
Polydatin Attenuates Intra-Uterine Growth Retardation-Induced Liver Injury and Mitochondrial Dysfunction in Weanling Piglets by Improving Energy Metabolism and Redox Balance


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Experimental Design
2.3. Sample Collection
2.4. Determination of Plasma Biochemical Parameters
2.5. Hepatic Apoptosis
2.6. Hepatic Caspase Activities
2.7. Electron Microscopy
2.8. Mitochondrial Separation
2.9. Mitochondrial Swelling
2.10. Mitochondrial Superoxide Anion
2.11. Mitochondrial DNA (mtDNA) Content
2.12. Immunofluorescence Staining of Hepatic SIRT1 Protein
2.13. Hepatic SIRT1 Activity
2.14. Mitochondrial Oxidative Enzyme Activities
2.15. Hepatic ATP Content
2.16. Hepatic Nicotinamide Adenine Dinucleotide (NAD+) and Its Reduced Form (NADH)
2.17. Evaluation of In Vitro Antioxidant Activity of Polydatin
2.18. Hepatic Antioxidant Enzyme Activities and Redox Metabolites
2.19. Total RNA Isolation and RT-qPCR Analysis
2.20. Statistical Analysis
3. Results
3.1. Effects of BW and Diet Factors on Plasma Biochemical Parameters of Weaned Piglets
3.2. Effects of BW and Diet Factors on Hepatic Apoptosis Rate and Caspase Activities of Weaned Piglets
3.3. Effects of BW and Diet Factors on Hepatic Energy Metabolism of Weaned Piglets
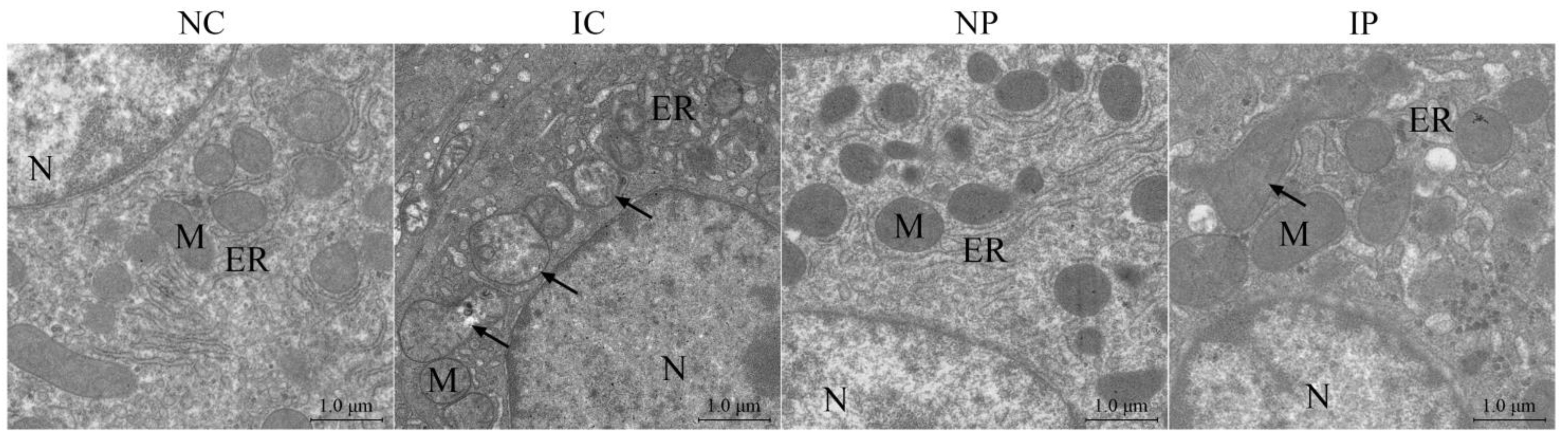
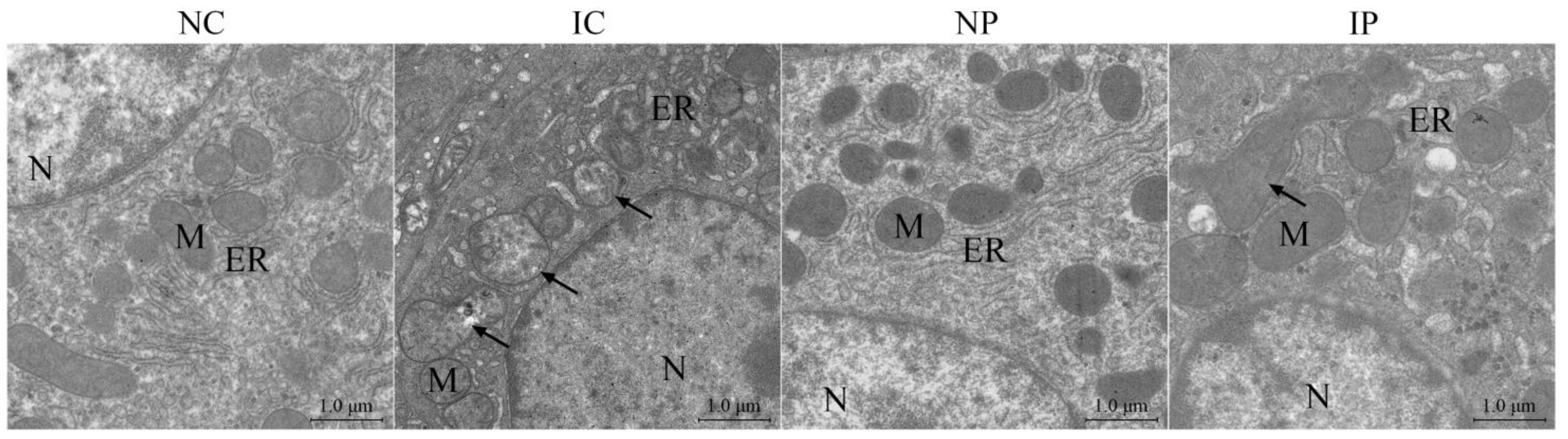
3.4. Effects of BW and Diet Factors on Hepatic Ultrastructure of Weaned Piglets
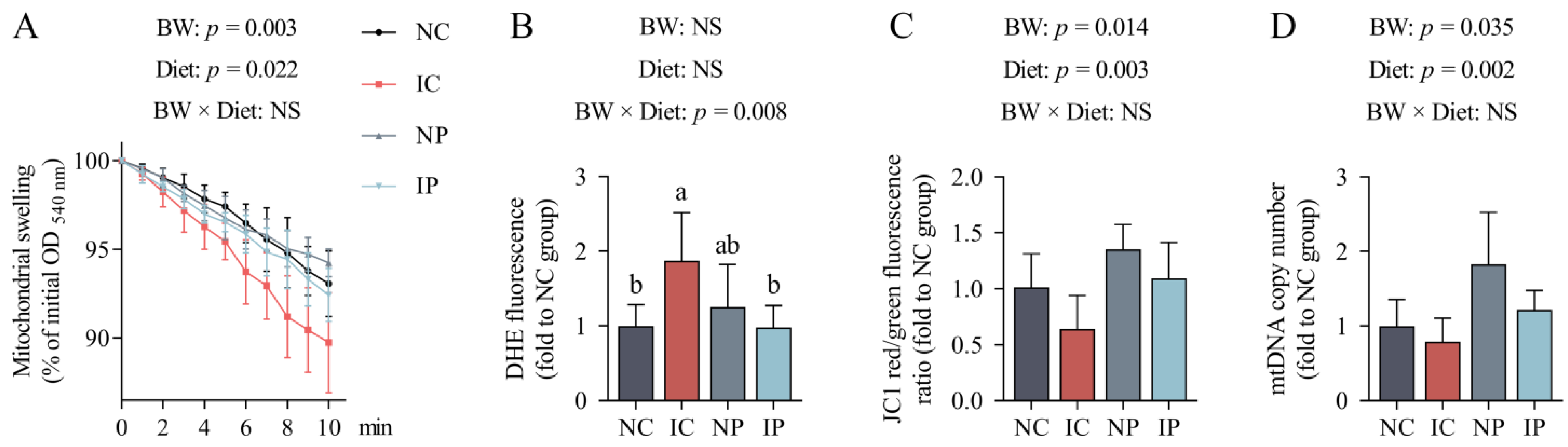
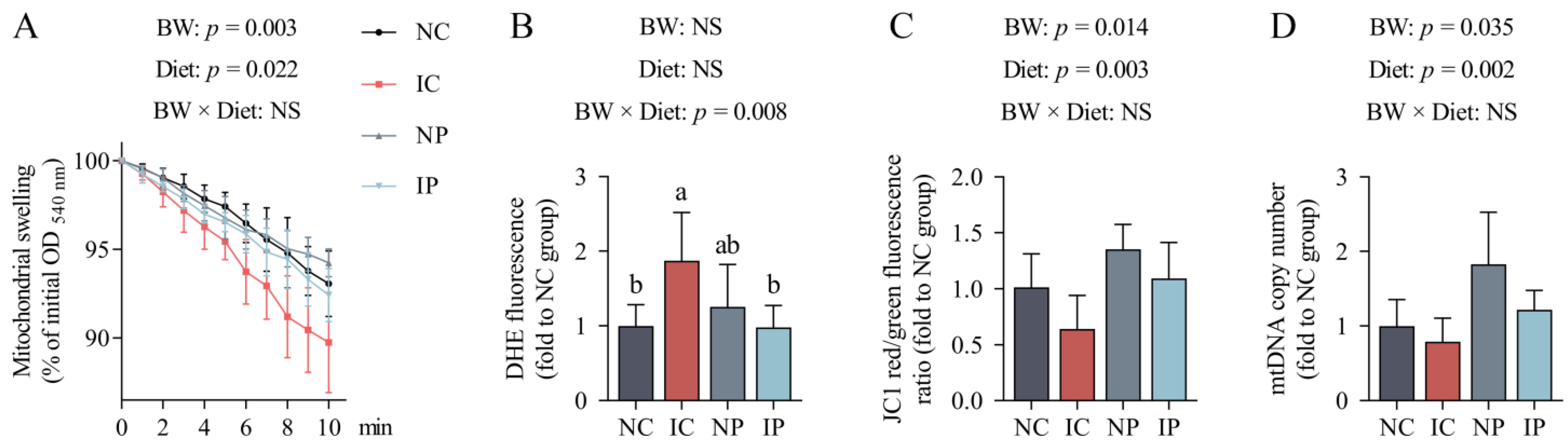
3.5. Effects of BW and Diet Factors on Hepatic Mitochondrial Injury of Weaned Piglets
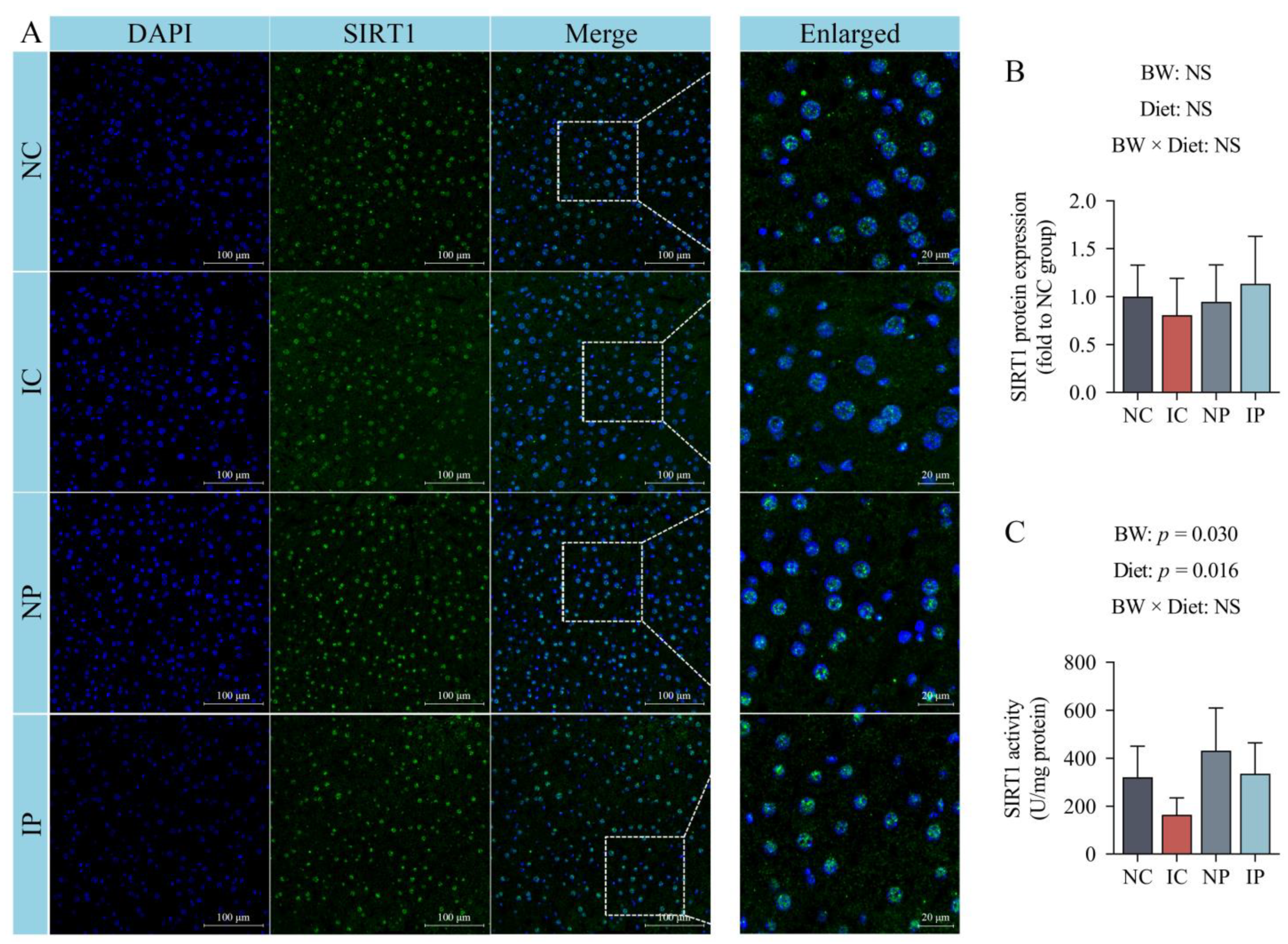
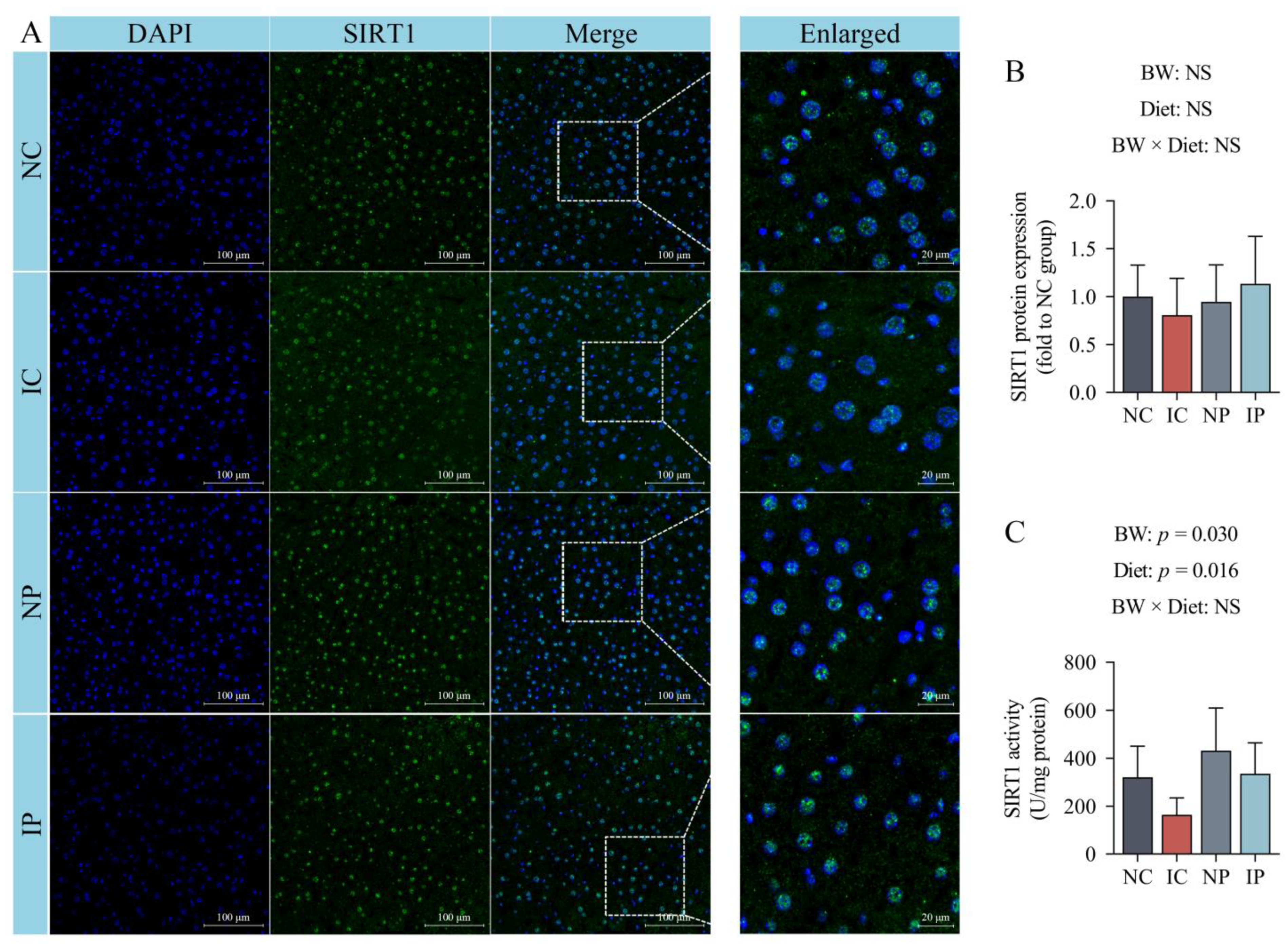
3.6. Effects of BW and Diet Factors on Hepatic SIRT1 Expression and Activity of Weaned Piglets
3.7. Effects of BW and Diet Factors on Hepatic Redox Status of Weaned Piglets
3.8. Effects of BW and Diet Factors on Hepatic Gene Expression of Weaned Piglets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wixey, J.A.; Chand, K.K.; Colditz, P.B.; Bjorkman, S.T. Neuroinflammation in intrauterine growth restriction. Placenta 2017, 54, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheson, S.M.; Walling, G.A.; Edwards, S.A. Genetic selection against intrauterine growth retardation in piglets: A problem at the piglet level with a solution at the sow level. Genet. Sel. Evol. 2018, 50, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hales, C.N.; Barker, D.J. Type 2 (non-insulindependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Agostoni, C.; Bedogni, G.; Canani, R.B.; Brambilla, P.; Nobili, V.; Pietrobelli, A. Effect of intrauterine growth retardation on liver and long-term metabolic risk. Int. J. Obes. 2012, 36, 1270–1277. [Google Scholar] [CrossRef] [Green Version]
- Milligan, B.N.; Fraser, D.; Kramer, D.L. Within-litter birth weight variation in the domestic pig and its relation to pre-weaning survival, weight gain, and variation in weaning weights. Livest. Prod. Sci. 2002, 76, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Quiniou, N.; Dagorn, J.; Gaudre, D. Variation of piglets’ birth weight and consequences on subsequent performance. Livest. Prod. Sci. 2002, 78, 63–70. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Su, W.; Ying, Z.; Zhou, L.; Zhang, L.; Wang, T. Resveratrol attenuates mitochondrial dysfunction in the liver of intrauterine growth retarded suckling piglets by improving mitochondrial biogenesis and redox status. Mol. Nutr. Food Res. 2017, 61, 1600653. [Google Scholar] [CrossRef]
- Rashid, C.S.; Bansal, A.; Simmons, R.A. Oxidative stress, intrauterine growth restriction, and developmental programming of type 2 diabetes. Physiology 2018, 33, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. BBA-Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Bio. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čáp, M.; Váchová, L.; Palková, Z. Reactive oxygen species in the signaling and adaptation of multicellular microbial communities. Oxid. Med. Cell. Longev. 2012, 2012, 976753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; He, J.; Ahmad, H.; Shen, M.; Zhao, Y.; Gan, Z.; Zhang, L.; Zhong, X.; Wang, C.; Wang, T. Dietary curcumin supplementation increases antioxidant capacity, upregulates Nrf2 and Hmox1 levels in the liver of piglet model with intrauterine growth retardation. Nutrients 2019, 11, 2978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Niu, Y.; He, J.; Zhang, L.; Wang, C.; Wang, T. Dietary dihydroartemisinin supplementation attenuates hepatic oxidative damage of weaned piglets with intrauterine growth retardation through the Nrf2/ARE signaling pathway. Animals 2019, 9, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akanbi, T.O.; Marshall, S.N.; Barrow, C.J. Polydatin-fatty acid conjugates are effective antioxidants for stabilizing omega 3-containing bulk fish oil and fish oil emulsions. Food Chem. 2019, 301, 125297. [Google Scholar] [CrossRef]
- Regev-Shoshani, G.; Shoseyov, O.; Bilkis, I.; Kerem, Z. Glycosylation of resveratrol protects it from enzymic oxidation. Biochem. J. 2003, 374, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Cheng, Y.; Liu, M.; Liu, D.; Cui, H.; Zhang, B.; Zhou, S.; Yang, T.; Mei, Q. Comparision of piceid and resveratrol in antioxidation and antiproliferation activities in vitro. PLoS ONE 2013, 8, e54505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şöhretoğlu, D.; Baran, M.Y.; Arroo, R.; Kuruüzüm-Uz, A. Recent advances in chemistry, therapeutic properties and sources of polydatin. Phytochem. Rev. 2018, 17, 973–1005. [Google Scholar] [CrossRef]
- Rocha, B.A.; Teixeira, C.S.; Silva-Filho, J.C.; Nóbrega, R.B.; Alencar, D.B.; Nascimento, K.S.; Freire, V.N.; Gottfried, C.J.S.; Nagano, C.S.; Sampaio, A.H.; et al. Structural basis of ConM binding with resveratrol, an anti-inflammatory and antioxidant polyphenol. Int. J. Biol. Macromol. 2015, 72, 1136–1142. [Google Scholar] [CrossRef]
- Fabris, S.; Momo, F.; Ravagnan, G.; Stevanato, R. Antioxidant properties of resveratrol and piceid on lipid peroxidation in micelles and monolamellar liposomes. Biophys. Chem. 2008, 135, 76–83. [Google Scholar] [CrossRef]
- Lanzilli, G.; Cottarelli, A.; Nicotera, G.; Guida, S.; Ravagnan, G.; Fuggetta, M.P. Anti-inflammatory effect of resveratrol and polydatin by in vitro IL-17 modulation. Inflammation 2012, 35, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.J.; Yu, H.W.; Yang, Y.Z.; Wu, W.Y.; Chen, T.Y.; Jia, K.K.; Kang, L.L.; Jiao, R.Q.; Kong, L.D. Polydatin prevents fructose-induced liver inflammation and lipid deposition through increasing miR-200a to regulate Keap1/Nrf2 pathway. Redox Biol. 2018, 18, 124–137. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Li, Y.; Wang, T. Protective effect of polydatin on jejunal mucosal integrity, redox status, inflammatory response, and mitochondrial function in intrauterine growth-retarded weanling piglets. Oxid. Med. Cell. Longev. 2020, 2020, 7178123. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, X.; Xu, Z. SIRT1 provides new pharmacological targets for polydatin through its role as a metabolic sensor. Biomed. Pharmacother. 2021, 139, 111549. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Yang, Y.; Dai, X.; Zeng, Z.; Yang, Y.; Dai, X.; Xu, S.; Li, T.; Zhang, Q.; Zhao, K.; et al. Polydatin ameliorates injury to the small intestine induced by hemorrhagic shock via SIRT3 activation-mediated mitochondrial protection. Expert Opin. Ther. Tar. 2016, 20, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Bheereddy, P.; Yerra, V.G.; Kalvala, A.K.; Sherkhane, B.; Kumar, A. SIRT1 activation by polydatin alleviates oxidative damage and elevates mitochondrial biogenesis in experimental diabetic neuropathy. Cell. Mol. Neurobiol. 2021, 41, 1563–1577. [Google Scholar] [CrossRef]
- Wang, T.; Huo, Y.J.; Shi, F.X.; Xu, R.J.; Hutz, R.J. Effects of intrauterine growth retardation on development of the gastrointestinal tract in neonatal pigs. Neonatology 2005, 88, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [Green Version]
- Ong, M.M.; Wang, A.S.; Leow, K.Y.; Khoo, Y.M.; Boelsterli, U.A. Nimesulide-induced hepatic mitochondrial injury in heterozygous Sod2+/− mice. Free Radic. Bio. Med. 2006, 40, 420–429. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Chen, M.; Fujita, T.; Xu, Q.; Peng, W.; Liu, W.; Jensen, T.K.; Pei, Y.; Wang, F.; Han, X.; et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 2010, 13, 883–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Shi, J.; Ibarra, A.C.; Kakuda, Y.; Xue, S.J. The scavenging capacity and synergistic effects of lycopene, vitamin E, vitamin C, and β-carotene mixtures on the DPPH free radical. LWT–Food Sci. Technol. 2008, 41, 1344–1349. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, B.; Mao, X.; He, J.; Yu, J.; Zheng, P.; Huang, Z.; Chen, D. Effects of intrauterine growth retardation and maternal folic acid supplementation on hepatic mitochondrial function and gene expression in piglets. Arch. Anim. Nutr. 2012, 66, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Hou, X.; Zhang, L.; Wang, T. Medium-chain TAG improve energy metabolism and mitochondrial biogenesis in the liver of intra-uterine growth-retarded and normal-birth-weight weanling piglets. Br. J. Nutr. 2016, 115, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, A.L.; Wesolowski, S.R.; Regnault, T.R.; Lynch, R.M.; Limesand, S.W. Dimming the powerhouse: Mitochondrial dysfunction in the liver and skeletal muscle of intrauterine growth restricted fetuses. Front. Endocrinol. 2021, 12, 515. [Google Scholar] [CrossRef] [PubMed]
- Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid. Redox Sign. 2019, 30, 375–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.; Wang, T.; Li, S.; Song, Z.; Zhang, H.; Zhang, L.; Wang, T. Effects of early resveratrol intervention on skeletal muscle mitochondrial function and redox status in neonatal piglets with or without intrauterine growth retardation. Oxid. Med. Cell. Longev. 2020, 2020, 4858975. [Google Scholar] [CrossRef]
- Luo, Z.; Luo, W.; Li, S.; Zhao, S.; Sho, T.; Xu, X.; Zhang, J.; Xu, W.; Xu, J. Reactive oxygen species mediated placental oxidative stress, mitochondrial content, and cell cycle progression through mitogen-activated protein kinases in intrauterine growth restricted pigs. Reprod. Biol. 2018, 18, 422–431. [Google Scholar] [CrossRef]
- Stremming, J.; Chang, E.; Knaub, L.A.; Armstrong, M.L.; Baker, P.R.; Wesolowski, S.R.; Reisdorph, N.; Reusch, J.E.B.; Brown, L.D. Lower citrate synthase activity, mitochondrial complex expression, and fewer oxidative myofibers characterize skeletal muscle from growth restricted fetal sheep. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2021, 322, R228–R240. [Google Scholar] [CrossRef] [PubMed]
- Thorn, S.R.; Regnault, T.R.; Brown, L.D.; Rozance, P.J.; Keng, J.; Roper, M.; Wilkening, R.B.; Hay, W.W.; Friedman, J.E. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces messenger ribonucleic acid translation initiation and nutrient sensing in fetal liver and skeletal muscle. Endocrinology 2009, 150, 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gronert, M.S.; Ozanne, S.E. Experimental IUGR and later diabetes. J. Intern. Med. 2007, 261, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.; Bollani, L.; Decembrino, L.; Comite, A.D.; Angelini, M.; Stronati, M. Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J. Matern-Fetal Neonatal Med. 2013, 26, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, Z.; Shen, M.; Zhang, Y.; Duan, J.; Guo, Y.; Zhang, D.; Hu, J.; Lin, J.; Man, W.; et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. Recent advances in pharmacotherapy for hypertriglyceridemia. Prog. Lipid Res. 2014, 56, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Deng, Z.; Sun, M.; Zhang, W.; Yang, Y.; Zeng, Z.; Wu, J.; Zhang, Q.; Liu, Y.; Chen, Z.; et al. Polydatin protects against lipopolysaccharide-induced endothelial barrier disruption via SIRT3 activation. Lab. Investig. 2020, 100, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Villarroya, F. SIRT3, a pivotal actor in mitochondrial functions: Metabolism, cell death and aging. Biochem. J. 2012, 444, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, M.; Zeng, X.; Yang, J.; Deng, H.; Yi, L.; Mi, M. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis. 2014, 5, e1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korla, K. Reactive oxygen species and energy machinery: An integrated dynamic model. J. Biomol. Struct. Dyn. 2016, 34, 1625–1640. [Google Scholar] [CrossRef]
- Petrosillo, G.; Francesca, M.R.; Di Venosa, N.; Paradies, A.G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: Role of reactive oxygen species and cardiolipin. FASEB J. 2003, 17, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Zoratti, M.; Szabò, I. The mitochondrial permeability transition. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001, 495, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Friberg, H.; Wieloch, T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie 2002, 84, 241–250. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Coskun, P.; Esposito, L.A.; Wallace, D.C. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. USA 2001, 98, 2278–2283. [Google Scholar] [CrossRef] [Green Version]
- James, A.M.; Collins, Y.; Logan, A.; Murphy, M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 2012, 23, 429–434. [Google Scholar] [CrossRef]
- Li, P.; Wang, X.; Zhao, M.; Song, R.; Zhao, K.S. Polydatin protects hepatocytes against mitochondrial injury in acute severe hemorrhagic shock via SIRT1-SOD2 pathway. Expert Opin. Ther. Tar. 2015, 19, 997–1010. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Feng, Q.; Shao, M.; Yuan, F.; Liu, F. Polydatin attenuates cadmium-induced oxidative stress via stimulating SOD activity and regulating mitochondrial function in Musca domestica larvae. Chemosphere 2020, 248, 126009. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Bio. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Qiu, T.; Kathayat, R.S.; Azizi, S.A.; Thorne, A.K.; Ahn, D.; Fukata, Y.; Fukata, M.; Rice, P.A.; Dickinson, B.C. ABHD10 is an S-depalmitoylase affecting redox homeostasis through peroxiredoxin-5. Nat. Chem. Biol. 2019, 15, 1232–1240. [Google Scholar] [CrossRef]
- Cui, X.; Brockman, D.; Campos, B.; Myatt, L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: Involvement in preeclampsia. Placenta 2006, 27, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gao, X.; Ma, L.; Liu, Z.; Li, L.; Wang, H.; Gao, L.; Xu, D.; Chen, Y.H. Obeticholic acid protects against gestational cholestasis-induced fetal intrauterine growth restriction in mice. Oxid. Med. Cell. Longev. 2019, 2019, 7419249. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Yen, G.C. Antioxidant activity and free radical-scavenging capacity of extracts from guava (Psidium guajava L.) leaves. Food Chem. 2007, 101, 686–694. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | GenBank ID | Sequence (5′-3′) | Length (bp) |
|---|---|---|---|
| mt D-loop | AF_276923 | GATCGTACATAGCACATATCATGTC | 198 |
| GGTCCTGAAGTAAGAACCAGATG | |||
| ACTB | DQ_452569 | CCCCTCCTCTCTTGCCTCTC | 74 |
| AAAAGTCCTAGGAAAATGGCAGAAG | |||
| SIRT1 | NM_001145750.2 | AGTTGAAAGATGGCGGACGA | 127 |
| CTCTCCGCGGTTTCTTGCG | |||
| SIRT3 | NM_001110057.1 | TGGTGTCGTTCATCTGTTGCTG | 117 |
| GGCACCGGGAGAAAAAGATATG | |||
| PGC1α | NM_213963.2 | TGTGCAACCAGGACTCTGTA | 152 |
| CCACTTGAGTCCACCCAGAAA | |||
| NRF1 | XM_021078993.1 | GAAGCTGTCCAGGGGCTTTA | 116 |
| ATCCATGCTCTGCTACTGGG | |||
| NRF2 | NM_001185152.1 | GGACAGCAGAAGTGATCCCC | 97 |
| CAAAACCGTATCACTGGCCG | |||
| ERRα | NM_001170521.1 | GTCGCTACCCTCTGTGACCT | 150 |
| GGCCACACCCAACACCAATA | |||
| TFAM | NM_001130211.1 | TGCTTTGTCTACGGGTGCAA | 100 |
| ACTTCCACAAACCGCACAGA | |||
| POLG | XM_001927064.4 | CTGTCAGATGAGGGCGAGTG | 133 |
| ACTTCTTCCGTCGTGACTTTCT | |||
| SSBP1 | XM_013985577.1 | CTTTGAGGTAGTGCTGTGTCG | 143 |
| CTCACCCCTGACGATGAAGAC | |||
| PPARA | NM_001044526.1 | GGCTGCTATCATTTGGTGCG | 80 |
| GCACGATACCCTCCTGCATT | |||
| CPT1A | NM_001129805.1 | TGGTGTCCAAATACCTCGCC | 145 |
| CCTCCGCTCGACACATACTC | |||
| FABP1 | NM_001004046.2 | AGGGGACATCGGAAATCGTG | 103 |
| TCACACTCCTCTCCCAAGGT | |||
| ACAA1 | XM_003132103.4 | TTCAAGGACACCACCCCTGA | 115 |
| GCTGAAGCACATTTCCCACG | |||
| ACOX1 | NM_001101028.1 | GCTGTCACCATGAACCAGGA | 166 |
| AGTTCAGGTCCTCATGCTGC | |||
| ACSL1 | NM_001167629.2 | AGAAGGAAACCGAAGCCTCC | 173 |
| AGAGTATCATCGGAGGAAGGACT | |||
| ACSL5 | NM_001195321.1 | ACTTCAGAGCAGCTCACACC | 123 |
| CCTTAGCTCTCCCCTCACCT | |||
| HADHA | NM_213962.2 | CGTCTACCAGGAGGGAGTGA | 90 |
| ATGTCAGGCCGAGAGGGTAT | |||
| Mn-SOD | NM_214127.2 | GGCCTACGTGAACAACCTGA | 126 |
| TGATTGATGTGGCCTCCACC | |||
| PRDX3 | NM_001244531.1 | CATGTGAGTGCCGTTCCTTG | 93 |
| AGACCACAGCACACTTGTCA | |||
| PRDX5 | NM_214144.1 | GTGGTGGCATGTCTGAGTGT | 170 |
| AGCCGTCGATTCCCAAAGAG | |||
| TXNRD2 | NM_001168702.1 | TGCTACGACCTCCTGGTGA | 110 |
| GGCGAAGGGCTCACATAGTC | |||
| GAPDH | NM_001206359.1 | CCAAGGAGTAAGAGCCCCTG | 125 |
| AAGTCAGGAGATGCTCGGTG |
| Items | NC | IC | NP | IP | p-Value | ||
|---|---|---|---|---|---|---|---|
| BW | Diet | BW × Diet | |||||
| GLU (mmol/L) | 6.18 ± 1.31 | 4.57 ± 1.20 | 6.63 ± 1.40 | 6.70 ± 1.82 | NS | 0.041 | NS |
| TG (mmol/L) | 0.438 ± 0.384 | 0.350 ± 0.195 | 0.987 ± 0.466 | 0.678 ± 0.310 | NS | 0.006 | NS |
| TC (mmol/L) | 1.90 ± 1.09 | 1.62 ± 0.325 | 2.03 ± 0.766 | 2.07 ± 0.535 | NS | NS | NS |
| TP (g/L) | 46.7 ± 7.63 | 44.5 ± 6.09 | 52.0 ± 8.51 | 51.3 ± 6.80 | NS | NS | NS |
| UN (mmol/L) | 4.10 ± 1.19 | 6.30 ± 1.93 | 4.18 ± 2.52 | 5.58 ± 2.19 | 0.041 | NS | NS |
| ALT (U/L) | 40.8 ± 21.2 | 103 ± 74.2 | 30.3 ± 8.89 | 64.7 ± 39.8 | 0.014 | NS | NS |
| AST (U/L) | 85.5 ± 43.9 | 172 ± 72.4 | 44.2 ± 22.8 | 61.2 ± 38.2 | 0.015 | 0.001 | NS |
| T-Bil (μmol/L) | 3.88 ± 2.74 | 9.03 ± 6.10 | 1.98 ± 3.51 | 3.50 ± 3.29 | NS | 0.039 | NS |
| Items | NC | IC | NP | IP | p-Value | ||
|---|---|---|---|---|---|---|---|
| BW | Diet | BW × Diet | |||||
| Apoptosis (%) | 1.18 ± 0.410 b | 3.86 ± 1.31 a | 1.06 ± 0.570 b | 1.89 ± 0.785 b | <0.001 | 0.006 | 0.014 |
| Caspase-3 (U/mg protein) | 23.9 ± 7.94 | 40.3 ± 8.57 | 26.7 ± 10.1 | 29.0 ± 7.78 | 0.016 | NS | NS |
| Caspase-8 (U/mg protein) | 5.41 ± 1.92 | 6.16 ± 1.68 | 4.78 ± 1.69 | 4.96 ± 2.00 | NS | NS | NS |
| Caspase-9 (U/mg protein) | 13.8 ± 1.73 | 18.0 ± 2.88 | 10.3 ± 4.41 | 13.1 ± 3.10 | 0.013 | 0.004 | NS |
| Items | NC | IC | NP | IP | p-Value | ||
|---|---|---|---|---|---|---|---|
| BW | Diet | BW × Diet | |||||
| Citrate cycle enzyme activities | |||||||
| CS (U/mg protein) | 20.9 ± 12.3 | 10.3 ± 3.48 | 27.5 ± 9.16 | 19.9 ± 5.22 | 0.014 | 0.026 | NS |
| ICDH (U/mg protein) | 8.13 ± 2.22 a,b | 5.56 ± 2.76 b | 7.52 ± 1.38 a,b | 9.65 ± 1.87 a | NS | NS | 0.013 |
| α-KGDH (U/mg protein) | 2.27 ± 0.802 | 1.38 ± 0.665 | 3.26 ± 1.38 | 2.22 ± 1.17 | 0.035 | 0.046 | NS |
| MDH (U/mg protein) | 5.28 ± 1.45 | 5.76 ± 1.84 | 5.50 ± 1.31 | 5.14 ± 3.52 | NS | NS | NS |
| Respiratory chain complex activities | |||||||
| Complex I (U/mg protein) | 15.7 ± 9.32 | 7.88 ± 3.97 | 23.2 ± 9.34 | 16.1 ± 5.50 | 0.023 | 0.017 | NS |
| Complex II (U/mg protein) | 8.11 ± 4.52 | 6.61 ± 5.25 | 10.5 ± 4.08 | 11.8 ± 6.40 | NS | NS | NS |
| Complex III (U/mg protein) | 7.39 ± 3.33 | 4.91 ± 1.65 | 8.66 ± 2.39 | 6.54 ± 2.58 | 0.039 | NS | NS |
| Complex IV (U/mg protein) | 31.0 ± 12.5 a,b | 18.6 ± 7.33 b | 25.1 ± 6.11 a,b | 32.6 ± 5.40 a | NS | NS | 0.008 |
| ATP synthase (U/mg protein) | 39.4 ± 10.6 | 29.3 ± 8.74 | 50.5 ± 12.6 | 43.7 ± 18.4 | NS | 0.027 | NS |
| Energy metabolite contents | |||||||
| ATP (μmol/g wet weight) | 25.6 ± 10.1 | 15.6 ± 6.96 | 30.3 ± 2.29 | 23.7 ± 7.77 | 0.012 | 0.045 | NS |
| NAD+ (μmol/g wet weight) | 1.33 ± 0.382 | 1.07 ± 0.184 | 1.66 ± 0.228 | 1.47 ± 0.309 | NS | 0.005 | NS |
| NADH (μmol/g wet weight) | 0.559 ± 0.363 | 0.614 ± 0.138 | 0.598 ± 0.142 | 0.550 ± 0.0895 | NS | NS | NS |
| NAD + /NADH (μmol/μmol) | 2.73 ± 0.829 | 1.77 ± 0.283 | 2.90 ± 0.686 | 2.73 ± 0.664 | 0.045 | 0.047 | NS |
| Items | NC | IC | NP | IP | p-Value | ||
|---|---|---|---|---|---|---|---|
| BW | Diet | BW × Diet | |||||
| Antioxidant enzyme activities | |||||||
| Cu/Zn-SOD (U/mg protein) | 66.5 ± 15.0 | 57.5 ± 15.5 | 87.3 ± 23.1 | 61.4 ± 9.89 | 0.018 | NS | NS |
| Mn-SOD (U/mg protein) | 36.0 ± 12.5 a,b | 19.6 ± 8.16 b | 37.1 ± 16.8 a,b | 48.6 ± 21.9 a | NS | 0.029 | 0.041 |
| GPx (U/mg protein) | 95.4 ± 44.4 | 85.2 ± 28.8 | 126 ± 42.3 | 88.1 ± 19.0 | NS | NS | NS |
| GR (U/g protein) | 9.54 ± 3.08 | 8.54 ± 2.66 | 11.3 ± 4.22 | 8.22 ± 2.82 | NS | NS | NS |
| CAT (U/g protein) | 115 ± 38.4 | 116 ± 44.0 | 106 ± 39.8 | 134 ± 47.5 | NS | NS | NS |
| Redox metabolite contents | |||||||
| GSH (μmol/g protein) | 5.94 ± 2.18 | 2.77 ± 1.09 | 5.80 ± 3.76 | 3.91 ± 2.83 | 0.030 | NS | NS |
| 8-OHdG (ng/mg DNA) | 1.69 ± 0.738 b | 3.04 ± 0.380 a | 1.77 ± 0.399 b | 1.76 ± 0.481 b | 0.005 | 0.011 | 0.004 |
| PC (nmol/mg protein) | 1.51 ± 0.443 | 2.39 ± 0.971 | 1.56 ± 0.306 | 1.61 ± 0.431 | NS | NS | NS |
| MDA (nmol/mg protein) | 2.56 ± 0.772 b | 5.28 ± 1.99 a | 2.89 ± 1.53 b | 2.70 ± 1.24 b | 0.046 | NS | 0.024 |
| Items | NC | IC | NP | IP | p-Value | ||
|---|---|---|---|---|---|---|---|
| BW | Diet | BW × Diet | |||||
| SIRT1 | 1.00 ± 0.758 | 0.877 ± 0.668 | 1.04 ± 0.891 | 1.26 ± 0.692 | NS | NS | NS |
| SIRT3 | 1.00 ± 0.366 a | 0.380 ± 0.0922 b | 0.819 ± 0.257 a,b | 1.14 ± 0.487 a | NS | 0.048 | 0.003 |
| PGC1α | 1.00 ± 0.358 | 0.567 ± 0.227 | 1.89 ± 0.898 | 1.23 ± 0.482 | 0.025 | 0.003 | NS |
| NRF1 | 1.00 ± 0.252 | 1.27 ± 0.454 | 2.39 ± 1.64 | 1.83 ± 0.978 | NS | 0.025 | NS |
| NRF2 | 1.00 ± 0.678 | 1.10 ± 0.394 | 1.24 ± 0.749 | 1.44 ± 0.777 | NS | NS | NS |
| ERRα | 1.00 ± 0.283 | 0.694 ± 0.324 | 1.29 ± 0.381 | 1.36 ± 0.594 | NS | 0.010 | NS |
| TFAM | 1.00 ± 0.424 | 0.538 ± 0.308 | 1.23 ± 0.248 | 0.871 ± 0.257 | 0.005 | 0.043 | NS |
| POLG | 1.00 ± 0.820 | 1.10 ± 1.06 | 1.49 ± 1.27 | 0.823 ± 0.344 | NS | NS | NS |
| SSBP1 | 1.00 ± 0.202 | 1.35 ± 0.817 | 0.939 ± 0.452 | 0.896 ± 0.267 | NS | NS | NS |
| PPARα | 1.00 ± 0.392 | 0.453 ± 0.106 | 2.59 ± 1.13 | 2.95 ± 2.05 | NS | <0.001 | NS |
| CPT1α | 1.00 ± 0.301 | 0.571 ± 0.252 | 2.03 ± 1.54 | 2.46 ± 1.10 | NS | 0.001 | NS |
| FABP1 | 1.00 ± 0.104 | 0.604 ± 0.178 | 1.69 ± 0.563 | 1.45 ± 0.273 | 0.028 | <0.001 | NS |
| ACAA1 | 1.00 ± 0.165 | 0.882 ± 0.333 | 0.951 ± 0.216 | 0.824 ± 0.180 | NS | NS | NS |
| ACOX1 | 1.00 ± 0.330 | 0.828 ± 0.396 | 0.785 ± 0.295 | 1.10 ± 0.409 | NS | NS | NS |
| ACSL1 | 1.00 ± 0.205 | 0.629 ± 0.214 | 1.07 ± 0.674 | 1.08 ± 0.168 | NS | NS | NS |
| ACSL5 | 1.00 ± 0.348 | 1.04 ± 0.529 | 1.70 ± 0.729 | 2.14 ± 0.640 | NS | 0.001 | NS |
| HADHA | 1.00 ± 0.306 | 0.811 ± 0.293 | 1.81 ± 0.621 | 1.03 ± 0.702 | 0.031 | 0.023 | NS |
| Mn-SOD | 1.00 ± 0.261 a,b | 0.560 ± 0.186 b | 0.943 ± 0.350 a,b | 1.10 ± 0.307 a | NS | 0.048 | 0.017 |
| PRDX3 | 1.00 ± 0.267 a,b | 0.414 ± 0.130 c | 0.815 ± 0.271 b,c | 1.36 ± 0.333 a | NS | 0.002 | <0.001 |
| PRDX5 | 1.00 ± 0.247 | 0.704 ± 0.222 | 1.46 ± 0.560 | 1.10 ± 0.194 | 0.028 | 0.006 | NS |
| TXNRD2 | 1.00 ± 0.296 | 0.806 ± 0.608 | 1.49 ± 0.635 | 0.881 ± 0.498 | NS | NS | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Li, Y.; Jia, P.; Ji, S.; Zhang, H.; Wang, T. Polydatin Attenuates Intra-Uterine Growth Retardation-Induced Liver Injury and Mitochondrial Dysfunction in Weanling Piglets by Improving Energy Metabolism and Redox Balance. Antioxidants 2022, 11, 666. https://doi.org/10.3390/antiox11040666
Chen Y, Li Y, Jia P, Ji S, Zhang H, Wang T. Polydatin Attenuates Intra-Uterine Growth Retardation-Induced Liver Injury and Mitochondrial Dysfunction in Weanling Piglets by Improving Energy Metabolism and Redox Balance. Antioxidants. 2022; 11(4):666. https://doi.org/10.3390/antiox11040666
Chicago/Turabian StyleChen, Yanan, Yue Li, Peilu Jia, Shuli Ji, Hao Zhang, and Tian Wang. 2022. "Polydatin Attenuates Intra-Uterine Growth Retardation-Induced Liver Injury and Mitochondrial Dysfunction in Weanling Piglets by Improving Energy Metabolism and Redox Balance" Antioxidants 11, no. 4: 666. https://doi.org/10.3390/antiox11040666
APA StyleChen, Y., Li, Y., Jia, P., Ji, S., Zhang, H., & Wang, T. (2022). Polydatin Attenuates Intra-Uterine Growth Retardation-Induced Liver Injury and Mitochondrial Dysfunction in Weanling Piglets by Improving Energy Metabolism and Redox Balance. Antioxidants, 11(4), 666. https://doi.org/10.3390/antiox11040666