbas, H.; Atmaca, G.; Erten, O.; Kaymak, K. Melatonin reduces nitric oxide via increasing arginase in rhabdomyolysis-induced acute renal failure in rats. Ren. Fail. 2006, 28, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, A.; Viegas, C.M.; Tonin, A.M.; Busanello, E.N.; Grings, M.; Moura, A.P.; Leipnitz, G.; Wajner, M. Disturbance of redox homeostasis by ornithine and homocitrulline in rat cerebellum: A possible mechanism of cerebellar dysfunction in HHH syndrome. Life Sci. 2013, 93, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Rose, C.F. Oxidative stress: A systemic factor implicated in the pathogenesis of hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 175–178. [Google Scholar] [CrossRef]
- Tunez, I.; Munoz, M.C.; Villavicencio, M.A.; Medina, F.J.; de Prado, E.P.; Espejo, I.; Barcos, M.; Salcedo, M.; Feijoo, M.; Montilla, P. Hepato- and neurotoxicity induced by thioacetamide: Protective effects of melatonin and dimethylsulfoxide. Pharmacol. Res. 2005, 52, 223–228. [Google Scholar] [CrossRef]
- Ochoa-Sanchez, R.; Rose, C.F. Pathogenesis of Hepatic Encephalopathy in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 262–271. [Google Scholar] [CrossRef]
- Tunez, I.; Munoz, M.C.; Medina, F.J.; Salcedo, M.; Feijoo, M.; Montilla, P. Comparison of melatonin, vitamin E and L-carnitine in the treatment of neuro- and hepatotoxicity induced by thioacetamide. Cell Biochem. Funct. 2007, 25, 119–127. [Google Scholar] [CrossRef]
- Lena, P.J.; Subramanian, P. Effects of melatonin on the levels of antioxidants and lipid peroxidation products in rats treated with ammonium acetate. Pharmazie 2004, 59, 636–639. [Google Scholar] [PubMed]
- Lena, P.J.; Subramanian, P. Evaluation of the antiperoxidative effects of melatonin in ammonium acetate-treated Wistar rats. Pol. J. Pharmacol. 2003, 55, 1031–1036. [Google Scholar]
- Morvaridzadeh, M.; Sadeghi, E.; Agah, S.; Nachvak, S.M.; Fazelian, S.; Moradi, F.; Persad, E.; Heshmati, J. Effect of melatonin supplementation on oxidative stress parameters: A systematic review and meta-analysis. Pharmacol. Res. 2020, 161, 105210. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Yang, Q.; Liu, Y.; Zhou, S.; Jiang, J.; Reiter, R.J.; Bhattacharya, P.; Cui, Y.; Yang, H.; Ma, H.; et al. The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic. Biol. Med. 2019, 130, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015, 127–128, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Montoliu, C.; Cauli, O.; Urios, A.; ElMlili, N.; Serra, M.A.; Giner-Duran, R.; Gonzalez-Lopez, O.; Del Olmo, J.A.; Wassel, A.; Rodrigo, J.M.; et al. 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am. J. Gastroenterol. 2011, 106, 1629–1637. [Google Scholar] [CrossRef]
- Cimen, B.; Turkozkan, N.; Unlu, A.; Erbil, M.K. Effects of melatonin on 3-nitrotyrosine formation and energy charge ratio in guinea pig kidney in LPS-induced stress. Cell Biochem. Funct. 2005, 23, 273–277. [Google Scholar] [CrossRef]
- Yin, J.; Liu, Y.H.; Xu, Y.F.; Zhang, Y.J.; Chen, J.G.; Shu, B.H.; Wang, J.Z. Melatonin arrests peroxynitrite-induced tau hyperphosphorylation and the overactivation of protein kinases in rat brain. J. Pineal. Res. 2006, 41, 124–129. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin metabolism in the central nervous system. Curr. Neuropharmacol. 2010, 8, 168–181. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Rama Rao, K.V.; Norenberg, M.D. Neuroinflammation in hepatic encephalopathy: Mechanistic aspects. J. Clin. Exp. Hepatol. 2015, 5, S21–S28. [Google Scholar] [CrossRef]
- Butterworth, R.F. Altered glial-neuronal crosstalk: Cornerstone in the pathogenesis of hepatic encephalopathy. Neurochem. Int. 2010, 57, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Sheeler, C.; Rosa, J.G.; Ferro, A.; McAdams, B.; Borgenheimer, E.; Cvetanovic, M. Glia in Neurodegeneration: The Housekeeper, the Defender and the Perpetrator. Int. J. Mol. Sci. 2020, 21, 9188. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Ott, P.; Larsen, F.S. Blood-brain barrier permeability to ammonia in liver failure: A critical reappraisal. Neurochem. Int. 2004, 44, 185–198. [Google Scholar] [CrossRef]
- Claeys, W.; Van Hoecke, L.; Lefere, S.; Geerts, A.; Verhelst, X.; Van Vlierberghe, H.; Degroote, H.; Devisscher, L.; Vandenbroucke, R.E.; Van Steenkiste, C. The neurogliovascular unit in hepatic encephalopathy. JHEP Rep. 2021, 3, 100352. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J. The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem. Res. 2012, 37, 2439–2455. [Google Scholar] [CrossRef] [PubMed]
- Goldbecker, A.; Buchert, R.; Berding, G.; Bokemeyer, M.; Lichtinghagen, R.; Wilke, F.; Ahl, B.; Weissenborn, K. Blood-brain barrier permeability for ammonia in patients with different grades of liver fibrosis is not different from healthy controls. J. Cereb. Blood Flow Metab. 2010, 30, 1384–1393. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Norenberg, M.D. Glutamine in the pathogenesis of hepatic encephalopathy: The trojan horse hypothesis revisited. Neurochem. Res. 2014, 39, 593–598. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Liu, M.; Moriyama, M.; Ramakrishnan, R.; Forbush, B., 3rd; Reddy, P.V.; Norenberg, M.D. Na-K-Cl Cotransporter-1 in the mechanism of ammonia-induced astrocyte swelling. J. Biol. Chem. 2008, 283, 33874–33882. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of astrocyte-mediated cerebral edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Gorg, B.; Becker, S.; Qvartskhava, N.; Bidmon, H.J.; Selbach, O.; Haas, H.L.; Schliess, F.; Haussinger, D. Hypoosmotic swelling and ammonia increase oxidative stress by NADPH oxidase in cultured astrocytes and vital brain slices. Glia 2007, 55, 758–771. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Tong, X.Y.; Ospel, J.; Norenberg, M.D. Role of cerebral endothelial cells in the astrocyte swelling and brain edema associated with acute hepatic encephalopathy. Neuroscience 2012, 218, 305–316. [Google Scholar] [CrossRef]
- Wright, G.; Jalan, R. Ammonia and inflammation in the pathogenesis of hepatic encephalopathy: Pandora’s box? Hepatology 2007, 46, 291–294. [Google Scholar] [CrossRef]
- Mehrotra, A.; Trigun, S.K. Moderate grade hyperammonemia activates lactate dehydrogenase-4 and 6-phosphofructo-2-kinase to support increased lactate turnover in the brain slices. Mol. Cell Biochem. 2013, 381, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Desjardins, P.; Chatauret, N.; Butterworth, R.F. Loss of expression of glial fibrillary acidic protein in acute hyperammonemia. Neurochem. Int. 2002, 41, 155–160. [Google Scholar] [CrossRef]
- Belanger, M.; Desjardins, P.; Chatauret, N.; Butterworth, R.F. Selectively increased expression of the astrocytic/endothelial glucose transporter protein GLUT1 in acute liver failure. Glia 2006, 53, 557–562. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Norenberg, M.D. Aquaporin-4 in hepatic encephalopathy. Metab. Brain Dis. 2007, 22, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rabaza, V.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Agusti, A.; Llansola, M.; Felipo, V. Hyperammonemia induces glial activation, neuroinflammation and alters neurotransmitter receptors in hippocampus, impairing spatial learning: Reversal by sulforaphane. J. Neuroinflamm. 2016, 13, 41. [Google Scholar] [CrossRef]
- Gorg, B.; Schliess, F.; Haussinger, D. Osmotic and oxidative/nitrosative stress in ammonia toxicity and hepatic encephalopathy. Arch. Biochem. Biophys. 2013, 536, 158–163. [Google Scholar] [CrossRef]
- Pierzchala, K.; Simicic, D.; Sienkiewicz, A.; Sessa, D.; Mitrea, S.; Braissant, O.; McLin, V.A.; Gruetter, R.; Cudalbu, C. Central nervous system and systemic oxidative stress interplay with inflammation in a bile duct ligation rat model of type C hepatic encephalopathy. Free Radic. Biol. Med. 2022, 178, 295–307. [Google Scholar] [CrossRef]
- Genovese, T.; Mazzon, E.; Muia, C.; Bramanti, P.; De Sarro, A.; Cuzzocrea, S. Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J. Pineal. Res. 2005, 38, 198–208. [Google Scholar] [CrossRef]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef]
- Ortiz, F.; Acuna-Castroviejo, D.; Doerrier, C.; Dayoub, J.C.; Lopez, L.C.; Venegas, C.; Garcia, J.A.; Lopez, A.; Volt, H.; Luna-Sanchez, M.; et al. Melatonin blunts the mitochondrial/NLRP3 connection and protects against radiation-induced oral mucositis. J. Pineal. Res. 2015, 58, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Cuzzocrea, S. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010, 8, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Permpoonputtana, K.; Govitrapong, P. The anti-inflammatory effect of melatonin on methamphetamine-induced proinflammatory mediators in human neuroblastoma dopamine SH-SY5Y cell lines. Neurotox. Res. 2013, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, Z.; Pan, S.; Zhang, H.; Fang, M.; Jiang, H.; Zhang, H.; Gao, Z.; Xu, K.; Li, Z.; et al. Melatonin protects against blood-brain barrier damage by inhibiting the TLR4/ NF-kappaB signaling pathway after LPS treatment in neonatal rats. Oncotarget 2017, 8, 31638–31654. [Google Scholar] [CrossRef] [PubMed]
- Gorg, B.; Qvartskhava, N.; Keitel, V.; Bidmon, H.J.; Selbach, O.; Schliess, F.; Haussinger, D. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology 2008, 48, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Galland, F.; Negri, E.; Da Re, C.; Froes, F.; Strapazzon, L.; Guerra, M.C.; Tortorelli, L.S.; Goncalves, C.A.; Leite, M.C. Hyperammonemia compromises glutamate metabolism and reduces BDNF in the rat hippocampus. Neurotoxicology 2017, 62, 46–55. [Google Scholar] [CrossRef]
- Rose, C. Increased extracellular brain glutamate in acute liver failure: Decreased uptake or increased release? Metab. Brain Dis. 2002, 17, 251–261. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Palomero-Gallagher, N.; Zilles, K. Neurotransmitter receptor alterations in hepatic encephalopathy: A review. Arch. Biochem. Biophys. 2013, 536, 109–121. [Google Scholar] [CrossRef]
- Vaquero, J.; Butterworth, R.F. The brain glutamate system in liver failure. J. Neurochem. 2006, 98, 661–669. [Google Scholar] [CrossRef]
- Escames, G.; Macias, M.; Leon, J.; Garcia, J.; Khaldy, H.; Martin, M.; Vives, F.; Acuna-Castroviejo, D. Calcium-dependent effects of melatonin inhibition of glutamatergic response in rat striatum. J. Neuroendocrinol. 2001, 13, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; McDowell, M.; Pava, M.J.; Smith, J.A.; Reiter, R.J.; Woodward, J.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-alpha toxicity involves membrane melatonin receptors. J. Pineal. Res. 2010, 48, 157–169. [Google Scholar] [CrossRef]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate Excitotoxicity and Oxidative Stress in Epilepsy: Modulatory Role of Melatonin. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 2002, 40, 475–486. [Google Scholar] [CrossRef]
- Yanase, H.; Shimizu, H.; Yamada, K.; Iwanaga, T. Cellular localization of the diazepam binding inhibitor in glial cells with special reference to its coexistence with brain-type fatty acid binding protein. Arch. Histol. Cytol. 2002, 65, 27–36. [Google Scholar] [CrossRef]
- Butterworth, R.F. Hepatic encephalopathy: A central neuroinflammatory disorder? Hepatology 2011, 53, 1372–1376. [Google Scholar] [CrossRef]
- Hazell, A.S.; Normandin, L.; Nguyen, B.; Kennedy, G. Upregulation of ‘peripheral-type’ benzodiazepine receptors in the globus pallidus in a sub-acute rat model of manganese neurotoxicity. Neurosci. Lett. 2003, 349, 13–16. [Google Scholar] [CrossRef]
- Haussinger, D. Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1187–1190. [Google Scholar] [CrossRef]
- Reddy, D.S. Neurosteroids: Endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 2010, 186, 113–137. [Google Scholar] [CrossRef]
- Garcia-Ayllon, M.S.; Cauli, O.; Silveyra, M.X.; Rodrigo, R.; Candela, A.; Compan, A.; Jover, R.; Perez-Mateo, M.; Martinez, S.; Felipo, V.; et al. Brain cholinergic impairment in liver failure. Brain 2008, 131, 2946–2956. [Google Scholar] [CrossRef]
- Li, F.; Endo, T.; Isa, T. Presynaptic muscarinic acetylcholine receptors suppress GABAergic synaptic transmission in the intermediate grey layer of mouse superior colliculus. Eur. J. Neurosci. 2004, 20, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Pakala, R.S.; Brown, K.N.; Preuss, C.V. Cholinergic Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Kalsbeek, A.; Garidou, M.L.; Palm, I.F.; Van Der Vliet, J.; Simonneaux, V.; Pevet, P.; Buijs, R.M. Melatonin sees the light: Blocking GABA-ergic transmission in the paraventricular nucleus induces daytime secretion of melatonin. Eur. J. Neurosci. 2000, 12, 3146–3154. [Google Scholar] [CrossRef]
- Rosenstein, R.E.; Cardinali, D.P. Central gabaergic mechanisms as targets for melatonin activity in brain. Neurochem. Int. 1990, 17, 373–379. [Google Scholar] [CrossRef]
- Prada, C.; Udin, S.B.; Wiechmann, A.F.; Zhdanova, I.V. Stimulation of melatonin receptors decreases calcium levels in xenopus tectal cells by activating GABA(C) receptors. J. Neurophysiol. 2005, 94, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.P.; Sun, H.; Ye, Z.Y.; Zhou, J.N. Melatonin modulates the GABAergic response in cultured rat hippocampal neurons. J. Pharmacol. Sci. 2012, 119, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Leng, Z. Melatonin inhibits GABAergic neurons in the hypothalamus consistent with a reduction in wakefulness. Neuroreport 2020, 31, 92–98. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Campillo, N.E.; Paez, J.A.; Gonzalez-Munoz, G.C.; Usan, P.; Garcia-Palomero, E.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; et al. Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antioxidant, and neuroprotective properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef]
- Sperlagh, B.; Vizi, E.S. The role of extracellular adenosine in chemical neurotransmission in the hippocampus and Basal Ganglia: Pharmacological and clinical aspects. Curr. Top. Med. Chem. 2011, 11, 1034–1046. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastiao, A.M.; de Mendonca, A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog. Neurobiol. 2002, 68, 377–392. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferre, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef]
- al Mardini, H.; Harrison, E.J.; Ince, P.G.; Bartlett, K.; Record, C.O. Brain indoles in human hepatic encephalopathy. Hepatology 1993, 17, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Lozeva, V.; Montgomery, J.A.; Tuomisto, L.; Rocheleau, B.; Pannunzio, M.; Huet, P.M.; Butterworth, R.F. Increased brain serotonin turnover correlates with the degree of shunting and hyperammonemia in rats following variable portal vein stenosis. J. Hepatol. 2004, 40, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Caride, A.; Pereiro, N.; Lafuente, A. Modulatory effects of melatonin on cadmium-induced changes in biogenic amines in rat hypothalamus. Neurotox. Res. 2011, 20, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Miguez, J.M.; Martin, F.J.; Aldegunde, M. Effects of single doses and daily melatonin treatments on serotonin metabolism in rat brain regions. J. Pineal. Res. 1994, 17, 170–176. [Google Scholar] [CrossRef]
- Agrawal, R.; Tyagi, E.; Shukla, R.; Nath, C. Effect of insulin and melatonin on acetylcholinesterase activity in the brain of amnesic mice. Behav. Brain Res. 2008, 189, 381–386. [Google Scholar] [CrossRef]
- Als-Nielsen, B.; Gluud, L.L.; Gluud, C. Dopaminergic agonists for hepatic encephalopathy. Cochrane Database Syst. Rev. 2004, CD003047. [Google Scholar] [CrossRef]
- Dhanda, S.; Sandhir, R. Role of dopaminergic and serotonergic neurotransmitters in behavioral alterations observed in rodent model of hepatic encephalopathy. Behav. Brain Res. 2015, 286, 222–235. [Google Scholar] [CrossRef]
- Chen, B.; Yang, Y.; Li, S.; Zhu, X.; Qi, Y.; Hong, F. The critical role of hippocampal dopamine in the pathogenesis of hepatic encephalopathy. Physiol. Res. 2021, 70, 101–110. [Google Scholar] [CrossRef]
- Junker, A.E.; Als-Nielsen, B.; Gluud, C.; Gluud, L.L. Dopamine agents for hepatic encephalopathy. Cochrane Database Syst. Rev. 2014, CD003047. [Google Scholar] [CrossRef]
- Zisapel, N. Melatonin-dopamine interactions: From basic neurochemistry to a clinical setting. Cell Mol. Neurobiol. 2001, 21, 605–616. [Google Scholar] [CrossRef]
- Alexiuk, N.A.; Vriend, J.P. Melatonin reduces dopamine content in the neurointermediate lobe of male Syrian hamsters. Brain Res. Bull. 1993, 32, 433–436. [Google Scholar] [CrossRef]
- Roberts, B.M.; Lopes, E.F.; Cragg, S.J. Axonal Modulation of Striatal Dopamine Release by Local gamma-Aminobutyric Acid (GABA) Signalling. Cells 2021, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Tenn, C.C.; Niles, L.P. Mechanisms underlying the antidopaminergic effect of clonazepam and melatonin in striatum. Neuropharmacology 1997, 36, 1659–1663. [Google Scholar] [CrossRef]
- Tenn, C.C.; Niles, L.P. Central-type benzodiazepine receptors mediate the antidopaminergic effect of clonazepam and melatonin in 6-hydroxydopamine lesioned rats: Involvement of a GABAergic mechanism. J. Pharmacol. Exp. Ther. 1995, 274, 84–89. [Google Scholar]
- Undieh, A.S. Pharmacology of signaling induced by dopamine D(1)-like receptor activation. Pharmacol. Ther. 2010, 128, 37–60. [Google Scholar] [CrossRef]
- Lozeva, V.; Tuomisto, L.; Tarhanen, J.; Butterworth, R.F. Increased concentrations of histamine and its metabolite, tele-methylhistamine and down-regulation of histamine H3 receptor sites in autopsied brain tissue from cirrhotic patients who died in hepatic coma. J. Hepatol. 2003, 39, 522–527. [Google Scholar] [CrossRef]
- Pham, L.; Baiocchi, L.; Kennedy, L.; Sato, K.; Meadows, V.; Meng, F.; Huang, C.K.; Kundu, D.; Zhou, T.; Chen, L.; et al. The interplay between mast cells, pineal gland, and circadian rhythm: Links between histamine, melatonin, and inflammatory mediators. J. Pineal. Res. 2021, 70, e12699. [Google Scholar] [CrossRef]
- Silva, C.L.; Tamura, E.K.; Macedo, S.M.; Cecon, E.; Bueno-Alves, L.; Farsky, S.H.; Ferreira, Z.S.; Markus, R.P. Melatonin inhibits nitric oxide production by microvascular endothelial cells in vivo and in vitro. Br. J. Pharmacol. 2007, 151, 195–205. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Damberg, G.S.; Tkac, I.; Gruetter, R. The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 2001, 50, 2203–2209. [Google Scholar] [CrossRef]
- Plum, L.; Belgardt, B.F.; Bruning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Investig. 2006, 116, 1761–1766. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, and dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M.; Alzheimer’s Disease Neuroimaging, I. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- Ampuero, J.; Ranchal, I.; del Mar Diaz-Herrero, M.; del Campo, J.A.; Bautista, J.D.; Romero-Gomez, M. Role of diabetes mellitus on hepatic encephalopathy. Metab. Brain Dis. 2013, 28, 277–279. [Google Scholar] [CrossRef]
- Machado, M.C.; Pinheiro da Silva, F. Hyperammonemia due to urea cycle disorders: A potentially fatal condition in the intensive care setting. J. Intensiv. Care 2014, 2, 22. [Google Scholar] [CrossRef]
- Alfadhel, M.; Mutairi, F.A.; Makhseed, N.; Jasmi, F.A.; Al-Thihli, K.; Al-Jishi, E.; AlSayed, M.; Al-Hassnan, Z.N.; Al-Murshedi, F.; Haberle, J.; et al. Guidelines for acute management of hyperammonemia in the Middle East region. Ther. Clin. Risk Manag. 2016, 12, 479–487. [Google Scholar] [CrossRef]
- Kelly, A.; Ng, D.; Ferry, R.J., Jr.; Grimberg, A.; Koo-McCoy, S.; Thornton, P.S.; Stanley, C.A. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3724–3728. [Google Scholar] [CrossRef]
- Visek, W.J. Ammonia: Its effects on biological systems, metabolic hormones, and reproduction. J. Dairy Sci. 1984, 67, 481–498. [Google Scholar] [CrossRef]
- Ivanovski, I.; Jesic, M.; Ivanovski, A.; Garavelli, L.; Ivanovski, P. Metabolically based liver damage pathophysiology in patients with urea cycle disorders—A new hypothesis. World J. Gastroenterol. 2017, 23, 7930–7938. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, H.; Ahmad, N.; Mishra, P.; Tiwari, A. The role of melatonin in diabetes: Therapeutic implications. Arch. Endocrinol. Metab. 2015, 59, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Muhlbauer, E.; Musshoff, U.; Csernus, V.J.; Chankiewitz, E.; Peschke, D. Receptor (MT(1)) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J. Pineal. Res. 2002, 33, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, I.; Bazwinsky, I.; Peschke, E. Modulation of the cGMP signaling pathway by melatonin in pancreatic beta-cells. J. Pineal. Res. 2009, 46, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.G.; Wolgast, S.; Muhlbauer, E.; Peschke, E. Melatonin stimulates inositol-1,4,5-trisphosphate and Ca2+ release from INS1 insulinoma cells. J. Pineal. Res. 2005, 39, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Peschke, E.; Bach, A.G.; Muhlbauer, E. Parallel signaling pathways of melatonin in the pancreatic beta-cell. J. Pineal. Res. 2006, 40, 184–191. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef]
- Liu, S.; Guo, Y.; Yuan, Q.; Pan, Y.; Wang, L.; Liu, Q.; Wang, F.; Wang, J.; Hao, A. Melatonin prevents neural tube defects in the offspring of diabetic pregnancy. J. Pineal. Res. 2015, 59, 508–517. [Google Scholar] [CrossRef]
- Ronn, T.; Wen, J.; Yang, Z.; Lu, B.; Du, Y.; Groop, L.; Hu, R.; Ling, C. A common variant in MTNR1B, encoding melatonin receptor 1B, is associated with type 2 diabetes and fasting plasma glucose in Han Chinese individuals. Diabetologia 2009, 52, 830–833. [Google Scholar] [CrossRef]
- Staiger, H.; Machicao, F.; Schafer, S.A.; Kirchhoff, K.; Kantartzis, K.; Guthoff, M.; Silbernagel, G.; Stefan, N.; Haring, H.U.; Fritsche, A. Polymorphisms within the novel type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS ONE 2008, 3, e3962. [Google Scholar] [CrossRef]
- Prokopenko, I.; Langenberg, C.; Florez, J.C.; Saxena, R.; Soranzo, N.; Thorleifsson, G.; Loos, R.J.; Manning, A.K.; Jackson, A.U.; Aulchenko, Y.; et al. Variants in MTNR1B influence fasting glucose levels. Nat. Genet. 2009, 41, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Amiry-Moghaddam, M.; Bergersen, L.H.; Bjaalie, J.G.; Eriksson, J.; Gundersen, V.; Leergaard, T.B.; Morth, J.P.; Storm-Mathisen, J.; Torp, R.; et al. The glia doctrine: Addressing the role of glial cells in healthy brain ageing. Mech. Ageing Dev. 2013, 134, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Haeger, P.; Bouchet, A.; Ossandon, C.; Bresky, G. Treatment with Melatonin Improves Cognitive Behavior and Motor Skills in a Rat Model of Liver Fibrosis. Ann. Hepatol. 2019, 18, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Brodersen, C.; Koen, E.; Ponte, A.; Sanchez, S.; Segal, E.; Chiapella, A.; Fernandez, M.; Torres, M.; Tripodi, V.; Lemberg, A. Cognitive function in patients with alcoholic and nonalcoholic chronic liver disease. J. Neuropsychiatry Clin. Neurosci. 2014, 26, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Schubert, C.M.; Heuman, D.M.; Wade, J.B.; Gibson, D.P.; Topaz, A.; Saeian, K.; Hafeezullah, M.; Bell, D.E.; Sterling, R.K.; et al. Persistence of cognitive impairment after resolution of overt hepatic encephalopathy. Gastroenterology 2010, 138, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Gorg, B.; Karababa, A.; Shafigullina, A.; Bidmon, H.J.; Haussinger, D. Ammonia-induced senescence in cultured rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Glia 2015, 63, 37–50. [Google Scholar] [CrossRef]
- Saba, J.; Turati, J.; Ramirez, D.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Astrocyte truncated tropomyosin receptor kinase B mediates brain-derived neurotrophic factor anti-apoptotic effect leading to neuroprotection. J. Neurochem. 2018, 146, 686–702. [Google Scholar] [CrossRef]
- Sobczyk, K.; Jordens, M.S.; Karababa, A.; Gorg, B.; Haussinger, D. Ephrin/Ephrin receptor expression in ammonia-treated rat astrocytes and in human cerebral cortex in hepatic encephalopathy. Neurochem. Res. 2015, 40, 274–283. [Google Scholar] [CrossRef]
- Gorg, B.; Karababa, A.; Schutz, E.; Paluschinski, M.; Schrimpf, A.; Shafigullina, A.; Castoldi, M.; Bidmon, H.J.; Haussinger, D. O-GlcNAcylation-dependent upregulation of HO1 triggers ammonia-induced oxidative stress and senescence in hepatic encephalopathy. J. Hepatol. 2019, 71, 930–941. [Google Scholar] [CrossRef]
- Csipo, T.; Lipecz, A.; Ashpole, N.M.; Balasubramanian, P.; Tarantini, S. Astrocyte senescence contributes to cognitive decline. Geroscience 2020, 42, 51–55. [Google Scholar] [CrossRef]
- Kennedy, M.B. Synaptic Signaling in Learning and Memory. Cold Spring Harb. Perspect. Biol. 2013, 8, a016824. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, S. From brain synapses to systems for learning and memory: Object recognition, spatial navigation, timed conditioning, and movement control. Brain Res. 2015, 1621, 270–293. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.I.; Tayler, H.M.; Love, S. Synaptic protein levels altered in vascular dementia. Neuropathol. Appl. Neurobiol. 2015, 41, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Terashima, A.; Pelkey, K.A.; Rah, J.C.; Suh, Y.H.; Roche, K.W.; Collingridge, G.L.; McBain, C.J.; Isaac, J.T. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 2008, 57, 872–882. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Ren, Y.; Yan, Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-D-aspartate receptors to calpain cleavage in cortical neurons. Mol. Pharmacol. 2008, 74, 360–370. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, T.X.; Hallett, P.J.; Watanabe, M.; Grant, S.G.; Isacson, O.; Yao, W.D. PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J. Neurosci. 2009, 29, 2948–2960. [Google Scholar] [CrossRef]
- Shah, F.A.; Liu, G.; Al Kury, L.T.; Zeb, A.; Abbas, M.; Li, T.; Yang, X.; Liu, F.; Jiang, Y.; Li, S.; et al. Melatonin Protects MCAO-Induced Neuronal Loss via NR2A Mediated Prosurvival Pathways. Front. Pharmacol. 2019, 10, 297. [Google Scholar] [CrossRef]
- Hu, B.R.; Park, M.; Martone, M.E.; Fischer, W.H.; Ellisman, M.H.; Zivin, J.A. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J. Neurosci. 1998, 18, 625–633. [Google Scholar] [CrossRef]
- Lin, L.; Huang, Q.X.; Yang, S.S.; Chu, J.; Wang, J.Z.; Tian, Q. Melatonin in Alzheimer’s disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef]
- Sumsuzzman, D.M.; Choi, J.; Jin, Y.; Hong, Y. Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer’s disease and insomnia: A systematic review and meta-analysis of randomized controlled trials. Neurosci. Biobehav. Rev. 2021, 127, 459–473. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Furio, A.M.; Brusco, L.I. Therapeutic application of melatonin in mild cognitive impairment. Am. J. Neurodegener. Dis. 2012, 1, 280–291. [Google Scholar] [PubMed]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal. Res. 2012, 52, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, L.; Ducrocq, C.; Gaudry-Talarmain, Y.M. Inhibition of acetylcholine synthesis and tyrosine nitration induced by peroxynitrite are differentially prevented by antioxidants. Mol. Pharmacol. 2001, 60, 838–846. [Google Scholar] [PubMed]
- Liu, S.J.; Wang, J.Z. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol. Sin. 2002, 23, 183–187. [Google Scholar] [PubMed]
- Wang, D.L.; Ling, Z.Q.; Cao, F.Y.; Zhu, L.Q.; Wang, J.Z. Melatonin attenuates isoproterenol-induced protein kinase A overactivation and tau hyperphosphorylation in rat brain. J. Pineal. Res. 2004, 37, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Zhang, J.; Yu, X.; Han, L.; Zhou, Z.T.; Zhang, Y.; Wang, J.Z. Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao 2005, 57, 7–12. [Google Scholar]
- Juan, W.S.; Huang, S.Y.; Chang, C.C.; Hung, Y.C.; Lin, Y.W.; Chen, T.Y.; Lee, A.H.; Lee, A.C.; Wu, T.S.; Lee, E.J. Melatonin improves neuroplasticity by upregulating the growth-associated protein-43 (GAP-43) and NMDAR postsynaptic density-95 (PSD-95) proteins in cultured neurons exposed to glutamate excitotoxicity and in rats subjected to transient focal cerebral ischemia even during a long-term recovery period. J. Pineal. Res. 2014, 56, 213–223. [Google Scholar] [CrossRef]
- Luo, Y.; Peng, M.; Wei, H. Melatonin Promotes Brain-Derived Neurotrophic Factor (BDNF) Expression and Anti-Apoptotic Effects in Neonatal Hemolytic Hyperbilirubinemia via a Phospholipase (PLC)-Mediated Mechanism. Med. Sci. Monit. 2017, 23, 5951–5959. [Google Scholar] [CrossRef]
- Sato, K.; Meng, F.; Francis, H.; Wu, N.; Chen, L.; Kennedy, L.; Zhou, T.; Franchitto, A.; Onori, P.; Gaudio, E.; et al. Melatonin and circadian rhythms in liver diseases: Functional roles and potential therapies. J. Pineal. Res. 2020, 68, e12639. [Google Scholar] [CrossRef]
- Chojnacki, C.; Blonska, A.; Chojnacki, J. The Effects of Melatonin on Elevated Liver Enzymes during Statin Treatment. Biomed. Res. Int. 2017, 2017, 3204504. [Google Scholar] [CrossRef]
- Velissaris, D.; Karanikolas, M.; Kalogeropoulos, A.; Solomou, E.; Polychronopoulos, P.; Thomopoulos, K.; Labropoulou-Karatza, C. Pituitary hormone circadian rhythm alterations in cirrhosis patients with subclinical hepatic encephalopathy. World J. Gastroenterol. 2008, 14, 4190–4195. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, M.; Cheraghpour, M.; Jafarirad, S.; Alavinejad, P.; Asadi, F.; Hekmatdoost, A.; Mohammadi, M.; Yari, Z. The effect of melatonin on treatment of patients with non-alcoholic fatty liver disease: A randomized double blind clinical trial. Complement. Ther. Med. 2020, 52, 102452. [Google Scholar] [CrossRef] [PubMed]
- Genario, R.; Cipolla-Neto, J.; Bueno, A.A.; Santos, H.O. Melatonin supplementation in the management of obesity and obesity-associated disorders: A review of physiological mechanisms and clinical applications. Pharmacol. Res. 2021, 163, 105254. [Google Scholar] [CrossRef] [PubMed]
- de Farias, T.; Cruz, M.M.; de Sa, R.; Severi, I.; Perugini, J.; Senzacqua, M.; Cerutti, S.M.; Giordano, A.; Cinti, S.; Alonso-Vale, M.I.C. Melatonin Supplementation Decreases Hypertrophic Obesity and Inflammation Induced by High-Fat Diet in Mice. Front. Endocrinol. 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed]
- Overberg, J.; Kalveram, L.; Keller, T.; Krude, H.; Kuhnen, P.; Wiegand, S. Interactions between nocturnal melatonin secretion, metabolism, and sleeping behavior in adolescents with obesity. Int. J. Obes. 2022. [Google Scholar] [CrossRef]
- Delpino, F.M.; Figueiredo, L.M. Melatonin supplementation and anthropometric indicators of obesity: A systematic review and meta-analysis. Nutrition 2021, 91–92, 111399. [Google Scholar] [CrossRef]
- Suriagandhi, V.; Nachiappan, V. Protective Effects of Melatonin against Obesity-Induced by Leptin Resistance. Behav. Brain Res. 2022, 417, 113598. [Google Scholar] [CrossRef]
- Patel, R.; Parmar, N.; Pramanik Palit, S.; Rathwa, N.; Ramachandran, A.V.; Begum, R. Diabetes mellitus and melatonin: Where are we? Biochimie 2022, in press. [Google Scholar] [CrossRef]
- Sun, H.; Huang, F.F.; Qu, S. Melatonin: A potential intervention for hepatic steatosis. Lipids Health Dis. 2015, 14, 75. [Google Scholar] [CrossRef]
- Naaz, S.; Mishra, S.; Pal, P.K.; Chattopadhyay, A.; Das, A.R.; Bandyopadhyay, D. Activation of SIRT1/PGC 1alpha/SIRT3 pathway by melatonin provides protection against mitochondrial dysfunction in isoproterenol induced myocardial injury. Heliyon 2020, 6, e05159. [Google Scholar] [CrossRef]
- Ling, L.; Alattar, A.; Tan, Z.; Shah, F.A.; Ali, T.; Alshaman, R.; Koh, P.O.; Li, S. A Potent Antioxidant Endogenous Neurohormone Melatonin, Rescued MCAO by Attenuating Oxidative Stress-Associated Neuroinflammation. Front. Pharmacol. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Tarocco, A.; Caroccia, N.; Morciano, G.; Wieckowski, M.R.; Ancora, G.; Garani, G.; Pinton, P. Melatonin as a master regulator of cell death and inflammation: Molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| No. | Model | Type of Liver Injury | Methods | Experimental Findings | References |
|---|---|---|---|---|---|
| 1 | Hepato- and neurotoxicity induced by TAA/Adult Wistar rats | Melatonin (3 mg·kg−1·day−1) TAA (150 mg·kg−1 IP) Vitamin E (20 mg·kg−1) L-carnitine (100 mg·kg−1) | Liver (AST, ALT, LDH) Kidney (urea, BUN) Brain (ammonia, GSH, LPO) | Melatonin is a potent antioxidant that protects against TAA-induced hepato- and neurotoxicity compared to vitamins C and E | (Túnez et al., 2007) [51] |
| 2 | Hepato- and neurotoxicity induced by TAA/Adult Wistar rats | Melatonin (3 mg·kg−1 day−1) TAA (150 mg·kg−1 IP) DMSO (2 g·kg−1·day−1) | Liver (AST, ALT, LDH) Kidney (urea, BUN) Brain (ammonia, GSH, LPO) | Reduced hyperammonemia. Melatonin acts as an antioxidant and exerts neuro-/hepato-protective effects against TAA-induced hepato- and neurotoxicity | (Túnez et al., 2005) [49] |
| 3 | Adult male Wistar rats/ammonium acetate-induced brain damage | Ammonium acetate (100 mg/kg IP)—45 days Melatonin (5 mg/kg IP)/45 days | Biochemical analysis of oxidative stress and antioxidant markers in brain | Antioxidant property of melatonin protects against brain damage induced by hyperammonemia | (Lena & Subramanian, 2004) [52] |
| 4 | Adult male Wistar rats/ammonium acetate-induced brain damage | Ammonium acetate (100 mg/kg IP)—45 days Melatonin (5 mg/kg IP)/45 days | Biochemical analysis of non-enzymatic antioxidant markers in the brain | Antioxidant property of melatonin protects against brain damage induced by hyperammonemia | (Subramanian, 2003) [53] |
| No. | Model | Type of Liver Injury | Methods | Clinical/Experimental Findings | References |
|---|---|---|---|---|---|
| 1 | CCl4-induced LF/Sprague–Dawley male rats | CCl4—0.2 mL twice per week via the intraperitoneal route for 5 months Melatonin-5 weeks after CCl4-induced LF (0.4 mg/kg/day) | Morris water maze | Melatonin treatment
| (Haeger et al., 2019) [165] |
| 2 | BDL/Young male Sprague–Dawley rats | BDL—5 weeks BDL + melatonin (release melatonin pellet (5 mg) implanted in peritoneum)—4 weeks | Morris water maze Plasma liver enzymes (ALT, AST, direct bilirubin, Total bilirubin) BDNF (Plasma, PFC, HI)—ELISA Anti-ADMA—IHC | Melatonin effectively
| (Hsu et al., 2018) [32] |
| 3 | Clinical | Liver cirrhosis patients | PHES: DST, NCT-A and NCT-B, SDT, and LTT, TAVEC, CVLT) Serum IL-6, IL-8, blood ammonia, plasma cGMP, MRI scan, HI subfield volumes, and resting FC analysis | Episodic memory (learning and long-term memory) impairments Poor performance related to verbal learning and long-term memory (delayed recall) Lower performance related to episodic verbal memory was more apparent In volumetric analysis Decreased right fimbria volume Efferent axons of the pyramidal cells in the hippocampus emerge and converge to form the fimbria, a prominent band of white matter. Alterations in the output of hippocampus information due to alterations in the integrity of the fimbria. Reduced FC | (García-García et al., 2018) [8] |
| 4 | Clinical | Liver cirrhosis patients | Psychometric tests (MMSE, WAIS, NCT, BNT) | Alteration of consciousness, speech disturbances, asterixis, tremor, increased tendon reflexes, muscle tone, and ataxic gait. Patients with MHE: subclinical cognitive alterations | (Brodersen et al., 2014) [166] |
| 5 | Clinical | Liver cirrhosis patients | psychometric tests (DS, BD, NCT-A&B, and ICT. | Persistent and cumulative deficits in working memory, response inhibition, and learning | (Bajaj et al., 2010) [167] |
| 6 | BDL/Young male Sprague–Dawley rats | BDL—2 weeks BDL + Melatonin (500 μg/kg/d)—2 weeks BDL + Melatonin (1000 μg/kg/d)—2 weeks | Morris water maze Plasma liver enzymes (AST, ALT, Creatinine, ALP, ammonia, MDA, GSH/GSSH) Liver, brain cortex, and HI (MDA, GSH/GSSG) | Melatonin treatment
| (Huang et al., 2009) [33] |
| 7 | Clinical | Patients with liver cirrhosis and HE | NCT-A, DST, and SIP test | Impaired cognition Elevated level of melatonin in plasma and diurnal variation | (Velissaris et al., 2009) [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arjunan, A.; Sah, D.K.; Jung, Y.D.; Song, J. Hepatic Encephalopathy and Melatonin. Antioxidants 2022, 11, 837. https://doi.org/10.3390/antiox11050837
Arjunan A, Sah DK, Jung YD, Song J. Hepatic Encephalopathy and Melatonin. Antioxidants. 2022; 11(5):837. https://doi.org/10.3390/antiox11050837
Chicago/Turabian StyleArjunan, Archana, Dhiraj Kumar Sah, Young Do Jung, and Juhyun Song. 2022. "Hepatic Encephalopathy and Melatonin" Antioxidants 11, no. 5: 837. https://doi.org/10.3390/antiox11050837
APA StyleArjunan, A., Sah, D. K., Jung, Y. D., & Song, J. (2022). Hepatic Encephalopathy and Melatonin. Antioxidants, 11(5), 837. https://doi.org/10.3390/antiox11050837