
Heparan Sulfate, Mucopolysaccharidosis IIIB and Sulfur Metabolism Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
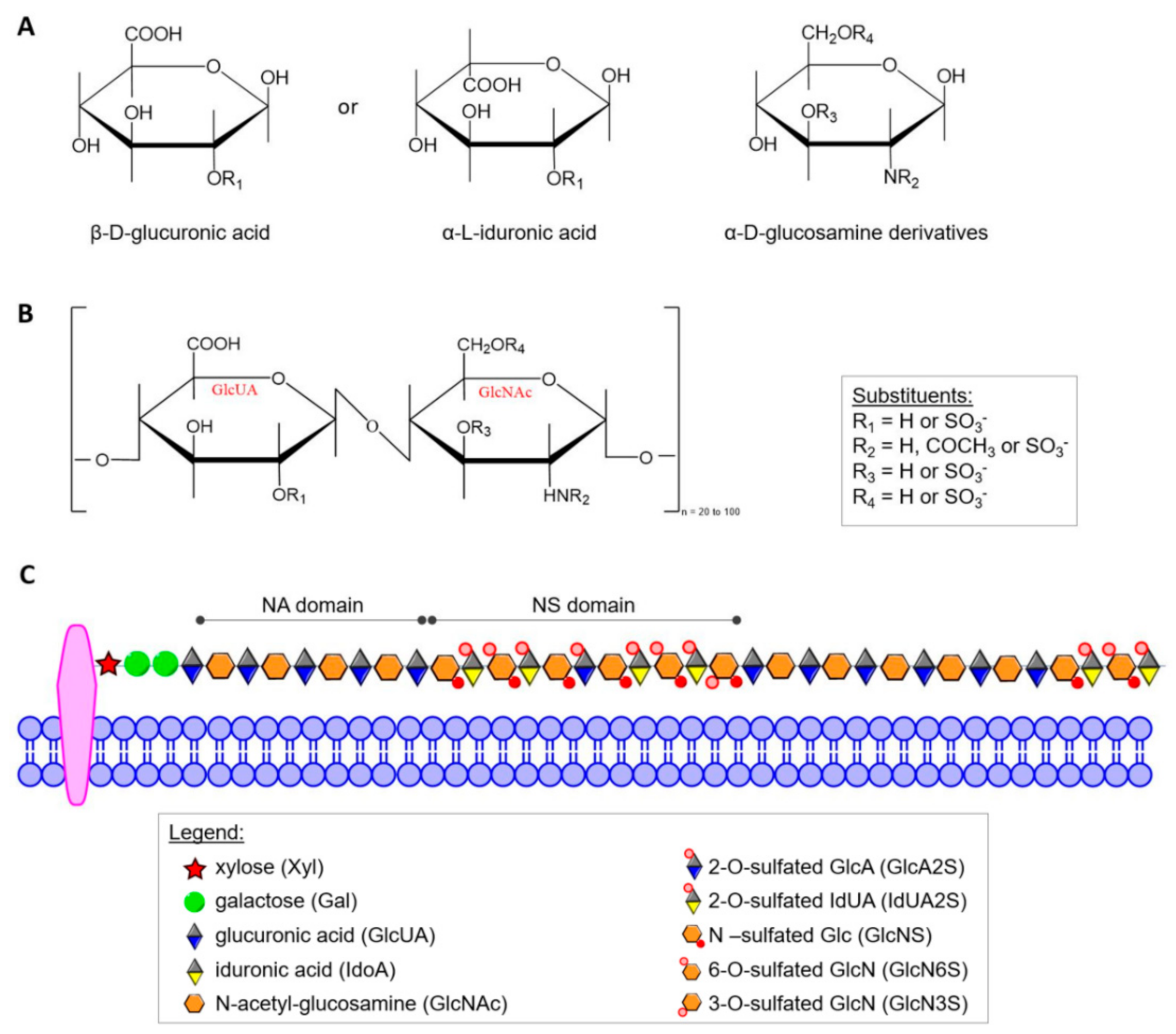
2. Glycosaminoglycans (GAGs)
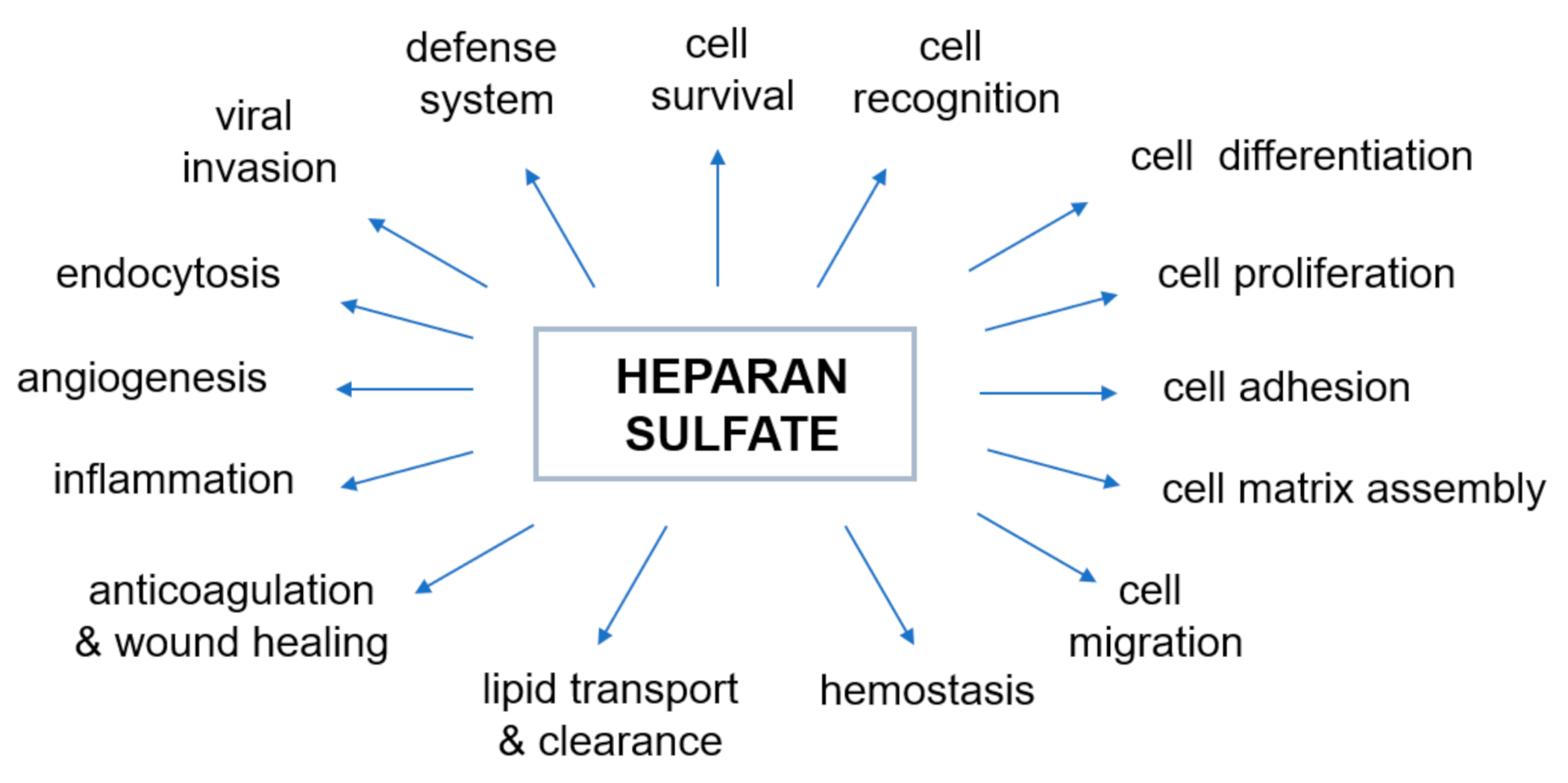
3. Heparan Sulfate (HS)
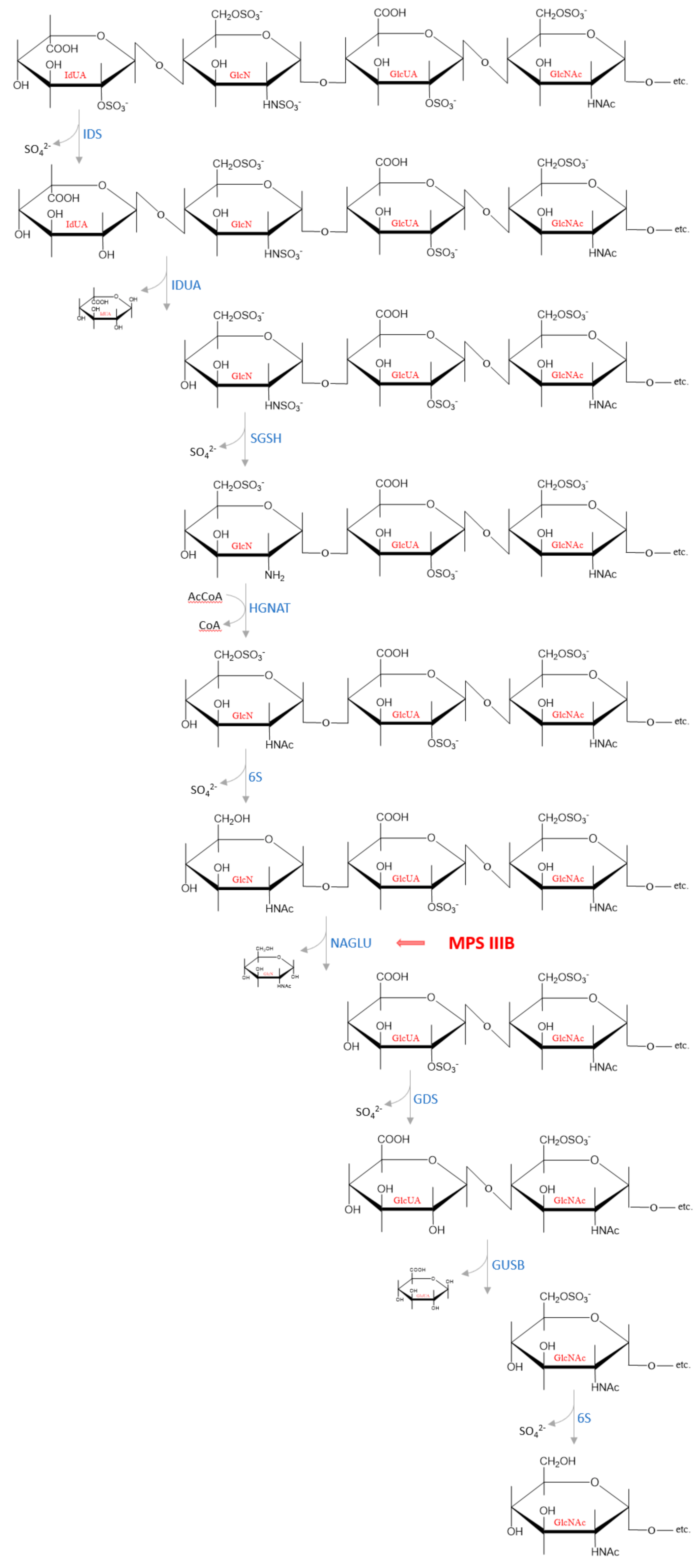
4. Decomposition of Heparan Sulfate
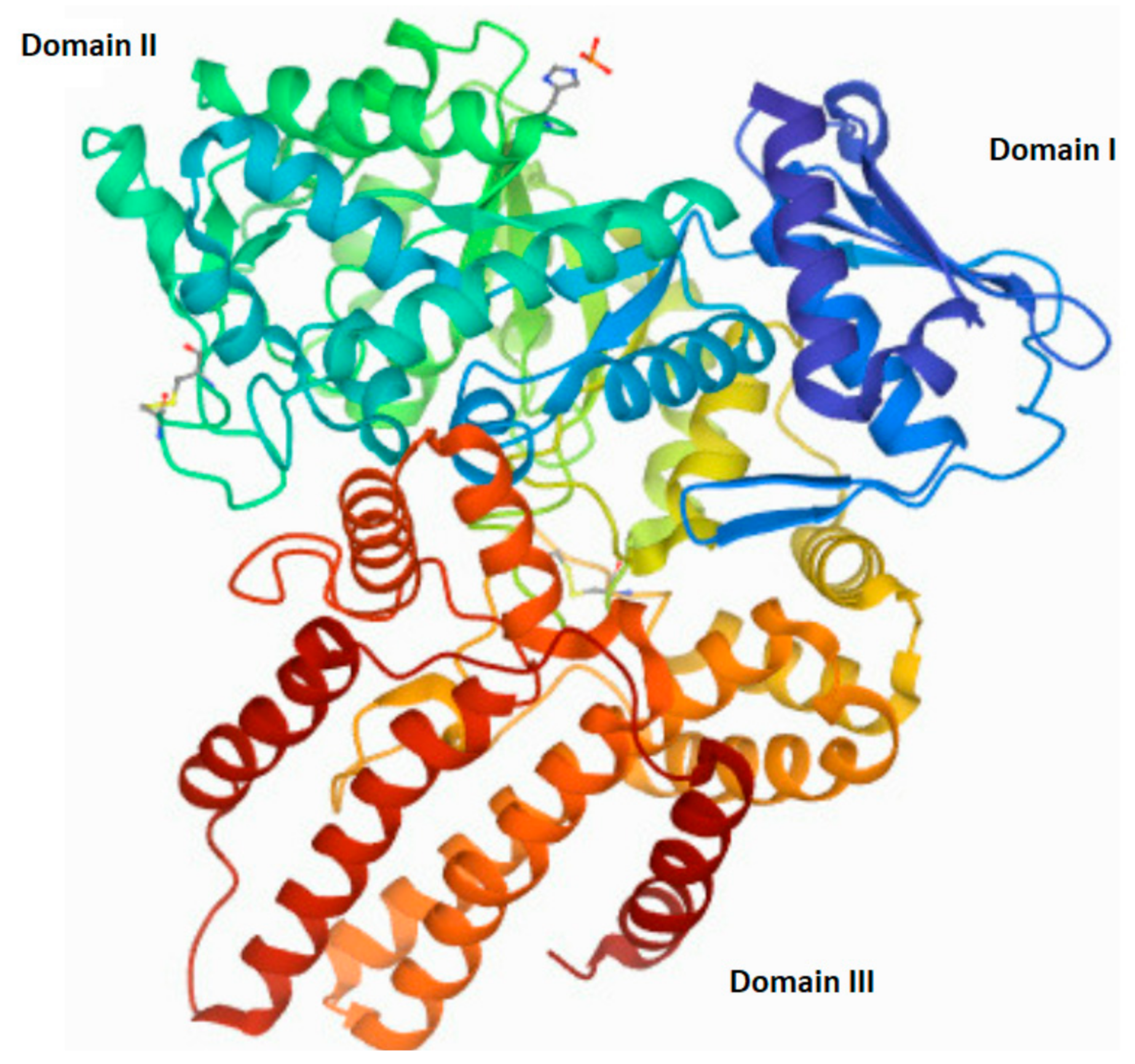
5. Mucopolysaccharidosis, Type IIIB
6. GAGs as a Source of Sulfate
7. Effects of GAGs/HS Accumulation on Sulfur Metabolism
8. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products; The Official Journal of the European Union; European Union: Maastricht, The Netherlands, 2000; pp. 1–5.
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Węgrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Dirosario, J.; Killedar, S.; Zarapse, K.; McCarty, D.M. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol. Ther. 2011, 19, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.H.; Aoyagai-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.N.; Brown, J.R.; et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. USA 2014, 111, 14870–14875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, S.H.; Troitskaya, L.A.; Sinow, C.S.; Haitz, K.; Todd, A.K.; Di Stefano, A.; Le, S.Q.; Dickson, P.I.; Tippin, B.L. Insulin-like growth factor II peptide fusion enables uptake and lysosomal delivery of α-N-acetylglucosaminidase to mucopolysaccharidosis type IIIB fibroblasts. Biochem. J. 2014, 458, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrey, D.A.; Naughton, B.J.; Duncan, F.J.; Meadows, A.S.; Ware, T.A.; Campbell, K.J.; Bremer, W.G.; Walker, C.M.; Goodchild, L.; Bolon, B.; et al. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: Toxicology, biodistribution, and immunological assessments in primates. Hum. Gene Ther. Clin. Dev. 2014, 25, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Marcó, S.; Haurigot, V.; Bosch, F. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon). Hum. Gene Ther. 2019, 30, 1211–1221. [Google Scholar] [CrossRef]
- Kong, W.; Yao, Y.; Zhang, J.; Lu, C.; Ding, Y.; Meng, Y. Update of treatment for mucopolysaccharidosis type III (Sanfilippo syndrome). Eur. J. Pharmacol. 2020, 888, 173562. [Google Scholar] [CrossRef]
- Tambuyzer, E.; Vandendriessche, B.; Austin, C.A.; Brooks, P.J.; Larsson, K.; Miller Needleman, K.I.; Valentine, J.; Davies, K.; Groft, S.C.; Preti, R.; et al. Therapies for rare diseases: Therapieutic modalities, progress and challenges ahead. Nat. Rev. Drug Discov. 2020, 19, 93–111. [Google Scholar] [CrossRef]
- Kaczor-Kamińska, M.; Kamiński, K.; Staińska, K.; Wróbel, M.; Feldman, A. Effect of glycosaminoglycans accumulation on the non-oxidative sulfur metabolism in mouse model of Sanfilippo syndrome, type B. Acta Biochim. Pol. 2019, 66, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Kaczor-Kamińska, M.; Stalińska, K.; Kamiński, K.; Pisarek, A.; Maziarz, U.; Feldman, A.; Wróbel, M. Murine cellular model of mucopolysaccharidosis, type IIIb (MPS (IIIB)—A preliminary study with particular emphasis on the non-oxidative L-cysteine metabolism. Biochimie 2020, 174, 84–94. [Google Scholar] [CrossRef]
- Breen, M.; Weindtein, H.G.; Johnson, R.L.; Veis, A.; Marshall, T.R. Acidic glycosaminoglycans in human skin during fatal development and adult life. Biochim. Biophys. Acta 1970, 201, 54–60. [Google Scholar] [CrossRef]
- Lee, T.Y.; Jamieson, A.; Schafer, I. Changes in the composition and structure of glycosaminoglycans in the human placenta during development. Pediatr. Res. 1973, 7, 965–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praus, R.; Brettschneider, I. Glycosaminoglycans in embryonic and postnatal human cornea. Ophthalmic Res. 1975, 7, 452–458. [Google Scholar] [CrossRef]
- Pepi, L.E.; Sasiene, Z.J.; Mendis, P.M.; Jackson, G.P.; Amster, J.I. Structural characterization of sulfated glycosaminoglycans using charge-transfer dissociation. J. Am. Soc. Mass. Spectrom. 2020, 31, 10. [Google Scholar] [CrossRef]
- Zaia, J.; Costello, C.E. Compositional Analysis of glycosaminoglycans by electrospray mass spectrometry. Anal. Chem. 2001, 73, 233–239. [Google Scholar] [CrossRef]
- Staples, G.O.; Zaia, J. Analysis of glycosaminoglycans using mass spectrometry. Curr. Proteomics 2011, 8, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Huxtable, R.J. Biochemistry of Sulfur; Frieden, E., Ed.; Plenum Press: New York, NY, USA, 1986. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Proteoglycans and sulfated glycosaminoglycans. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017. [Google Scholar] [CrossRef]
- Laurent, T.C.; Laurent, U.B.; Fraser, J.R. The structure and function of hyaluronan: An overview. Immunol. Cell Biol. 1996, 74, A1–A7. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Mizumoto, S.; Fujita, M. Novel insight into glycosaminoglycan biosynthesis based on gene expression profiles. Front. Cell Dev. Biol. 2021, 9, 709018. [Google Scholar] [CrossRef]
- Mattson, J.M.; Turcotte, R.; Zhang, Y. Glycosaminoglycans contribute to extracellular matrix fiber recruitment and arterial wall mechanics. Biomech. Model. Mechanobiol. 2017, 16, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.C.; Carvalho, M.S.; Han, X.; Xia, K.; Mikael, P.E.; Cabral, J.M.S.; Ferreira, F.C.; Linhardt, R.J. Compositional and structural analysis of glycosaminoglycans in cell-derived extracellular matrices. Glycoconj. J. 2019, 36, 141–154. [Google Scholar] [CrossRef]
- Puri, S.; Coulson-Thomas, Y.M.; Gesteira, T.F.; Coulson-Thomas, V.J. Distribution and function of glycosaminoglycans and proteoglycans in the development, homeostasis and pathology of the ocular surface. Front. Cell Dev. Biol. 2020, 8, 731. [Google Scholar] [CrossRef] [PubMed]
- Rienks, M.; Papageorgiou, A.-P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard-Blum, S. Glycosaminoglycans: Major biological players. Glycoconj. J. 2017, 34, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Köwitsch, A.; Zhou, G.; Growth, T. Medical application of glycosaminoglycans: A review. J. Tissue Regen. Med. 2018, 12, e23–e41. [Google Scholar] [CrossRef] [PubMed]
- Banachowicz, P.; Buda, S. Gram-scale carbasugar synthesis via intramolecular seleno-Michael/aldol reaction. RSC Adv. 2019, 9, 12928. [Google Scholar] [CrossRef] [Green Version]
- Łopatkiewicz, G.; Buda, S.; Młynarski, J. Application of the EF and GH fragments to the synthesis of idraparinux. J. Org. Chem. 2017, 82, 12701–12714. [Google Scholar] [CrossRef]
- Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M.B.; Schepers, U.; Bräse, S. Chemical synthesis of glycosaminoglycans. Chem. Rev. 2016, 116, 8193–8255. [Google Scholar] [CrossRef]
- Miura, Y.; Fukuda, T.; Seto, H.; Hoshino, Y. Development of glycosaminoglycan mimetrics using glycopolymers. Polym. J. 2016, 48, 229–237. [Google Scholar] [CrossRef]
- Kalaska, B.; Kamiński, K.; Miklosz, J.; Nakai, K.; Yusa, S.-I.; Pawlak, D.; Nowakowska, M.; Mogielnicki, A.; Szczubiałka, K. Anticoagulant properties of poly(sodium 2-(acrylamido)-2-methylpropanesulfonate)-based di- and triblock polymers. Biomacromolecules 2018, 17, 3104–3118. [Google Scholar] [CrossRef]
- Gray, E.; Hogwood, J.; Mulloy, B. The anticoagulant and antithrombotic mechanisms of heparin. In Heparin—A Century of Progress. Handbook of Experimental Pharmacology; Lever, R., Mulloy, B., Page, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207. [Google Scholar] [CrossRef]
- Sharma, S.K. Low molecular weight heparins. Med. J. Armed Forces India 1998, 54, 285–286. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V. Mechanism of action and pharmacology of unfractionated heparin. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1094–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelberg, H. Heparin and the removal of triglyceride from the blood stream. Am. J. Cardiol. 1964, 14, 8–17. [Google Scholar] [CrossRef]
- Capila., I.; Linhardt, R. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Soares da Costa, D.; Reis, R.L.; Pushkuleva, I. Sulfation of glycosaminoglycans and its implications in human health and disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariga, T.; Miyatake, T.; Yu, R.K. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: Amyloidogenesis and therapeutic strategies—A review. J Neurosci. Res. 2010, 88, 2303–20315. [Google Scholar] [CrossRef]
- Jorpes, J.E.; Gardell, S. On heparin monosulfuric acid. J. Biol. Chem. 1948, 176, 267–275. [Google Scholar] [CrossRef]
- Kalaska, B.; Miklosz, J.; Kamiński, K.; Musielak, B.; Yusa, S.-I.; Pawlak, D.; Nowakowska, M.; Szczubiałka, K.; Mogielnicki, A. The neutralization of heparan sulfate by heparin-binding copolymer as a potential therapeutic target. RSC Adv. 2019, 9, 3020–3029. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, J.T. Heparan sulfate: Growth control with a restricted sequence menu. J. Clin. Investig. 2001, 108, 357–361. [Google Scholar] [CrossRef]
- Li, J.-P.; Kusche-Gullberg, M. Heparan sulfate: Biosynthesis, structure, and function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [CrossRef]
- Gill, V.L.; Aich, U.; Rao, S.; Pohl, C.; Zaia, J. Disaccharide analysis of glycosaminoglycans using hydrophilic interaction chromatography and mass spectrometry. Anal. Chem. 2013, 15, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan sulfate and heparan sulfate proteoglycans in cancer initiation and progression. Front. Endocrinol. 2018, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Reis, C.A.; Vivès, R.R.; Magalhães, A. Heparan sulfate biosynthesis and sulfation profiles as modulators of cancer signalling and progression. Front. Oncol. 2021, 11, 778752. [Google Scholar] [CrossRef] [PubMed]
- Tekotte, H.; Engel, M.; Margolis, R.U.; Margolis, R.K. Disaccharide composition of heparan sulfates: Brain, nervous tissue storage organelles, kidney and lung. J. Neurochem. 1994, 62, 1126–1130. [Google Scholar] [CrossRef]
- Gordon, P.B.; Choi, H.U.; Conn, G.; Ahmed, A.; Ehrmann, B.; Rosenberg, L.; Hatcher, V.B. Extracellular matrix heparan sulfate proteoglycans modulate the mitogenic capacity of acidic fibroblast growth factor. J. Cell. Physiol. 1989, 140, 584–592. [Google Scholar] [CrossRef]
- Lindahl, U.; Kjellén, L. Pathophysiology of heparan sulfate: Many diseases, few drugs. J. Intern. Med. 2013, 273, 555–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoler-Barak, L.; Moussion, C.; Shezen, E.; Hatzav, M.; Sixt, M.; Alon, R. Blood vessels pattern heparan sulfate gradients between their apical and basolateral aspects. PLoS ONE 2014, 9, e85699. [Google Scholar] [CrossRef] [PubMed]
- Lindhal, B.; Lindhal, U. Amyloid-specific heparan sulfate from human liver and spleen. J. Biol. Chem. 1997, 272, 26091–26094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindhal, B.; Eriksson, L.; Lindhal, U. Structure of heparan sulfate from human brain, with special regard to Alzheimer’s disease. Biochem. J. 1995, 306, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Masri, R.E.; Crétinon, Y.; Gout, E.; Vivès, R.R. HS and inflammation: A potential playground for the Sulfs? Front. Immunol. 2020, 11, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, S.E.; Gallagher, J.T. Specific binding of the chemokine platelet factor 4 to heparan sulfate. J. Biol. Chem. 1997, 272, 20508–20514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of teansglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, A.D.; Selleck, S.B. The elusive functions of proteoglycans: In vivo veritas. J. Cell. Biol. 2000, 148, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Perrimon, N.; Bernfield, M. Specificities of heparan sulfate proteoglycans in development processes. Nature 2000, 404, 725–728. [Google Scholar] [CrossRef]
- Selleck, S.B. Proteoglycans and pattern formation: Sugar biochemistry meets development genetics. Trends Genet. 2000, 16, 206–212. [Google Scholar] [CrossRef]
- Onyeisi, J.O.S.; Ferreira, B.Z.F.; Nader, H.B.; Lopes, C.C. Heparan sulfate proteoglycans as targets for cancer therapy: A review. Cancer Biol. Ther. 2020, 21, 1087–1094. [Google Scholar] [CrossRef]
- Kresse, H.; Cantz, M.; von Figura, K.; Glössl, J.; Paschke, E. The Mucopolysaccharidoses: Biochemistry and clinical symptoms. Klin. Wochenschr. 1981, 59, 867–876. [Google Scholar] [CrossRef]
- Griffin, L.S.; Gloster, T.M. The enzymatic degradation of heparan sulfate. Protein Pept. Lett. 2017, 24, 710–722. [Google Scholar] [CrossRef]
- Bame, K.J. Heparanases: Endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology 2001, 11, 91R–98R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valstar, M.J.; Ruijter, G.J.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo syndrome: A mini-review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Kloska, A.; Tylki-Szymańska, A.; Węgrzyn, G. Mucopolysaccharidosis—Biochemical mechanisms of diseases and therapeutic possibilities. Postępy Biochem. 2011, 57, 133–147, (Article in Polish). [Google Scholar] [PubMed]
- Birrane, G.; Dassier, A.-L.; Romashko, A.; Lundberg, D.; Holmes, K.; Cottle, T.; Norton, A.W.; Zhang, B.; Concino, M.F.; Meiyappan, M. Structural characterization of the α-N-acetylglucosaminidase, a key enzyme in the pathogenesis of Sanfilippo syndrome B. J. Struct. Biol. 2019, 205, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Nidiffer, F.D.; Kelly, T.E. Developmental and degenerative patterns associated with cognitive, behavioral and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J. Ment. Defic. Res. 1983, 27, 185–203. [Google Scholar] [CrossRef]
- Wraith, J.E.; Danks, D.M.; Rogers, J.G. Mild Sanfilippo syndrome: A further cause of hyperactivity and behavioral disturbance. Med. J. Aust. 1987, 147, 450–451. [Google Scholar] [CrossRef]
- Bax, M.C.; Colville, G.A. Behavior in mucopolysaccharide disorders. Arch. Dis. Child. 1995, 73, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Valstar, M.J.; Bruggenwirth, H.T.; Olmer, R.; Wevers, R.A.; Verheijen, F.W.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. J. Inherit. Metab. Dis. 2010, 33, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Malm, G.; Månsson, J.-E. Mucopolysaccharidosis type III (Sanfilippo disease) in Sweden: Clinical presentation of 322 children diagnosed during a 30-year period. Acta Paediatr. 2010, 99, 1253–1257. [Google Scholar] [CrossRef]
- White, K.K.; Karol, L.A.; White, D.R.; Hale, S. Musculoskeletal manifestations of Sanfilippo Syndrome (mucopolysaccharidosis type III). J. Pediatr. Orthop. 2011, 31, 594. [Google Scholar] [CrossRef]
- Van Schrojenstein-de Valk, H.M.; van de Kamp, J.J. Follow-up on seven adults patients with mild Sanfilippo B-disease. Am. J. Med. Genet. 1987, 28, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, W.M.A.; Csepán, R.; Marcelis, C.L.M.; Lefeber, D.J.; Egger, J.I.M.; Tuinier, S. Sanfilippo B in an elderly female psychiatric patient: A rare but relevant diagnosis in presenile dementia. Acta Psychiatr. Scand. 2009, 122, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Walkey, S.U. Secondary accumulation of gangliosides in lysosomal storage disorder. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.; Gargiulo, N.; Faraonio, R.; Castalodo, S.; Gonzales, Y.; Reyero, E.; Di Natale, P. Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J. Neurosci. Res. 2007, 85, 612–622. [Google Scholar] [CrossRef]
- Baydakova, G.; Iiyushkina, A.; Gaffke, L.; Pierzynowska, K.; Bychov, I.; Ługowska, A.; Węgrzyn, G.; Tylki-Szymanska, A.; Zakharova, E. Elevated LysoGb3 concentration in the neuronopathic forms of mucopolysaccharidoses. Diagnostics 2020, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Walkey, S.U.; Vanier, M.T. Pathomechanisms in lysosomal storage disorders. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Pearse, Y.; Iacovino, M. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med. Res. Arch. 2020, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Fuller, M.; Hopwood, J.J. Lysosomal Degradation of Heparin and Heparan Sulfate. In Chemistry and Biology of Heparin and Heparan Sulfate; Garg, H.G., Linhardt, R.J., Hales, C.A., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2005; pp. 285–311. [Google Scholar] [CrossRef]
- Stromberg, P.E.; Cumpstion, K.L. Sulfates. In Encyclopedia of Toxicology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 413–415. [Google Scholar] [CrossRef]
- Miller, C.G.; Schmidt, E.E. Sulfur metabolism under stress. Antioxid. Redox Sign. 2020, 33, 1158–1173. [Google Scholar] [CrossRef]
- Jonas, A.J.; Jobe, H. Sulfate transport by rat liver lysosomes. J. Biol. Chem. 1990, 265, 17545–17549. [Google Scholar] [CrossRef]
- Chou, H.F.; Passage, M.; Jonas, A.J. ATP stimulates lysosomal sulphate transport at neutral pH: Evidence for phosphorylation of the lysosomal sulphate carrier. Biochem. J. 1997, 327, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Klaasen, C.D.; Boles, J.W. Sulfation and sulfotransferases 5: The importance of 3’-phosphoadenosine-5’-phosphosulfate (PAPS) in the regulation of sulfation. FASEB J. 1997, 11, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Paganini, C.; Tota, C.G.; Superti-Furga, A.; Rossi, A. Skeletal dysplasias caused by sulfation defects. Int. J. Mol. Sci. 2020, 21, 2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiselli, G. Drug-mediated regulation of glycosaminoglycan biosynthesis. Med. Res. Rev. 2017, 37, 1051–1094. [Google Scholar] [CrossRef] [PubMed]
- Elgavish, A.; Meezan, E. Sulfation by human lung fibroblasts: SO4(2−) and sulfur-containing amino acids as a source for macromolecular sulfation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1991, 260, L450–L456. [Google Scholar] [CrossRef]
- Baranczyk-Kuzma, A.; Borchardt, R.T.; Schasteen, C.S.; Pinnick, C.L. Phenolsulfotransferase: Purification and characterization of the rat brain enzyme. Psychopharmacol. Bull. 1981, 17, 50–51. [Google Scholar]
- Hanna, E.; Ng, K.F.; MacRae, I.J.; Bley, C.J.; Fisher, A.J.; Segel, I.H. Kinetic and stability properties of Penicillium chrysogenum ATP sulfurylase missing the C-terminal regulatory domain. J. Bio. Chem. 2004, 279, 4415–4424. [Google Scholar] [CrossRef] [Green Version]
- Markovich, D. Physiological roles and regulation of mammalian sulfate transporters. Physiol. Rev. 2001, 81, 1499–1533. [Google Scholar] [CrossRef]
- Priyamvada, S.; Saksena, S.; Alrefai, W.A.; Dudeja, P.K. Intestinal anion absorption. In Physiology of the Gastrointestinal Track, 6th ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1317–1362. [Google Scholar] [CrossRef]
- Langford, R.; Hurrion, E.; Dawson, P.A. Genetics and pathophysiology of mammalian sulfate biology. J. Genet. Genomics 2017, 44, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.R.; Norbeck, K.; Moldeus, P. The effectiveness of different sulfate precursors in supporting extrahepatic sulfate conjugation. Biochem. Pharm. 1983, 32, 1789–1791. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Metabolism of sulfur-containing amino acids: How the body copes with excess methionine, cysteine and sulfide. J. Nutr. 2020, 150, 2494S–2505S. [Google Scholar] [CrossRef]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.; Cranfold, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by a transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Sawada, N. The mercaptopyruvate pathway in cysteine metabolism: A physiologic role and related disease of the multifunctional 3-mercaptopyruvate sulfurtransferase. Curr. Med. Chem. 2006, 13, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Nagano, M.; Ito, T.; Suzuki, H. Redox regulation of mammalian 3-mercaptopyruvate sulfurtransferase. Meth. Enzymol. 2015, 554, 229–254. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Sulfur amino acid metabolism: Pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 2004, 24, 539–577. [Google Scholar] [CrossRef]
- Kaczor-Kamińska, M.; Kamiński, K.; Wróbel, M. The expression and activity of rhodanese, 3-mercaptopyruvate sulfurtransferase, cystathionine γ-lyase in the most frequently chosen cellular research models. Biomolecules 2021, 11, 1859. [Google Scholar] [CrossRef]
- Wróbel, M.; Jurkowska, H.; Kaczor, M.; Uchacz, T. Rodanese and 3-mercaptopyruvate sulfurtransferase—Evolutionary related enzymes. KOSMOS 2012, 61, 233–244. [Google Scholar]
- Jurkowska, H.; Kaczor-Kamińska, M.; Bronowicka-Adamska, B.; Wróbel, M. Cystathionine γ-lyase. Adv. Hyg. Exp. Med. 2014, 68, 1–9. [Google Scholar] [CrossRef]
- Kaczor-Kamińska, M.; Sura, P.; Wróbel, M. Multidirectional changes in parameters related to sulfur metabolism in frog tissues exposed to heavy metal-related stress. Biomolecules 2020, 14, 574. [Google Scholar] [CrossRef] [Green Version]
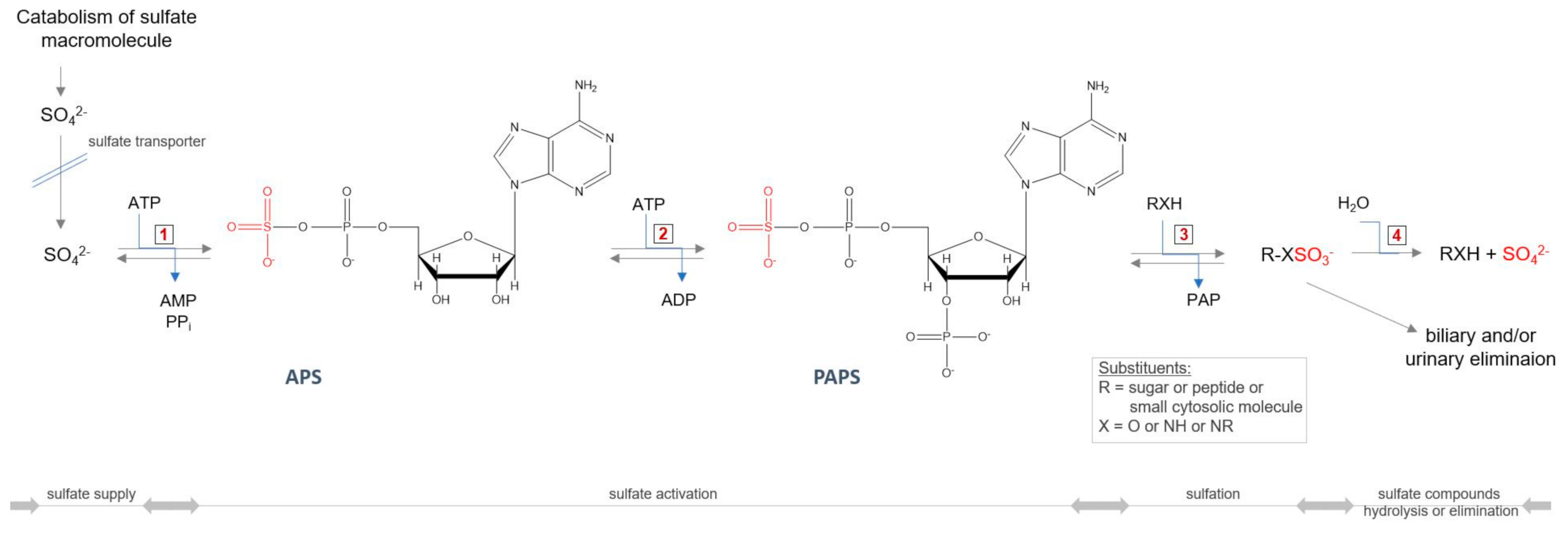
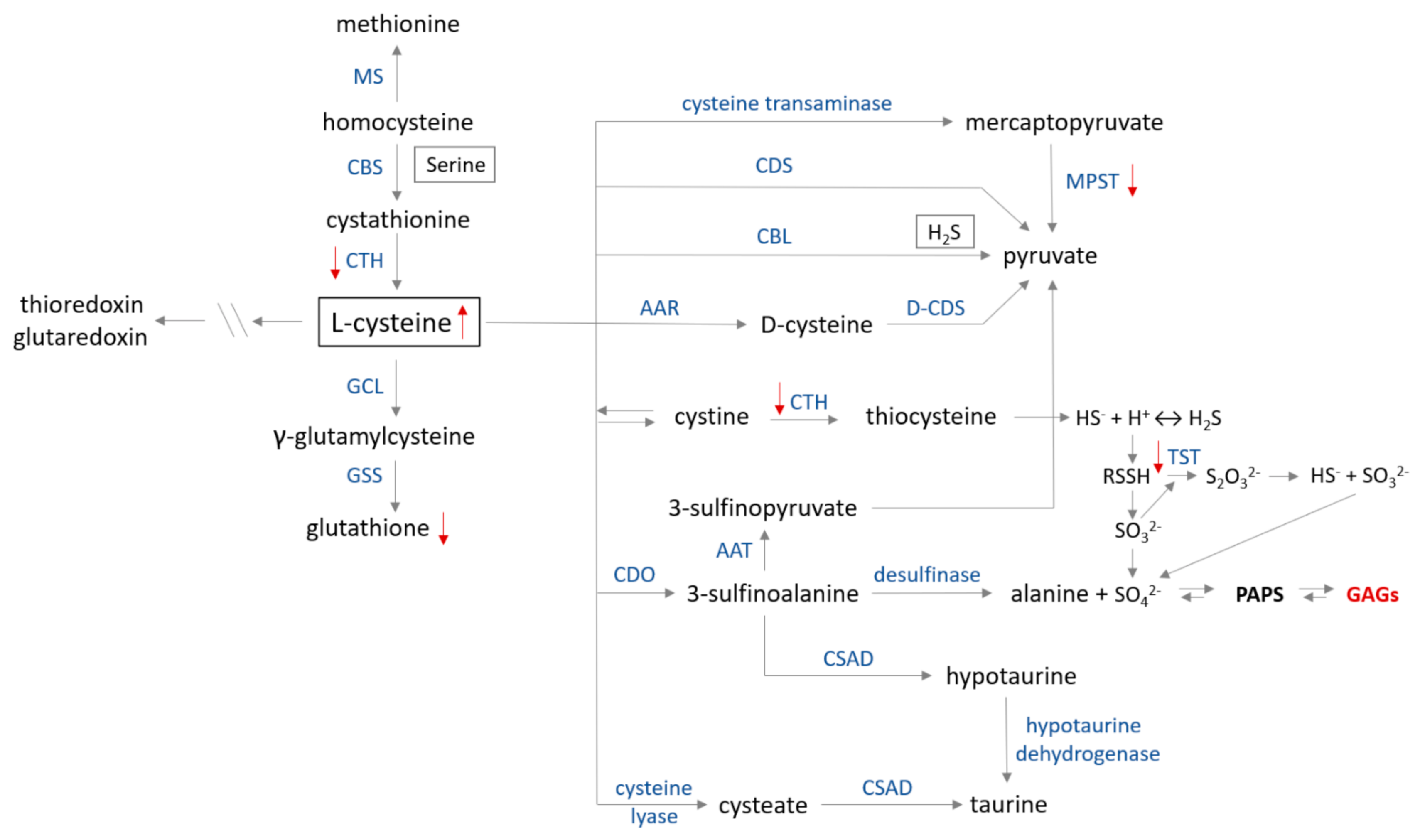
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczor-Kamińska, M.; Kamiński, K.; Wróbel, M. Heparan Sulfate, Mucopolysaccharidosis IIIB and Sulfur Metabolism Disorders. Antioxidants 2022, 11, 678. https://doi.org/10.3390/antiox11040678
Kaczor-Kamińska M, Kamiński K, Wróbel M. Heparan Sulfate, Mucopolysaccharidosis IIIB and Sulfur Metabolism Disorders. Antioxidants. 2022; 11(4):678. https://doi.org/10.3390/antiox11040678
Chicago/Turabian StyleKaczor-Kamińska, Marta, Kamil Kamiński, and Maria Wróbel. 2022. "Heparan Sulfate, Mucopolysaccharidosis IIIB and Sulfur Metabolism Disorders" Antioxidants 11, no. 4: 678. https://doi.org/10.3390/antiox11040678
APA StyleKaczor-Kamińska, M., Kamiński, K., & Wróbel, M. (2022). Heparan Sulfate, Mucopolysaccharidosis IIIB and Sulfur Metabolism Disorders. Antioxidants, 11(4), 678. https://doi.org/10.3390/antiox11040678