Abstract
Oxidative stress is defined as an unbalance between pro-oxidants and antioxidants, as evidenced by an increase in reactive oxygen and reactive nitrogen species production over time. It is important in the pathophysiology of retinal disorders such as diabetic retinopathy, age-related macular degeneration, retinal detachment, and proliferative vitreoretinopathy, which are the focus of this article. Although the human organism’s defense mechanisms correct autoxidation caused by endogenous or exogenous factors, this may be insufficient, causing an imbalance in favor of excessive ROS production or a weakening of the endogenous antioxidant system, resulting in molecular and cellular damage. Furthermore, modern lifestyles and environmental factors contribute to increased chemical exposure and stress induction, resulting in oxidative stress. In this review, we discuss the current information about oxidative stress and the vitreous proteome with a special focus on vitreoretinal diseases. Additionally, we explore therapies using antioxidants in an attempt to rescue the body from oxidation, restore balance, and maximize healthy body function, as well as new investigational therapies that have shown significant therapeutic potential in preclinical studies and clinical trial outcomes, along with their goals and strategic approaches to combat oxidative stress.
1. Introduction
Oxidative Stress in Retinal Diseases
Oxidative stress (OS) is a common factor in many disorders, including retinal diseases, and is defined as an unbalance between antioxidants and pro-oxidants in favor of the former, leading to a disturbance of redox signaling and/or molecular damage [1]. Despite low amounts of reactive oxygen species (ROS) being necessary for cell homeostasis and redox signaling, increased intracellular concentrations can cause OS [1]. In addition to ROS, other reactive species, such as reactive nitrogen species (RNS) [2], reactive sulfur species [3], reactive electrophile species [4], and reactive halogen species [5], also play an important role in metabolic regulation. The most common ROS and RNS are hydrogen peroxide (H2O2), superoxide (O2−), hydroxyl (OH−), nitric oxide (NO), and peroxynitrite (ONOO−) [1,6]. ROS are produced continuously in mitochondrial oxidative metabolism during cell respiration as a consequence of the use of O2 as the final electron acceptor in aerobic organisms [7]. Nevertheless, the excess ROS/RNS production results in an imbalance between oxidants and antioxidants (preventive, sequestering, and repairing) systems, weakens the organism’s defense, and damages essential biomolecules, such as lipids, proteins, and DNA, with negative effects on a variety of organs. Thus, OS can be considered physiological (eustress) when it has specific targets in redox signaling, with a low exposure of organisms. On the other hand, it is considered toxic OS (oxidative distress) when the exposure of non-specific targets to supraphysiological levels of oxidant agents leads to the interruption of redox signaling, with subsequent pathophysiological repercussions [8]. In fact, OS is strongly associated with the onset of different pathologies, including cancer, diabetes, neurodegeneration, and cardiovascular and eye diseases [9]. Retinal diseases, such as diabetic retinopathy (DR), age-related macular degeneration (AMD), retinal detachment (RD), and proliferative vitreoretinopathy (PVR) have been related to higher levels of ROS, affecting a multiplicity of physiological processes, including vascular reactivity and neuronal function. Under various pathological conditions in the retina, ROS-producing systems are activated, which can include enzymes such as NADPH oxidase (NOX), xanthine oxidoreductase, cytochrome P450, mitochondrial cytochrome oxidase, and uncoupled endothelial NOS (eNOS) [10]. The retina is particularly susceptible to OS and lipid peroxidation due to its high metabolic and oxidative phosphorylation rates, high concentration of polyunsaturated fatty acids, and continuous light exposure [11,12]. ROS production imbalance impairs the delicate homeostasis and dynamics of the retinal neurovascular unit since the cells they compose, including vascular cells (pericytes and endothelial cells), the retinal pigment epithelium (RPE), glia, and neuronal cells, are highly susceptible to OS [13]. Coupling these cells as a single and intricate structural network is essential for supplying oxygen and nutrients to the highly metabolically demanding retina, ensuring their normal function and adaptation to varying physiological/pathological conditions [13,14,15]. Under stress conditions such as OS, activated microglial cells and RPE mount an adaptative low-grade inflammatory response, termed para-inflammation, through the release of growth factors and cytokines, and complement activation to restore tissue homeostasis and function. However, in an environment that has sustained oxidative damage, dysregulated para-inflammation gives rise to chronic inflammation mediated by pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1-beta (IL1β), and interleukin-6, which further increase the production of ROS in RPE cells [16,17,18]. Therefore, although para-inflammation is initially activated in response to OS to maintain rescue photoreceptors, stress-induced chronic inflammatory is the main factor responsible for photoreceptor degeneration [16,19]. Considering the role of OS in the pathophysiology of ocular diseases, the use of compounds with direct or indirect antioxidant activity could represent a beneficial therapeutic strategy to simultaneously reduce inflammation, neurodegeneration, and retinal and vascular dysfunction [18,20,21].
2. Oxidative Stress and Vitreous Proteome in Vitreoretinal Diseases
2.1. Diabetic Retinopathy
DR is one of the major complications in patients with diabetes mellitus and a leading cause of blindness and visual impairment in middle-aged adults [22]. According to the World Health Organization (WHO), 146 million (34.6%) out of the 422 million adults with diabetes suffer some form of DR [23], but these numbers are expected to grow in parallel with the increased prevalence of DR in middle- and low-income countries [24,25]. The standard treatment for DR is the intravitreal injection of anti-vascular endothelial growth factor (VEGF) agents, or corticosteroids when the response to anti-VEGFs is insufficient [26]. Combination therapy with laser photocoagulation may also be considered. Vitreoretinal surgery is applied for advanced phases of DR [27]. However, these treatments only ameliorate the symptoms.
For many years, DR was considered as a merely microvascular and macrovascular disorder [28,29]. In the early stages (non-proliferative DR), several microvascular changes occur in the eye in response to hyperglycemia, including microaneurysms, thickening of the retinal capillary basement membrane, and subsequent loss of pericytes [30]. Capillary non-perfusion and occlusion gradually drive retinal ischemia, which in turn triggers molecular mechanisms that lead to pathological intraretinal and intravitreal neovascularization in the proliferative phase of DR (PDR) [22,25,30]. Nevertheless, new evidence suggests that neuroglial degeneration may precede microvascular changes as a result of crosstalk between OS, inflammation, glial reactivity, and an imbalance production of neurotrophic factors [29,31]. Due to its multifactorial nature, the pathophysiological mechanisms underlying DR are not fully understood, but it has been suggested that OS plays a central role in metabolic changes triggered in response to chronic hyperglycemia [29,32]. Multiple biochemical pathways have been implicated in ROS production in DR, including the activation of protein kinase C (PKC) and hexosamine pathways, the increased glycolytic flux in the polyol pathway, the production of advanced glycation end products (AGEs), and the upregulation of its receptors (RAGE), as recently reviewed by several authors [32,33,34]. The overproduction of ROS promotes endothelial and mitochondrial dysfunction, inflammatory responses, the activation of microglial cells, and retinal cell apoptosis, which ultimately lead to the appearance of DR clinical features, such as capillary basement membrane thickness, increased vascular permeability, blood–retinal barrier (BRB) breakdown, and neurodegeneration [32,33,35]. On the other hand, it has been shown that photoreceptor cells are a major source of ROS and can contribute to the development of microvascular abnormalities by promoting a pro-inflammatory environment in the diabetic retina [35,36]. Nonetheless, the loss of stressed photoreceptors reversed the OS and inflammation in the retina and attenuated the deterioration of retinal capillaries, thus reducing the severity of DR [35,36]. This supports the role of metabolic stress of photoreceptors on retinal capillary degeneration, which appears to be mediated by the activation and recruitment of nearby cells (e.g., endothelial cells and circulating leukocytes) and release of pro-inflammatory factors [37]. This suggests that the microvascular changes in DR could result from oxidative and metabolic stress in the neural retina [38]. Therefore, the neuroprotection of photoreceptor cells against OS could offer an opportunity for an earlier treatment of DR, even before the appearance of microvascular lesions [35,37]. Despite more clinical trials being required to assess the efficacy of antioxidants as a neuroprotective therapy, they could potentially benefit DR patients (see Section 3) [21,39].
Like other ocular tissues, DR vitreous is affected by metabolic and functional modifications associated with OS. AGEs trigger abnormal crosslinks between collagen fibrils, causing the dissociation from hyaluronan and the destabilization of the gel structure. As a matter of fact, structural and molecular changes in vitreous and at the vitreoretinal surface might exert pathological effects in retinal capillaries, contributing to the progression to PDR [40,41]. Several authors have studied the OS-triggered changes in the vitreous humor proteome in DR/PDR. Higher levels of OS markers such as NO [42,43,44] and AGEs [45,46] were found in PDR vitreous, as well as increased levels of markers of OS damage, including protein carbonylation [45] and lipid peroxidation [47,48]. More recently, Suzuki and co-workers reported the upregulation of a new potential oxidative biomarker, OS-responsive apoptosis-inducing protein, in the vitreous of patients with PDR compared to a non-diabetic group [49]. Furthermore, some authors suggested that a decrease in the antioxidant defenses may be associated with the progression of DR, evidenced by decreased levels of catalase (CAT) [50], superoxide dismutase (SOD) [47], reduced glutathione (GSH) [43], and total antioxidant capacity [51]. On the other hand, Izuta and colleagues detected an increased antioxidant capacity in PDR vitreous and a positive association between it and the levels of VEGF, suggesting that these controversial results may be due to the different types of antioxidant enzymes analyzed in this study [48].
Proteomics [52,53,54,55,56,57,58,59,60,61,62,63,64] and multiplex ELISA [65,66,67,68,69] studies reported similar outcomes, confirming the impaired antioxidant capacity of vitreous in DR/PDR. Considering the proteins found differentially expressed in these studies, a functional enrichment analysis was performed using STRING v11.5 [70] to gain new insights into the role of OS in DR/PDR and to found potential markers (Figure 1 and Supplementary Table S1). This analysis suggests that the OS increase in DR may be countered by the activation of protective mechanisms through the upregulation of several antioxidant enzymes. Minamoto and co-workers detected CAT only in PDR vitreous, whereas glutathione peroxidase (GPX) was found only in the Macular hole (MH) vitreous [57]. Gao and co-workers reported significantly increased levels of the antioxidant peroxiredoxin-1 (PRDX1) in PDR vitreous and a trend for the upregulation of GPX3 and CAT but, in turn, extracellular SOD (SOD3) was not detected [60]. On the other hand, Zou and co-workers only found superoxide dismutase [Cu-Zn] (SOD1) in PDR vitreous compared with patients with MH [56], which suggests that SOD1 and SOD3 could contribute to different protective mechanisms in DR despite having the same catalytic activity. In addition to SOD1, Zou and co-workers reported the upregulation of other antioxidant proteins (CAT, PRDX) in PDR vitreous after treatment with anti-VEGF drug ranibizumab [56].
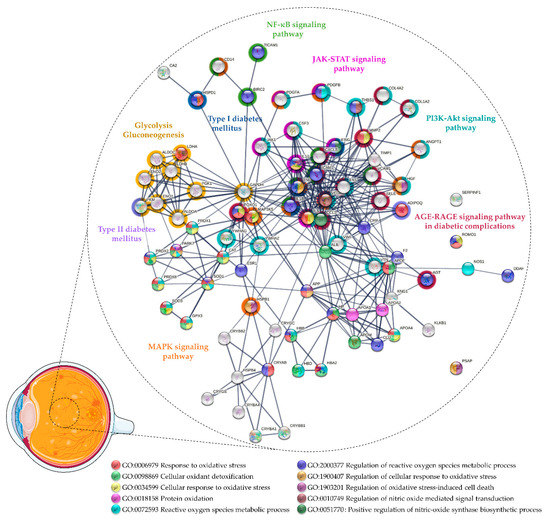
Figure 1.
Protein–protein interaction network of the proteins related to oxidative stress found differentially expressed in vitreous collected from patients with DR and proliferative DR (PDR). The network was predicted using STRING 11.5 based on high confidence interaction score and clustered using the Markov Cluster algorithm clustering (inflation parameter: 3).
Wang and co-workers found lower levels of antioxidants in PDR vitreous, including GPX3 and Parkinson disease protein 7 (PARK7) [62,63]. Loukovaara and co-workers detected higher levels of several OS markers in PDR versus NPDR vitreous, including reactive species modulator 1 and NO synthase (NOS), as well as several antioxidant proteins, such as PRDX2, PRDX6, and CAT [64]. Proteomics analysis also uncovered other antioxidant and neurotrophic proteins in DR/PDR, such as pigment epithelium-derived factor (PEDF), crystallins, or apolipoproteins (Supplementary Table S1 and Figure 1). PEDF is an anti-angiogenic, antioxidant, and neurotropic factor produced by retinal pigment epithelium (RPE) and Müller cells under physiological conditions [71]. Several authors reported unbalanced intravitreal levels of PEDF in DR/PDR; however, the results were inconsistent among studies [52,53,54,58,59,62]. PEDF was initially highlighted for its role as an inhibitor of angiogenesis [72], but its importance as an antioxidant and neurotrophic factor has been demonstrated in several animal models where the lack of this factor leads to an increased susceptibility to retinal degeneration and OS [71,73,74]. It was recently reported that the downregulation of PEDF induced in response to hyperglycemia is countered by the inhibition of AGE-RAGE signaling [75]. In turn, increased levels of PEDF could inhibit retinal hyperpermeability, leukostasis, and angiogenesis by repressing the generation of ROS and the AGE-signaling pathway [76,77,78]. Furthermore, it has been demonstrated that PEDF levels in patients with PDR are associated with the full antioxidant capacity of the vitreous, which suggests that PEDF has a protective role against OS and could be a therapeutic agent for DR/PDR [76].
Crystallins are lens structural molecules belonging to the small heat shock proteins (HSPs) family that also could have a neuroprotective role in DR [79]. Nevertheless, crystallins in DR/PDR could be linked to OS since their expression seems to be induced/repressed at mild and chronic OS conditions, respectively [80]. Accordingly, several members of the crystallin protein family were found to be differentially expressed in PDR vitreous, with some authors reporting their downregulation compared to healthy individuals [62,63], whereas others reported their upregulation in PDR [55,56,81,82]. The link between crystallins expression and OS was further confirmed by data suggesting that αA-crystallin was upregulated in mice after intravitreal injection of a recombinant AGEs protein, suggesting that αA-crystallin may protect photoreceptors against AGE-induced retinal injury [83]. In addition, other studies demonstrated that αB-crystallin could protect retinal cells against degeneration induced by OS [84,85]. More recently, Ghosh and co-workers reported that the loss of βA1-crystallin regulation in glucose metabolism and mitochondrial function of retinal astrocytes induces pathological features similar to DR, such as metabolic abnormalities, OS, and inflammation [82]. Nevertheless, diabetes can induce molecular changes in αA-crystallin, in particular at the phosphorylation level, which can affect its neuroprotective function [86,87]. So, crystallins undoubtedly play a role in DR pathogenesis and offer a potential therapeutic option, although more research will be required to fully understand their mechanism [82,86].
Clusterin (CLU), also known as Apolipoprotein J, is another extracellular chaperone produced by RPE cells that may have a protective role in DR by promoting cell survival and protection from apoptosis [88,89]. CLU showed to protect RPE cells from OS-induced damage, which contributes to cell survival via the PI3K/Akt pathway [90]. In an animal model in diabetes research, CLU expression decreased the levels of pro-inflammatory cytokines, including intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion protein 1 (VCAM1), and reduced the mitochondrial production of ROS, ameliorating vascular complications associated with diabetes [91]. Nevertheless, CLU has been found to be downregulated in PDR vitreous, suggesting the inhibition of its protective function [56,62,63]. Apolipoprotein E (APOE) could also have a protective role in DR by preventing the aggregation of toxic proteins. Recently, it has been shown that APOE is capable of protecting pericytes against amyloid-induced cytotoxicity linked to the development of type-2 diabetes [92]. Moreover, the lack of APOE has been correlated with an increase in ROS, which reinforces the potential role of OS in DR [93].
In addition to providing potential biomarkers of the vitreous antioxidant state in DR, many proteomics studies highlighted pathways related to OS, evidencing its role in PDR progression and as potential therapeutic targets (Figure 1 and Supplementary Table S1). Glycolysis/gluconeogenesis (KEGG entry (hsa): 00010) is one of the metabolic pathways affected in DR/PDR that seems to be related to OS. As mentioned above, multiple biochemical pathways, which are activated in response to chronic hyperglycemia, have been implicated in ROS production [32,33,34,94]. The glycolytic intermediates accumulated under hyperglycemia/ischemic conditions lead to the dysregulation of glycolysis and oxidative phosphorylation and, eventually, to other neural and microvascular complications observed in DR (reviewed by Yumnamcha and co-workers) [95]. For example, the activation of the polyol pathway increases the uptake of NADPH and NAD+, which is required for GSH regeneration, thus weakening the antioxidant defenses and increasing OS [32,34,94,95]. Meanwhile, the activation of PKC induces the expression of VEGF, endothelin-1, and plasminogen activator inhibitor-1 and the activation of NOX and nuclear factor kappa B (NF-κB) signaling (hsa: 04064), thus contributing to pericyte apoptosis, ROS production, inflammation, and vascular dysfunction [21,32,34,94,95]. AGE-RAGE signaling (hsa: 04933), one of the more enriched pathways according to Kyoto Encyclopedia of Genes and Genomes (KEGG) functional analysis, is another pathway activated in response to chronic hyperglycemia. AGE-RAGE signaling triggers several routes, which ultimately lead to the activation of signaling pathways such as phosphatidylinositol 3-kinase/protein kinase B (PI3K-Akt) (hsa: 04151), Janus kinase-signal transducer and activator of transcription (JAK-STAT) (hsa: 04630), and NF-κB signaling (hsa: 04064) [96]. NF-κB activation induces the expression of a series of pro-inflammatory cytokines [97], many of which were found to be upregulated in PDR vitreous, as described by Schori and colleagues [55]. Likewise, other authors reported the upregulation of TNF-α) [68], TIR domain-containing adapter molecule 1 (TICAM1) [64], baculoviral IAP repeat-containing protein 2 [64], IL1β [68,69], interleukin 8 [65,66,68], monocyte differentiation antigen CD14, and adhesion molecules (ICAM1, VCAM1) [55,66,67]. Many of these pro-inflammatory cytokines (TICAM1, IL1β, ICAM1), among other proteins (e.g., angiotensinogen, AGT), were related to the regulation of NO biosynthetic process (gene ontology (GO) entry: 0045428) in a functional analysis. The NO biosynthetic process is enhanced in response to the activation of NF-κB, which eventually leads to the apoptosis of endothelial cells exposed to hyperglycemia-induced OS [98]. The biosynthesis of NO is catalyzed by different isoforms of NOS, including constitutively expressed neuronal NOS (nNOS) and eNOS, and also inducible NOS (iNOS) [99], which is triggered under inflammatory and pathological conditions [99,100,101]. Under OS, cytokines such as IL1β, TNF-α, and interferon-gamma induce the synthesis of iNOS, a process that was shown to be reversed by antioxidants (e.g., CAT) [102]. Nevertheless, in proteomics studies, only nNOS, the main source of NO from retinal neurons in early DR, was found to be upregulated in PDR vitreous [64]. On the other hand, it has been suggested that eNOS, mainly expressed in the vascular endothelium, could trigger neovascularization by inducing VEGF and PGE2 [103]. Despite the increased levels of eNOS found in active diabetic fibrovascular epiretinal membranes (ERM) [104], it has been detected neither in the PDR vitreous nor in non-diabetic controls [103]. In any case, impaired eNOS expression has been linked to DR features such as changes in vascular permeability and the breakdown of the BRB [105,106]. Additionally, several pieces of evidence reinforce the idea that increased activity and levels of iNOS drive the increased BRB permeability, glial reactivity, and retinal degeneration observed in DR [44,107,108,109,110]. Enhanced NO production by iNOS has been implicated in various pathological processes, including inflammation, tissue damage, and cell apoptosis in an inflammatory and/or ischemic environment [101]. In addition, it was reported that the inhibition of iNOS by blocking angiotensin II (Ang II) type 1 receptor (AT1R) ameliorated glial activation and OS in animal models in diabetes [109]. These findings suggest a link between the NO and renin–angiotensin system (RAS), which are implicated in the progression of DR, and provide a potential therapeutic target against OS [100].
To counteract the negative effects of the activation of these pathways in response to OS, protective mechanisms seem to be triggered to modulate key pathological processes in DR. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-sensitive transcription factor that regulates the expression of genes encoding many antioxidant enzymes and phase II detoxifying enzymes, representing one of the major cellular defenses against OS [111]. It has been reported that diabetes increases retinal Nrf2 but decreases Nrf2 DNA-binding activity [112]. Gardner and Sundstrom [113] showed evidence that the Nrf2-mediated OS response is significantly activated in PDR, which is reinforced by the increased intravitreal expression of SOD2. However, other antioxidant proteins such as CAT, PRDX, and SOD1 and SOD3 were found to be downregulated in PDR vitreous compared to MH/ERM [113]. The potential therapeutic effect of Nrf2 was also supported by Deliyanti and co-workers, who showed that its stimulation prevents the increase in vascular permeability and the upregulation of angiogenic and inflammatory mediators induced by hyperglycemia [114].
2.2. Age-Related Macular Degeneration
AMD is a multifactorial ocular disease that affects the central retina and represents the leading cause of blindness in developed countries [115,116]. According to the WHO, the global burden of AMD is predicted to increase, foreseeing that 288 million people will be affected by the disease by 2040 [23,115]. AMD is categorized into two distinct forms, a “dry” or non-exudative form, and a “wet” or neovascular AMD (nAMD) [117]. In the early phase, yellowish deposits (drusen) are accumulated underneath the retina, accompanied by the infiltration of microglia and choroidal macrophages, thickening of Bruch’s membrane, and changes in RPE pigmentation [116,118]. The non-exudative form develops at more advanced stages of dry AMD and is characterized by overlying regions of RPE and photoreceptor cell degeneration [117,118]. The nAMD form accounts for only 10–15% of all cases but it is responsible of the most severe cases. At this stage, the patients present choroidal neovascularization (CNV) associated with vascular leakage and the breakthrough of these brittle vessels, which eventually leads to outer BRB breakdown, retinal and vitreous hemorrhage, fibrosis, and photoreceptor loss [116,118]. Currently, the treatment of these patients at this phase is based on anti-angiogenic drugs, but no therapy is yet available for “dry” AMD [119,120].
Even though AMD pathogenesis is not completely understood, OS has been identified as a key factor in the onset of this disease. As matter of fact, factors that contribute to OS, such as polymorphisms in antioxidant enzyme genes, cigarette smoke, and exposure to sunlight, are considered risk factors for AMD [120,121]. In recent years, multiple mechanisms have been evaluated to explain the role of OS in AMD [122,123,124]. The combination of a high demand for energy and oxygen, intensive light exposure, the rich content in polyunsaturated fatty acids (PUFAs) of the outer segment of photoreceptors, and the presence of photosensitizers makes the retina very susceptible to lipid peroxidation [17,125]. RPE is particularly important for maintaining retinal homeostasis and protecting from OS since it captures the excess light and removes the oxidized PUFAs and other waste products through different mechanisms [126,127,128]. Under acute OS, the autophagic activity becomes augmented in RPE cells as a protection mechanism. Nevertheless, chronic OS can in turn contribute to dysfunctional autophagy by blocking the function of lysosomal enzymes and the hyperpermeabilization of lysosomal membranes [128,129,130]. Impaired autophagy contributes to the formation of drusen [131,132], preventing the influx of oxygen and nutrients to the photoreceptors and the removal of the waste between RPE and choroid [133]. This disturbs the metabolic co-dependence ecosystem between the RPE and the photoreceptors, which ensures their proper function, suggesting that metabolic reprogramming could underlie retinal degeneration in AMD [134,135]. Several reports indicate that the overactivation of the mammalian target of rapamycin (mTOR) pathway may be the link between OS response, autophagic dysfunction, and metabolic reprogramming in AMD [129]. Increased activation of mTOR has been reported in RPE from AMD donors when compared with RPE cells from non-AMD eyes [136]. Although the mTOR pathway is activated in response to stress and starvation to promote RPE cells survival, its overactivation inhibits autophagy, induces mitochondria damages, and RPE dedifferentiation [129,136]. In turn, the chronic activation of mTOR prevents the return to baseline of glycolytic metabolism in RPE, leading to the nutrient deprivation of photoreceptors and further neurodegeneration [137,138]. Therefore, targeting the pathways associated with OS by reinforcing the supply of antioxidants and energy to photoreceptors could tip the balance toward the activation of antioxidant defense mechanisms in AMD [123]. Although therapies for “dry” AMD are still an unmet requirement, the consumption of antioxidant supplements in the context of a healthy diet has been recommended for the prevention of risk factors [139,140,141].
Although few studies have focused on vitreous proteomics in AMD, some potential biomarkers were provided [55,142,143]. Thus, the proteins found differentially expressed in these studies were considered for functional enrichment by STRING v11.5 [70] to identify potential biomarkers of OS and functional partnerships (Figure 2 and Supplementary Table S2). Specific pathways were found to be enriched, suggesting that compensatory mechanisms are triggered in response to OS. This fact is more evident at advanced stages of AMD, since several proteins associated with cellular oxidant detoxification were found to be upregulated in nAMD. Koss and co-workers reported higher levels of GPX3 and haptoglobin (HP) in the nAMD vitreous compared to patients with idiopathic floaters, proposing that their upregulation is due to the activation of detoxification mechanisms in response to OS [142]. GPX3 protects the retina from oxidative damage through the GSH metabolism (hsa:00480), which has been found to be affected in AMD according to a functional analysis based on the KEGG database. As recently reviewed, mitochondrial GSH removal in RPE and retina has been related to mitochondrial dysfunction and increased RPE cell death, emphasizing that the supply of GSH and the reinforcement of the glutathione antioxidant system could provide a potential therapeutic strategy for maintaining retinal homeostasis in AMD [144]. HP is an acute-phase protein that plays a relevant detoxification role by capturing free hemoglobin, protecting tissues against OS-induced damages [145]. It has been suggested that HP is expressed in the retina as an early protective mechanism against iron-mediated ROS formation, regulated by cytokines, such as IL1 and IL6 [146]. Therefore, HP could be also involved in the mediation of inflammatory processes in AMD, which is suggested by a functional analysis considering their role in immune system signaling (Reactome entry: R-HSA:168256). HP levels were found to be significantly increased in the macula with AMD when compared to controls [147], but its increase was non-significant in the serum of AMD patients [148]. Furthermore, although the HP phenotype 1–1, recognized for its better binding efficiency to hemoglobin and antioxidant action, has reported to be protective against DR, the same has not been verified for nAMD [149].
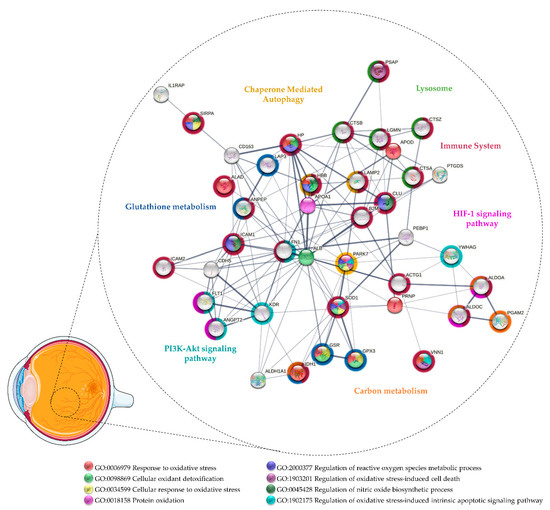
Figure 2.
Protein–protein interaction network of the proteins related to oxidative stress found differentially expressed in vitreous collected from patients with age-related macular degeneration. The network was predicted using STRING 11.5 based on medium confidence interaction.
Nobl and colleagues identified four potential biomarkers by comparing nAMD vitreous with different degrees of CNV [143]. Of these, they validated the upregulation of CLU and PEDF in nAMD [143], proteins that can play a protective role against OS and, hence, could be therapeutic agents in AMD such as in DR/PDR (see Section 3.1). Other authors reported decreased intravitreal levels of PEDF associated with CNV in AMD patients [150,151]. CLU has a cytoprotective effect, reducing apoptosis and OS in the retina [88,89,90]. Beyond vitreous, CLU was found to be upregulated in the aqueous humor [152], aqueous humor exosomes [153], macular Bruch membrane/Choroid [154], and drusen [155] of AMD donors. CLU has also been detected in the drusen from healthy/older donors [156,157], but it has been suggested that this accumulation is a response to injury or stress of the RPE or choriocapillaris [88,155]. As matter of fact, it was reported that RPE cells from AMD donors secrete more CLU compared to healthy control donors [158]. Although their role in ocular diseases is not well understood, multiple functions have been attributed to CLU [88,89]. This apolipoprotein is capable of inhibiting the formation of toxic beta-amyloid (Aβ) fibrils [159,160,161], known to increase the secretion of cytokines, triggering a pro-inflammatory environment in the retina and destabilization of RPE tight junctions [162,163,164,165]. More recently, it has been proposed that the autophagy activation in RPE cells may be a protective mechanism to reduce the accumulation of aggregated toxic proteins in the retina, such as Aβ [165]. This process can be assisted by chaperones, such as CLU, through chaperone-mediated autophagy (CMA; R-HSA:9613829) [132,166], which is one of the biological processes inferred in the functional analysis. Furthermore, both Aβ and CLU [88] can further contribute to AMD pathogenesis through their potential role in the regulation of the complement system. While Aβ peptides have been implicated in the activation of complement cascades [162,167,168], CLU is a negative regulator of the cytolysis mediated by complement activation [88,89]. Indeed, several complement components were detected in drusen and some genetic variants (e.g., Complement C3, factor H, and factor B) are considered to be risk factors for AMD [169,170,171,172,173]. Complement activation contributes to the recruitment and activation of immune cells, expression, and secretion of several pro-inflammatory cytokines and growth factors (e.g., VEGF), increased OS, and the accumulation of lipids in the retina [172,173,174]. On the other hand, it was hypothesized that CLU secretion may contribute to Aβ production by RPE cells and drusen formation [175]. Nevertheless, CLU seems to play a relevant role in AMD pathogenesis, but further studies are required to understand if it is a protective or pathogenic factor.
Interestingly, Schori and colleagues compared the vitreous from dry AMD and nAMD patients with ERM controls using a label-free LC-MS/MS quantitative method [55]. They found that several antioxidant proteins were significantly upregulated in nAMD, such as SOD1, glutathione reductase (GSR), and PARK7. However, SOD1 and other antioxidant enzymes such as GPX3, SOD3, and CAT only showed a trend for upregulation in dry AMD, which suggests that protective mechanisms (e.g., Nrf2 pathway) are activated with the progression of the disease [55]. As seen in Supplementary Table S2 and Figure 2, SOD1 and PARK7 were associated in the functional analysis as positive regulators of the OS-induced intrinsic apoptotic signaling pathways (GO: 1902175; GO: 1902177). As previously mentioned, PARK7 protects photoreceptors and RPE cells against OS by transcription regulation and ROS scavenging. The lack of PARK7 has also been linked to retinal/RPE degeneration in response to OS [176,177,178]. SOD1-deficient mice also exhibit pathological features found in AMD, including drusen, thickened Bruch’s membrane, and CNV [179]. Therefore, the loss of gene function of these antioxidant proteins provides a link between OS and neurodegeneration in the retina [180]. It was demonstrated recently by Zhu and co-workers that SOD1/PARK7/Parkin triple knockout exhibits retinal degeneration with aging [181]. Moreover, Nrf2-deficient mice also developed pathological hallmarks similar to human AMD, including deregulated autophagy, the accumulation of drusen, and RPE degeneration [182]. Considering that PARK7 and SOD1 have been correlated with the Nrf2 pathway [180], their upregulation in nAMD implies the activation of this defense system against OS [180,183,184]. In addition to proteins related to the Nrf2 pathway, Schori and colleagues [55] reported differentially expressed proteins implicated in processes associated with OS, including CMA (R-HSA: 9613829), GSH metabolism (hsa: 00480), HIF-1 signaling (hsa:04066), and PI3K-Akt signaling (hsa: 04151). Lysosome-associated membrane glycoprotein 2 (LAMP2), upregulated in nAMD, is a lysosome marker that may play a protective role in AMD as it mediates autophagy/CMA clearance through the regulation of autophagosomes and lysosomes (autolysosome) [185]. Under OS, an increase in the autophagic activity is required to assist the digestion and removal of damaged and oxidized material within autolysosomes, performed by lysosomal acid hydrolases such as cathepsins [166], which are also upregulated in nAMD [55]. In addition, the loss of LAMP2 function compromises phagocytic and lysosomal degradation, leading to the increased exocytosis of defective materials in RPE cells and the accumulation of basal laminar deposits [185]. Regarding the proteins involved in glutathione metabolism, GSR has been found to be upregulated in nAMD, but isocitrate dehydrogenase [NADP] cytoplasmic (IDH1) and aminopeptidases were found to be downregulated in dry AMD. IDH1 is essential for efficient glutathione recycling since it provides NADPH to form GSH by GSR [186]. Indeed, the activation of the Nrf2 pathway regulates the NADPH-producing enzymes, such as IDH1, thus modulating its levels [187,188]. An IDH1 increase in nAMD compared to ERM controls and dry AMD was detected, but this difference was not statistically significant. Considering the contribution of OS in AMD, it is likely that the Nrf2 represents a chief regulator of antioxidant protective mechanisms [55]. Thus, as in PDR, the activation of the Nrf2 pathway could be a suitable therapeutic approach, particularly in dry AMD, for which no therapy is available.
2.3. Retinal Detachment and Proliferative Vitreoretinopathy
RD is a potentially blinding disease characterized by a physical separation between the neurosensory retina and the RPE [189]. Rhegmatogenous retinal detachment (RRD) represents the most common type of RD [189], with an incidence of 13 cases per 100,000 people per year [190,191]. In developed countries, 3.9% to 13.7% of all patients with RD develop PVR, a complication that represents the most common cause of failure in RRD surgery [192,193]. PVR results of exacerbated vitreoretinal wound-healing in response to trauma, RD, and surgical interventions at the vitreoretinal interface, leading to the formation of fibrovascular membranes and vitreoretinal traction [193,194]. Currently, the management of these diseases is exclusively surgical, with primary success rates of up to 90% [195,196]. Nevertheless, when RRD is associated with PVR, patients experience worse visual recovery and reattachment rates [197]. Numerous drugs have been proposed for the prevention of PVR or recurrence after surgery, including corticosteroids, anti-neoplastic/anti-proliferative, anti-VEGF, and antioxidant agents but, until now, no proven pharmacologic agents have been incorporated routinely into clinical practice [193,198].
In most cases, RRD arises as a consequence of the liquefaction and degeneration of the vitreous, which is initiated by several factors (e.g., aging or trauma) that induce the disintegration of collagen structural network and reduce vitreoretinal adhesion, leading to the accumulation of vitreous fluid in the subretinal space [199,200]. OS is one of these factors, suggesting that an imbalance between the levels of ROS and antioxidants can be underlying vitreous liquefaction and RRD [201]. Some authors reported higher levels of OS biomarkers [202], but lower antioxidant capacity [203], in the vitreous of patients with RRD when compared to MH patients. Higher levels of OS markers in the vitreous have been related to RRD severity [202], whereas the extent of the detached area in RRD was positively correlated with biological antioxidant potential in the vitreous fluid [203]. Although no significant differences in the antioxidative capacity of the vitreous between patients with RRD and ERM/MH were found, the increase in the total antioxidant status of vitreous is correlated with the duration of RRD and probably with the progression to PVR [204]. Beyond this, the physical separation of the neurosensory retina and the underlying RPE interferes with the supply of oxygen and nutrients, thus leading to an ischemic environment, lack of neurotrophic factors and, eventually, the death of photoreceptors and neurons. Under hypoxic and metabolic stress, photoreceptors undergo mitochondrial dysfunction, which contributes to the overproduction of ROS and a higher susceptibility to OS [194,205]. Furthermore, the BRB breakdown leads to an influx of growth factors and inflammatory mediators, which increases the chemotactic and mitogenic activity in vitreous and foments leukocyte infiltration into the injury site [192,194,206]. The oxidative burst of neutrophils and macrophages further increases the release of ROS, neurotoxic enzymes, and cytokines [193]. In turn, cytokines stimulate the activation of macrophages, Muller cells, and microglia, which also contribute to the cytotoxic effect mediated by OS on the photoreceptors after RD [194,207]. The exposition to cytokines and growth factors, changes in cell adhesion, and the loss of signaling resultant from retinal cells death provoke the epithelial–mesenchymal transition (EMT), an essential pathological process in the development of PVR [193,208] that results in ECM remodeling, preventing the reattachment of the retina [209,210]. As in other pathological processes, OS plays a role in EMT, which contributes to the pathogenesis of PVR [211]. A synergistic effect between transforming growth factor, macrophage migration inhibitory factor, and H2O2 induces EMT through the upregulation of α-smooth muscle actin, vimentin, and fibronectin and the downregulation of cadherins [211,212]. So, considering the central role of OS in many of these pathological mechanisms, it has been suggested that antioxidants may attenuate photoreceptor death after RD and delay its progression to PVR [213,214,215].
The first studies on vitreous proteome were focused on PVR [59,216,217,218] and only recently more attention has been directed to the pathological mechanisms underlying RRD pathogenesis [219,220,221,222]. These studies shed some light on the role of OS in these pathologies (see Supplementary Table S3 and Figure 3). Yu and co-workers conducted some of the first proteomics approaches in the study of human vitreous samples collected from RRD patients with PVR [216,217,218]. Detoxification proteins, such as SOD1, PARK7, PRDX1, and PRDX2, were only detected in vitreous from healthy donors, whereas SOD3, GPX3, and HP were only detected at more severe PVR grades [216]. This suggests that a reduction in the antioxidant capacity in the eye could be involved in the onset of PVR, but protective mechanisms against OS can be triggered in more advanced states. For example, SOD1 was only detected in healthy donors, while SOD3 was only detected in severe PVR, which reinforces, like in PDR (see Section 3.1), that these proteins play different roles despite having the same catalytic activity. Yu and co-workers also found differential levels of Aβ, AGT, and neutrophil collagenase (MMP8), proteins associated with the regulation of ROS biosynthetic process (GO: 1903426), including the NO biosynthetic process [216]. High intravitreal levels of NO and NO pathway metabolites have been implicated in RD/PVR [43,223] and associated with various pathological processes (see Section 3.1). Therefore, the inhibition of NOS and scavenging of ROS could provide a neuroprotection mechanism against OS by potentiating the effect of neurotrophic factors [224]. Aβ was only detected in moderate PVR in this study, while AGT and MMP8 were found in severe PVR [216]. It has been shown that Aβ is expressed in response to elevated ROS [225]. In turn, Aβ stimulates NO biosynthesis through the induction of iNOS, providing a correlation between OS and Aβ-induced damages, such as mitochondrial dysfunction, neurotoxicity, inflammation, and neuronal apoptosis [225,226,227].
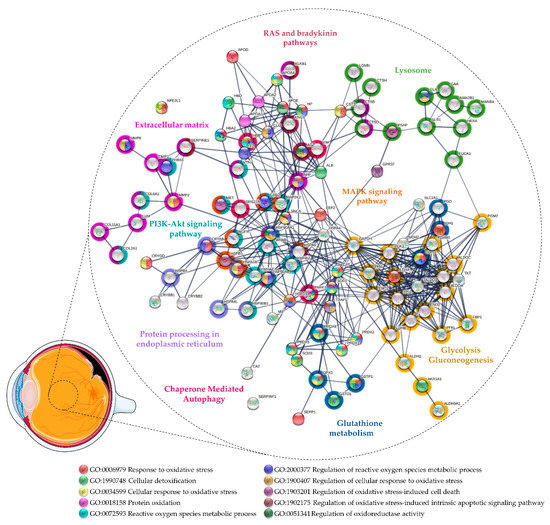
Figure 3.
Protein–protein interaction network of the proteins related to oxidative stress found differentially expressed in vitreous from patients with rhegmatogenous retinal detachment (RRD) and proliferative vitreoretinopathy (PVR). The network was predicted using STRING 11.5 based on high confidence interaction score and clustered using the Markov Cluster algorithm clustering (inflation parameter: 3).
MMP8 is constitutively expressed and stored in the secretory granules of neutrophils and eosinophils [228,229]. MMP8 is mainly present in its latent form (pro-MMP8) [229], but NO and its reactive intermediates are capable of converting MMP8 into its active form, which could be inhibited by antioxidant proteins such as SOD or CAT [230]. Once activated, MMP8 cleaves a wide range of substrates, including fibrillar collagens (types I, II, III), proteoglycans, serine protease inhibitors, and several chemokines [228,229]. An imbalance in the levels of matrix metalloproteins and their inhibitors (e.g., TIMP) within the detached retina and vitreous has been suggested to have a role in RRD and PVR membrane formation [231,232]. Indeed, the levels of MMP8 in vitreous/subretinal fluid have been correlated with pathological features, such as the duration and extent of RD [233,234] and the grade of postoperative PVR [235]. AGT is an important component of the RAS, which is responsible for the control of retinal vasculature (recently reviewed by Phipps [236] and Holappa [237]) and, therefore, its dysfunction has been correlated with some vitreoretinal diseases. AGT is cleaved into several effector peptides through different cleavage pathways, but the best known is the conversion to Ang II via the renin/angiotensin-converting enzyme (ACE). Ang II has a vasoconstrictor effect, through cell proliferation and fibrosis, or a vasodilator effect, depending on whether the activation is via AT1R or receptor 2 (AT2R), respectively. Even thus, its role in the regulation of retinal vasculature and the maintenance of the neurovascular unit is not fully understood [236,237]. Nevertheless, Hoerster and co-workers hypothesized that the inhibition of ACE could counteract EMT and, eventually, fibrosis in PVR. However, the administration of ACE inhibitors did not show any effect on the levels of pro-fibrotic cytokines/growth factors [238]. Notwithstanding this, ACE inhibitors can also affect the bradykinin breakdown in the kallikrein–kinin system [239,240] which has also been implicated in PVR by Yu and co-workers [216,217,218]. They found higher levels of kininogen 1 in severe PVR when compared to moderate cases, suggesting a potential biomarker of the disease severity [216,217,218]. Kininogen is converted by kallikrein into kinin-free kininogen, which has anti-proliferative and anti-angiogenic properties, and bradykinin, which promotes vasodilation through the release of tissue plasminogen activator, NO, and prostacyclin from endothelial cells [239,240]. Thus, it may have beneficial effects by preventing PVR events, such as ECM deposition, cell proliferation, and by protecting the retina against ischemic damage [241,242,243]. On the other hand, bradykinin could further contribute to the pathology by inducing the expression of inflammatory mediators and NO formation and increasing retinal vascular permeability, which could lead to retinal edema and intraocular hemorrhage [241,243]. So, further studies are needed to understand the role of the RAS and kallikrein–kinin systems in PVR.
Regarding vitreous studies on RRD, Wu and co-workers found 103 proteins differentially expressed by comparing samples from patients with RRD, associated with choroidal detachment, using iTRAQ labeling. Of these, proteins associated with response to OS (GO:0006979) and cellular detoxification (GO: 1990748) were found to be upregulated, including HP and apolipoproteins A4 and D. On the other hand, proteins with chaperone activity such as APOE and CLU were downregulated. These proteins may have a protective role against OS-induced damage and amyloid-induced cytotoxicity in retinal diseases (see Section 3.1). Of note, Aβ was found to be downregulated in this study. Higher levels of kininogen 1 and plasma kallikrein were found in RRD associated with choroidal detachment than in RRD, reinforcing the potential role of kallikrein–kinin systems in PVR pathogenesis [244]. Our research group compared the vitreous proteome in RRD with ERM using iTRAQ labeling [219]. Of the 150 proteins reported as differentially expressed, several detoxification enzymes were found to be upregulated in RRD, including PRDX1, PRDX2, glutathione S-transferase (GSTP1), and aldo-keto reductase family 1 member A1 (AKR1A1). GSTP1 is an intracellular detoxification enzyme expressed in the retina, iris, and cornea that plays an important role in glutathione metabolism (see Section 3.2) [144,245,246,247]. Its overexpression in RPE cells protects them against H2O2-induced mitochondrial damages and death [248]. We also found higher levels of several proteins of the crystallin family, including α- and β-crystallins, which could have a role in the response to OS. We hypothesized that their overexpression could represent the activation of a protective mechanism against retinal cell death in RD [219], as mentioned above in Section 3.1 [84,85]. Recently, Hamadmad and co-workers confirmed our results, both at the transcriptional and translational levels, showing that αA-crystallin is upregulated after RD and that its increase is time dependent [249]. More recently, Öhman and co-workers compared the vitreous proteome of RRD patients with other vitreoretinal diseases, including MH, ERM, and PDR using SWATH-mass spectrometry [220]. They identified and quantified a total of 1177 proteins across all 151 vitreous samples, which represents, to our knowledge, the largest dataset of the RRD vitreous proteome. They confirmed our previous results, finding higher levels of AKR1A1, PRDX1, PRDX5, PRDX6, and glutathione metabolism-associated enzymes (GSTP1, GSTO1) in RRD when compared to other diseases. Additionally, they reported the upregulation of other detoxification enzymes including CAT, SOD1, and PARK7. This study also confirmed our reported upregulation of HSPs, which can represent a mechanism to protect retinal cells against ischemia and OS. The upregulation of HSPs can prevent apoptosis, promoting cell survival by several mechanisms, including the activation of the PI3K-Akt signaling pathway (hsa: 04151) or the increase in cell antioxidant potential (e.g., higher GSH levels) [250,251,252]. The upregulation of HSPs and other proteins associated with CMA (R-HSA: 9613829) and the ubiquitin–proteasome system (WikiPathways entry: WP2359) in both studies [219,220] may suggest the triggering of autophagy during RRD. The downregulation of autophagy has been associated with a reduction in photoreceptor survival after RD [253]. Recently, Xiao and co-workers showed that the inhibition of autophagy in rod photoreceptors increased their apoptosis and necroptosis during RD and diminished the expression of key aerobic glycolysis intermediates. This suggests that functional autophagy maintains photoreceptors’ homeostasis, glycolysis, and survival under oxygen and nutrient deprivation [254]. Interestingly, several glycolytic enzymes were reported as upregulated in RRD vitreous both by our research group and Öhman and co-workers [219,220], which implies that retinal cells increase their metabolism after RD. On the other hand, Yu and co-workers reported lower levels of several glycolytic enzymes in moderate and severe PVR [216]. Therefore, this suggests that the glycolytic process is activated after RD to compensate for the metabolic stress of retinal cells, but its reduction may lead to the death of photoreceptors at more severe stages of PVR. In conclusion, the regulation of the glycolytic process may represent a potential therapeutic strategy to limit photoreceptor death after DR.
3. Targeting Oxidative Stress/the Treatment by Inhibition of Oxidative Stress
Several pathways are implicated in ROS production and increased OS in retinal diseases. RAS is a significant route pathway in retinal diseases, and drugs targeting Ang II are under research. High levels of prorenin and Ang II have been identified in the vitreous of diabetic patients [255]. Ang II contributes to diabetic retinal complications by activating the AT1R/AT2R in blood vessels and retinal neurons and may trigger NOX via AT1R, causing ROS production in retinal blood vessels and Müller cells, resulting in retinal vascular dysfunction and neurodegeneration [256]. Other causes of an increase in ROS include structural changes in macromolecules (DNA, proteins, and lipids), which generate permanent chemical reactions that cause oxidative damage to the human body. However, unexpected electron release and interaction with molecular oxygen are major contributors to ROS generation [257]. Other ROS are produced in excess by the action of exogenous factors representing stress to the human organism, such as environmental pollution (air, soil, and water), radiation of all types (UV, gamma, etc.), toxic habits (tobacco, alcohol, and drugs), poor diet, the exposure to toxic substances (fertilizers and pesticides), metabolism of some medicines and high physical or psychological stress [258]. Although controversial, there is another way of ROS production as a consequence of genetic alterations (hereditary or not) or physiological disorders of the organism. However, some authors consider the excessive production of ROS, either endogenous or exogenous, as a consequence of genetic alterations or physiological disorders [259]. Although antioxidant mechanisms are available to reduce the excess of ROS, when these defenses are insufficient an imbalance occurs in favor of excessive ROS production or the weakening of endogenous antioxidant systems [260]. To summarize, reducing OS can be achieved by decreasing exposure to environmental pollutants with oxidative properties, by increasing levels of endogenous and exogenous antioxidants, or by decreasing the generation of ROS. In addition to the elimination of toxic habits, such as alcohol and tobacco consumption, and the increased consumption of vegetables and fruit, the introduction of supplements with antioxidant properties in the diet has increased lately with the aim of counteracting the harmful effects of OS. In fact, high antioxidant levels may eliminate or prevent the formation of ROS However, studies on the effect of antioxidant diet supplements are inconclusive or inconsistent, suggesting that they do not offer sufficient protection against OS. We are left with the third option of suppressing oxidative damage, which appears to be the key to success in the fight against OS.
3.1. Antioxidants
An antioxidant is a compound that can reduce or inhibit the oxidation of other molecules. Oxidation is a chemical reaction in which electrons or hydrogen are transferred to an oxidizing agent. Free radicals produced during oxidation reactions initiate chain reactions that, when occurring inside the cells, might harm or kill them. Antioxidants remove the free radicals and inhibit additional oxidation processes [261]. The optimal antioxidant treatment would be one preventing the production of excess ROS, stimulating the reparation processes of tissues damaged by ROS attack, and increasing the antioxidant capacity of the human organism by supplying components capable of removing excess ROS. Eating fresh fruits and vegetables regularly is one of the strategies to prevent excessive ROS production and should be the first step in the treatment approach; nevertheless, it is not always a sufficient response to excessive ROS production [262]. The topic of antioxidant therapy is still a matter of debate, being the discussion focused on the administration of food supplements containing antioxidant compounds. Experimental evidence on the relationship between OS and disease progression is overwhelming, particularly in chronic disease. However the therapeutic administration of antioxidant products is frequently regarded as supplemental or secondary [260]. Current regulations consider antioxidants as nutritional supplements or natural health products rather than medications because OS is not recognized as a clinical category [263]. Studies show that dietary supplementation with foods rich in carotenoids (especially lutein and zeaxanthin) can increase macular pigment concentration and density. Their protective effects include absorption of harmful wavelengths that are associated with photochemical damage to the sensory retina as well as the removal of free radicals and ROS generated by metabolic activity [264]. However, studies on the anatomical and functional benefits appear to be controversial. A five-year follow-up study (The Beaver Dam Eye Study), showed a modest link between zinc intake and decreased risk of early AMD [265]. In a similar study (Blue Mountains Eye Study), there a significant association between antioxidant intake and the incidence of early AMD was not found [266]. Noteworthy, antioxidant doses in this study were lower than in a third study (Age-Related Eye Disease Study, AREDS) [267]. AREDS demonstrated that a combination of antioxidants, such as vitamin C 500 mg, Vitamin E 400IU, Beta-carotene 15 mg (or Vitamin A 25,000 IU) plus zinc oxide 80 mg, and cupric oxide 2 mg, decreased the risk of progression to the advanced form of AMD by 25% and reduced the risk of decreased vision by 27% [267].
It is worthwhile to acknowledge that the arsenal of antioxidant therapy does not only include antioxidant substances, but also non-classical agents, such as metal chelators (for example, serum EDTA, D-spherrosamine, among others), oxidant agents whose mechanism is based on the stimulation of endogenous antioxidants. Table 1 provides a brief overview of antioxidants, molecules essential for the action of various antioxidant enzymes, and enzymatic systems for reducing OS.

Table 1.
Summary of antioxidants, molecules essential for the activity of some antioxidant enzymes and enzymatic systems for reducing oxidative stress (OS).
3.2. Investigational Therapies
Several antioxidant therapy approaches are currently being researched, some of which are in the clinical trial stage. These new therapies aim to prevent the formation of ROSor/and promote its removal. The elimination of O2− prevents the formation of ONOO−, while the remotion of H2O2 prevents the formation of OH− and HOX. Other investigational therapies consist of the administration of enzymes (e.g., SOD) that remove H2O2 and lipid hydroperoxides, preventing the formation of other ROS. Several different strategies are being investigated to combat OS, such as the increase in GSH using its precursors, the stimulation of the synthesis of antioxidant enzymes through the activation of Nrf2, the inhibition of NOX, and the supplementation of dietary antioxidants, among others [284].
Clinical trials have shown that preventing ROS formation can reduce the retinal damage associated with the vascular breakdown and neurodegeneration observed in DR and AMD [285]. Numerous molecules have been evaluated as antioxidants in preclinical studies, as summarized in Table 2, suggesting that there is an important therapeutic potential. The effectiveness of activators of Nrf2, such as PB125, in decreasing OS in age-related diseases has been studied in clinical assays [286]. As previously mentioned, the activation of Nrf2 has great potential as a therapeutic approach for the treatment of retinal diseases, particularly neurodegenerative diseases [111]. In addition to protecting the retina against the OS, Nrf2 activators demonstrated great efficiency in reducing inflammation, vascular permeability, and neuronal degeneration in animal/cells models of glaucoma [287,288], DR [114,289], and AMD [290]. However, the results of clinical trials with antioxidants are inconclusive to date. One of the main reasons for this may be the use of a single agent in clinical trials with antioxidants, challenging the ability to therapeutically apply antioxidant strategies. One limitation of the single use of antioxidants depends on whether OS plays a primary or secondary role in a specific disease. If secondary, the antioxidant agent may not significantly impact disease progression, and therefore, the extent to which these agents may be used to ameliorate some symptoms should be assessed [263]. Another important limitation is the fact that certain molecules do not work per “si” but in a chain of reactions, leading to them potentially not being so efficacious in vivo [263]. Lastly, as retinal diseases are typically multifactorial diseases, patients could benefit from the combined administration of several exogenous antioxidant compounds or antioxidants with other drugs [291]. However, since these formulations could have synergistic therapeutic effects, it is difficult to isolate the effect of a specific antioxidant molecule by itself or generate certain antioxidant therapy combinations, which can make it difficult to adjust the recommended dose for each patient. Therefore, the safety and effectiveness of these formulations and/or purified natural antioxidants should be assessed in clinical trials or pharmacovigilance studies [291]. All these limitations make conducting a clinical trial very challenging.
Table 2.
Summary of the therapeutic strategies against oxidative stress (OS) and their use for certain diseases, including diabetes and its complications.
4. Conclusions
Research on oxidative stress has been of increasing importance for understanding the development of vitreoretinal diseases. This review summarizes the results of proteomics studies with the focus on biomarkers of the vitreous antioxidant state in DR, AMD, RD, and PVR. These biomarkers are potential hallmarks to evaluate vitreoretinal disease progression and to provide new and innovative therapeutic targets for vitreoretinal disorders. Proteins found to be differentially expressed in these studies were used to perform functional enrichment analyses in STRING v11.5 to gather new insights into the role of ROS in these diseases and to find potential markers for the detection of early stages of OS, essential to prevent further retinal harmful effects. Additionally, several molecules were described as antioxidants with therapeutic potential in preclinical studies. Although the results of clinical trials are inconclusive to date, they offer great hope for the future development of new therapeutic strategies based on antioxidative treatments.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/antiox11030505/s1, Table S1: List of proteins found differently expressed in DR vitreous and functional analysis; Table S2: List of proteins found differently expressed in AMD vitreous and functional analysis; Table S3: List of proteins found differently expressed in RRD/PVR vitreous and functional analysis.
Author Contributions
Conceptualization, F.M.S., J.M., A.P. and C.T.T.; validation, F.M.S., J.M., A.P., S.C., J.P.C.-d.-S. and C.T.T.; data curation, F.M.S. and S.C.; writing—original draft preparation, F.M.S., J.M. and C.T.T.; writing—review and editing, F.M.S., J.M., A.P., S.C., J.P.C.-d.-S. and C.T.T.; supervision, J.P.C.-d.-S., A.P. and C.T.T.; project administration, C.T.T.; funding acquisition, C.T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was developed within the scope of the CICS-UBI projects UIDB/00709/2020 and UIDP/00709/2020, financed by national funds through the Portuguese Foundation for Science and Technology/MCTES. It was also funded by the European Regional Development Fund through the “Programa Operacional Regional do Centro (Centro 2020)—Sistema de Apoio à Investigação Científica e Tecnológica—Programas Integrados de IC&DT”, Centro-01-0145-FEDER-000019—C4—Centro de Competências em Cloud Computing.
Acknowledgments
F.M.S. acknowledges a doctoral fellowship [SFRH/BD/112526/2015] from FCT. CNB-CSIC proteomics lab is a member of Proteored, PRB3-ISCIII, supported by grant PT17/0019/0001. C.T.T. acknowledges COMBINE project “CombinatiOn aptaMer-drug Based therapeutics for ocular neovascular dIsease: wheN two are bettEr than one”, funded by BInov 2021, Innovation Grant of the Southern Regional Section and Autonomous Regions (SRSRA) of Portuguese Pharmacists Order.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACE | Angiotensin-Converting Enzyme |
| AGEs | Advanced glycation end products |
| AGT | Angiotensinogen |
| AKR1A1 | Aldo-Keto Reductase Family 1 Member A1 |
| AMD | Age-Related Macular Degeneration |
| Ang II | Angiotensin II |
| APOE | Apolipoprotein E |
| AT1R | Angiotensin II type 1 receptor |
| AT2R | Angiotensin II type 2 receptor |
| Aβ | Beta Amyloid |
| BRB | Blood–retinal barrier |
| CAT | Catalase |
| CLU | Clusterin |
| CMA | Chaperone-Mediated Autophagy |
| CNV | Choroidal Neovascularization |
| DR | Diabetic Retinopathy |
| EMT | Epithelial–Mesenchymal Transition |
| eNOS | Endothelial Nitric Oxide Synthase |
| ERM | Epiretinal membranes |
| GPX | Glutathione Peroxidase |
| GSH | Reduced Glutathione |
| GSTP1 | Glutathione S-Transferase |
| H2O2 | Hydrogen Peroxide |
| HP | Haptoglobin |
| HSPs | Heat Shock Proteins |
| ICAM1 | Intercellular Adhesion Molecule 1 |
| IDH1 | Isocitrate dehydrogenase [NADP] cytoplasmic |
| IL1β | Interleukin-1 beta |
| iNOS | Inducible Nitric Oxide Synthase |
| JAK-STAT | Janus Kinase-Signal Transducer and Activator of Transcription |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| hsa | Kyoto Encyclopedia of Genes and Genomes Entry |
| LAMP2 | Lysosome-associated membrane glycoprotein 2 |
| MH | Macular hole |
| MMP8 | Neutrophil Collagenase |
| mTOR | Mammalian Target of Rapamycin |
| nAMD | “Wet” or Neovascular Age-Related Macular Degeneration |
| NF-κB | Nuclear Factor Kappa B |
| nNOS | Neuronal Nitric Oxide Synthase |
| NO | Nitric Oxide |
| NOS | Nitric Oxide Synthase |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| O2− | Superoxide |
| OH− | Hydroxyl |
| ONOO− | Peroxynitrite |
| OS | Oxidative Stress |
| PARK7 | Parkinson Disease Protein 7 |
| PDR | Proliferative Diabetic Retinopathy |
| PEDF | Pigment Epithelium-Derived Factor |
| PI3K-Akt | Phosphatidylinositol 3-Kinase/Protein Kinase B |
| PKC | Protein Kinase C |
| PRDX | Peroxiredoxin |
| PRDX1 | Peroxiredoxin-1 |
| PRDX2 | Peroxiredoxin-2 |
| PRDX5 | Peroxiredoxin-5 |
| PRDX6 | Peroxiredoxin-6 |
| PUFAs | Polyunsaturated Fatty Acids |
| R-HSA | Reactome Entry |
| PVR | Proliferative Vitreoretinopathy |
| RAGE | Receptor for advanced glycation end products |
| RAS | Renin–Angiotensin System |
| RD | Retinal Detachment |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| RPE | Retinal Pigment Epithelium |
| RRD | Rhegmatogenous Retinal Detachment |
| SOD | Superoxide Dismutase |
| SOD1 | Superoxide Dismutase [Cu-Zn] |
| SOD3 | Extracellular Superoxide Dismutase [Cu-Zn] |
| TICAM1 | TIR Domain-Containing Adapter Molecule 1 |
| TNF-α | Tumor Necrosis Factor |
| VCAM1 | Vascular Cell Adhesion Protein 1 |
| VEGF | Vascular endothelial growth factor |
| WHO | World Health Organization |
References
- Sies, H. Oxidative stress: Concept and some practical aspects. Antioxidants 2020, 9, 852. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Nitric Oxide-Dependent Protein Post-Translational Modifications Impair Mitochondrial Function and Metabolism to Contribute to Neurodegenerative Diseases. Antioxid. Redox Signal. 2020, 32, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Reactive oxygen species or reactive sulfur species: Why we should consider the latter. J. Exp. Biol. 2020, 223, jeb196352. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L. The Role of Myeloperoxidase in Biomolecule Modification, Chronic Inflammation, and Disease. Antioxid. Redox Signal. 2020, 32, 957–981. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2018, 7, 122–126. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A.; Iordache, F.; Stanca, L.; Predoi, G.; Serban, A.I. Oxidative stress mitigation by antioxidants—An overview on their chemistry and influences on health status. Eur. J. Med. Chem. 2021, 209, 112891. [Google Scholar] [CrossRef]
- Mahajan, N.; Arora, P.; Sandhir, R. Perturbed Biochemical Pathways and Associated Oxidative Stress Lead to Vascular Dysfunctions in Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2019, 2019, 8458472. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Durán, M.D.; Gallego-Pinazo, R.; García-Medina, J.J.; Zanón-Moreno, V.; Nucci, C.; Dolz-Marco, R.; Martínez-Castillo, S.; Galbis-Estrada, C.; Marco-Ramírez, C.; López-Gálvez, M.I.; et al. Oxidative stress and its downstream signaling in aging eyes. Clin. Interv. Aging 2014, 9, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’angelo, R.; Sidoti, A. Oxidative stress and the neurovascular unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Sun, Y.; Cakir, B.; Tomita, Y.; Huang, S.; Wang, Z.; Liu, C.-H.; Cho, S.S.; Britton, W.; Kern, T.S.; et al. Targeting Neurovascular Interaction in Retinal Disorders. Int. J. Mol. Sci. 2020, 21, 1503. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.H.; Ma, J. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. Circ. Physiol. 2016, 311, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Domènech, E.B.; Marfany, G. The relevance of oxidative stress in the pathogenesis and therapy of retinal dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative stress and vascular dysfunction in the retina: Therapeutic strategies. Antioxidants 2020, 9, 761. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 1–11. [Google Scholar] [CrossRef]
- Jabbehdari, S.; Handa, J.T. Oxidative stress as a therapeutic target for the prevention and treatment of early age-related macular degeneration. Surv. Ophthalmol. 2021, 66, 423–440. [Google Scholar] [CrossRef]
- Li, C.; Miao, X.; Li, F.; Wang, S.; Liu, Q.; Wang, Y.; Sun, J. Oxidative Stress-Related Mechanisms and Antioxidant Therapy in Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2017, 2017, 9702820. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A.C.Y. Diabetic retinopathy: Pathophysiology and treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Report on Vision; World Health Organization: Geneva, Switzerland, 2019; Volume 214. [Google Scholar]
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI insight 2017, 2, e93751. [Google Scholar] [CrossRef]
- Figueira, J.; Henriques, J.; Carneiro, Â.; Marques-Neves, C.; Flores, R.; Castro-Sousa, J.P.; Meireles, A.; Gomes, N.; Nascimento, J.; Amaro, M.; et al. Guidelines for the management of center-involving diabetic macular edema: Treatment options and patient monitorization. Clin. Ophthalmol. 2021, 15, 3221–3230. [Google Scholar] [CrossRef]
- Sharma, T.; Fong, A.; Lai, T.Y.; Lee, V.; Das, S.; Lam, D. Surgical treatment for diabetic vitreoretinal diseases: A review. Clin. Exp. Ophthalmol. 2016, 44, 340–354. [Google Scholar] [CrossRef]
- Simó-Servat, O.; Hernández, C.; Simó, R. Diabetic Retinopathy in the Context of Patients with Diabetes. Ophthalmic Res. 2019, 62, 211–217. [Google Scholar] [CrossRef]
- Rossino, M.G.; Dal Monte, M.; Casini, G. Relationships between Neurodegeneration and Vascular Damage in Diabetic Retinopathy. Front. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vis. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef]
- Pillar, S.; Moisseiev, E.; Sokolovska, J.; Grzybowski, A. Recent Developments in Diabetic Retinal Neurodegeneration: A Literature Review. J. Diabetes Res. 2020, 2020, 5728674. [Google Scholar] [CrossRef]
- Rodríguez, M.L.; Pérez, S.; Mena-Mollá, S.; Desco, M.C.; Ortega, Á.L. Oxidative Stress and Microvascular Alterations in Diabetic Retinopathy: Future Therapies. Oxid. Med. Cell. Longev. 2019, 2019, 4940825. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrósio, A.F.; Fernandes, R. Sweet Stress: Coping with Vascular Dysfunction in Diabetic Retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef]
- Liu, H.; Tang, J.; Du, Y.; Saadane, A.; Tonade, D.; Samuels, I.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells influence retinal vascular degeneration in mouse models of retinal degeneration and diabetes. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4272–4281. [Google Scholar] [CrossRef]
- Kern, T.S. Do photoreceptor cells cause the development of retinal vascular disease? Vis. Res. 2017, 139, 65–71. [Google Scholar] [CrossRef]
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic retinopathy: The role of mitochondria in the neural retina and microvascular disease. Antioxidants 2020, 9, 905. [Google Scholar] [CrossRef]
- Payne, A.J.; Kaja, S.; Naumchuk, Y.; Kunjukunju, N.; Koulen, P. Antioxidant drug therapy approaches for neuroprotection in chronic diseases of the retina. Int. J. Mol. Sci. 2014, 15, 1865–1886. [Google Scholar] [CrossRef]
- Nawaz, I.M.; Rezzola, S.; Cancarini, A.; Russo, A.; Costagliola, C.; Semeraro, F.; Presta, M. Human vitreous in proliferative diabetic retinopathy: Characterization and translational implications. Prog. Retin. Eye Res. 2019, 109, 110–119. [Google Scholar] [CrossRef]
- Sebag, J. Vitreous and Vision Degrading Myodesopsia. Prog. Retin. Eye Res. 2020, 79, 100847. [Google Scholar] [CrossRef]
- Yilmaz, G.; Esser, P.; Kociek, N.; Aydin, P.; Heimann, K. Elevated vitreous nitric oxide levels in patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 2000, 130, 87–90. [Google Scholar] [CrossRef]
- Cicik, E.; Tekin, H.; Akar, S.; Ekmekçi, Ö.B.; Donma, O.; Koldaş, L.; Özkana, Ş. Interleukin-8, nitric oxide and glutathione status in proliferative vitreoretinopathy and proliferative diabetic retinopathy. Ophthalmic Res. 2003, 35, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Desmet, S.; Meersschaert, A.; Dralands, L.; Missotten, L.; Geboes, K. Expression of the inducible isoform of nitric oxide synthase in the retinas of human subjects with diabetes mellitus. Am. J. Ophthalmol. 2001, 132, 551–556. [Google Scholar] [CrossRef]
- Géhl, Z.; Bakondi, E.; Resch, M.D.; Hegedus, C.; Kovács, K.; Lakatos, P.; Szabó, A.; Nagy, Z.; Virág, L. Diabetes-induced oxidative stress in the vitreous humor. Redox Biol. 2016, 9, 100–103. [Google Scholar] [CrossRef]
- Katagiri, M.; Shoji, J.; Inada, N.; Kato, S.; Kitano, S.; Uchigata, Y. Evaluation of vitreous levels of advanced glycation end products and angiogenic factors as biomarkers for severity of diabetic retinopathy. Int. Ophthalmol. 2018, 38, 607–615. [Google Scholar] [CrossRef]
- Brzović-Šarić, V.; Landeka, I.; Šarić, B.; Barberić, M.; Andrijašević, L.; Cerovski, B.; Oršolić, N.; Đikić, D. Levels of selected oxidative stress markers in the vitreous and serum of diabetic retinopathy patients. Mol. Vis. 2015, 21, 649–664. [Google Scholar]
- Izuta, H.; Matsunaga, N.; Shimazawa, M.; Sugiyama, T.; Ikeda, T.; Hara, H. Proliferative diabetic retinopathy and relations among antioxidant activity, oxidative stress, and VEGF in the vitreous body. Mol. Vis 2010, 16, 130–136. [Google Scholar] [PubMed]
- Suzuki, Y.; Yao, T.; Okumura, K.; Seko, Y.; Kitano, S. Elevation of the vitreous body concentrations of oxidative stress-responsive apoptosis-inducing protein (ORAIP) in proliferative diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 1519–1525. [Google Scholar] [CrossRef]
- Bokhary, K.; Aljaser, F.; Abudawood, M.; Tabassum, H.; Bakhsh, A.; Alhammad, S.; Aleyadhi, R.; Almajed, F.; Alsubki, R. Role of Oxidative Stress and Severity of Diabetic Retinopathy in Type 1 and Type 2 Diabetes. Ophthalmic Res. 2021, 64, 613–621. [Google Scholar] [CrossRef]
- Mancino, R.; Di Pierro, D.; Varesi, C.; Cerulli, A.; Feraco, A.; Cedrone, C.; Pinazo-Duran, M.D.; Coletta, M.; Nucci, C. Lipid peroxidation and total antioxidant capacity in vitreous, aqueous humor, and blood samples from patients with diabetic retinopathy. Mol. Vis. 2011, 17, 1298–1304. [Google Scholar]
- Yamane, K.; Minamoto, A.; Yamashita, H.; Takamura, H.; Miyamoto-Myoken, Y.; Yoshizato, K.; Nabetani, T.; Tsugita, A.; Mishima, H.K. Proteome Analysis of Human Vitreous Proteins. Mol. Cell. Proteom. 2003, 2, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, S.J.; Park, J.; Lee, H.K.; Park, K.S.; Yu, H.G.; Kim, Y. Differential Expression of Vitreous Proteins in Proliferative Diabetic Retinopathy. Curr. Eye Res. 2006, 31, 231–240. [Google Scholar] [CrossRef]
- Balaiya, S.; Zhou, Z.; Chalam, K.V. Characterization of vitreous and aqueous proteome in humans with proliferative diabetic retinopathy and its clinical correlation. Proteom. Insights 2017, 8, 1178641816686078. [Google Scholar] [CrossRef] [PubMed]
- Schori, C.; Trachsel, C.; Grossmann, J.; Zygoula, I.; Barthelmes, D.; Grimm, C. The Proteomic Landscape in the Vitreous of Patients with Age-Related and Diabetic Retinal Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Han, C.; Zhao, M.; Yu, J.; Bai, L.; Yao, Y.; Gao, S.; Cao, H.; Zheng, Z. Change of ranibizumab-induced human vitreous protein profile in patients with proliferative diabetic retinopathy based on proteomics analysis. Clin. Proteom. 2018, 15, 12. [Google Scholar] [CrossRef]
- Minamoto, A.; Yamane, K.; Yokoyama, T. Proteomics of Vitreous Fluid. In Proteomics of Human Body Fluids; Thongboonkerd, V., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 495–507. ISBN 978-1-58829-657-3. [Google Scholar]
- García-Ramírez, M.; Canals, F.; Hernández, C.; Colomé, N.; Ferrer, C.; Carrasco, E.; García-Arumí, J.; Simó, R. Proteomic analysis of human vitreous fluid by fluorescence-based difference gel electrophoresis (DIGE): A new strategy for identifying potential candidates in the pathogenesis of proliferative diabetic retinopathy. Diabetologia 2007, 50, 1294–1303. [Google Scholar] [CrossRef]
- Shitama, T.; Hayashi, H.; Noge, S.; Uchio, E.; Oshima, K.; Haniu, H.; Takemori, N.; Komori, N.; Matsumoto, H. Proteome profiling of vitreoretinal diseases by cluster analysis. Proteom.-Clin. Appl. 2008, 2, 1265–1280. [Google Scholar] [CrossRef]
- Gao, B.-B.; Chen, X.; Timothy, N.; Aiello, L.P.; Feener, E.P. Characterization of the Vitreous Proteome in Diabetes without Diabetic Retinopathy and Diabetes with Proliferative Diabetic Retinopathy. J. Proteome Res. 2008, 7, 2516–2525. [Google Scholar] [CrossRef]
- Kim, K.; Kim, S.J.; Yu, H.G.; Yu, J.; Park, K.S.; Jang, I.J.; Kim, Y. Verification of biomarkers for diabetic retinopathy by multiple reaction monitoring. J. Proteome Res. 2010, 9, 689–699. [Google Scholar] [CrossRef]
- Wang, H.; Feng, L.; Hu, J.; Xie, C.; Wang, F. Characterisation of the vitreous proteome in proliferative diabetic retinopathy. Proteome Sci. 2012, 10, 15. [Google Scholar] [CrossRef]
- Wang, H.; Feng, L.; Hu, J.; Xie, C.; Wang, F. Differentiating vitreous proteomes in proliferative diabetic retinopathy using high-performance liquid chromatography coupled to tandem mass spectrometry. Exp. Eye Res. 2013, 108, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Nurkkala, H.; Tamene, F.; Gucciardo, E.; Liu, X.; Repo, P.; Lehti, K.; Varjosalo, M. Quantitative Proteomics Analysis of Vitreous Humor from Diabetic Retinopathy Patients. J. Proteome Res. 2015, 14, 5131–5143. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Nakazawa, M.; Suzuki, K.; Yamazaki, H.; Miyagawa, Y. Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn. J. Ophthalmol. 2011, 55, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Koskela, U.E.; Kuusisto, S.M.; Nissinen, A.E.; Savolainen, M.J.; Liinamaa, M.J. High vitreous concentration of IL-6 and IL-8, but not of adhesion molecules in relation to plasma concentrations in proliferative diabetic retinopathy. Ophthalmic Res. 2013, 49, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; de Vries, E.W.; Vogels, I.M.C.; van Kampen, A.H.C.; Bosscha, M.I.; Steel, D.H.W.; Van Noorden, C.J.F.; Lesnik-Oberstein, S.Y.; Schlingemann, R.O. Identification of proteins associated with clinical and pathological features of proliferative diabetic retinopathy in vitreous and fibrovascular membranes. PLoS ONE 2017, 12, e0187304. [Google Scholar] [CrossRef]
- Srividya, G.; Jain, M.; Mahalakshmi, K.; Gayathri, S.; Raman, R.; Angayarkanni, N. A novel and less invasive technique to assess cytokine profile of vitreous in patients of diabetic macular oedema. Eye 2018, 32, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Kuehn, S.; Tsiampalis, N.; Vu, M.-K.; Kakkassery, V.; Stute, G.; Dick, H.B.; Joachim, S.C. Anti-inflammatory cytokine and angiogenic factors levels in vitreous samples of diabetic retinopathy patients. PLoS ONE 2018, 13, e0194603. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, 607–613. [Google Scholar] [CrossRef]
- Tsao, Y.P.; Ho, T.C.; Chen, S.L.; Cheng, H.C. Pigment epithelium-derived factor inhibits oxidative stress-induced cell death by activation of extracellular signal-regulated kinases in cultured retinal pigment epithelial cells. Life Sci. 2006, 79, 545–550. [Google Scholar] [CrossRef]
- Park, K.; Jin, J.; Hu, Y.; Zhou, K.; Ma, J.-X.X. Overexpression of Pigment Epithelium-Derived Factor Inhibits Retinal Inflammation and Neovascularization. AJPA 2011, 178, 688–698. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Ren, Y.; Liu, Y.; Han, S.; Zhao, J.; Gou, X.; He, Y. PEDF protects human retinal pigment epithelial cells against oxidative stress via upregulation of UCP2 expression. Mol. Med. Rep. 2019, 19, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Polato, F.; Samardzija, M.; Abu-Asab, M.; Grimm, C.; Crawford, S.E.; Becerra, S.P. PEDF deficiency increases the susceptibility of rd10 mice to retinal degeneration. Exp. Eye Res. 2020, 198, 108121. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-K.; Lee, E.-J.; Kim, Y.-H.; Kim, D.; Oh, H.; Kim, S.-I.; Kang, Y.-H. Chrysin Ameliorates Malfunction of Retinoid Visual Cycle through Blocking Activation of AGE-RAGE-ER Stress in Glucose-Stimulated Retinal Pigment Epithelial Cells and Diabetic Eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, M.; Yamagishi, S.-i.; Saito, A.; Yoshida, Y.; Matsui, T.; Saito, W.; Hirose, S.; Ohgami, K.; Kase, M.; Ohno, S. Positive association of pigment epithelium-derived factor with total antioxidant capacity in the vitreous fluid of patients with proliferative diabetic retinopathy. Br. J. Ophthalmol. 2007, 91, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.I.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef]
- Fort, P.E.; Lampi, K.J. New focus on alpha-crystallins in retinal neurodegenerative diseases. Exp. Eye Res. 2011, 92, 98–103. [Google Scholar] [CrossRef]
- Kannan, R.; Sreekumar, P.G.; Hinton, D.R. Novel roles for α-crystallins in retinal function and disease. Prog. Retin. Eye Res. 2012, 31, 576–604. [Google Scholar] [CrossRef]
- Chen, W.; Lu, Q.; Lu, L.; Guan, H. Increased levels of alphaB-crystallin in vitreous fluid of patients with proliferative diabetic retinopathy and correlation with vascular endothelial growth factor. Clin. Exp. Ophthalmol. 2017, 45, 379–384. [Google Scholar] [CrossRef]
- Ghosh, S.; Liu, H.; Yazdankhah, M.; Stepicheva, N.; Shang, P.; Vaidya, T.; Hose, S.; Gupta, U.; Calderon, M.J.; Hu, M.-W.; et al. βA1-crystallin regulates glucose metabolism and mitochondrial function in mouse retinal astrocytes by modulating PTP1B activity. Commun. Biol. 2021, 4, 248. [Google Scholar] [CrossRef]
- Kase, S. Increased expression of αA-crystallin in human diabetic eye. Int. J. Mol. Med. 2011, 28, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Kannan, R.; Spee, C.; Sreekumar, P.G.; Dou, G.; Hinton, D.R. Protection of Retina by αB Crystallin in Sodium Iodate Induced Retinal Degeneration. PLoS ONE 2014, 9, e98275. [Google Scholar] [CrossRef]
- Yaung, J.; Jin, M.; Barron, E.; Spee, C.; Wawrousek, E.F.; Kannan, R.; Hinton, D.R. α-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol. Vis. 2007, 13, 566–577. [Google Scholar] [PubMed]
- Ruebsam, A.; Dulle, J.E.; Myers, A.M.; Sakrikar, D.; Green, K.M.; Khan, N.W.; Schey, K.; Fort, P.E. A specific phosphorylation regulates the protective role of αA-crystallin in diabetes. JCI Insight 2018, 3, e97919. [Google Scholar] [CrossRef]
- Losiewicz, M.K.; Fort, P.E. Diabetes Impairs the neuroprotective properties of retinal Alpha-crystallins. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5034–5042. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.E.; Bauskar, A.; Jeong, S.; Wilson, M.R. Clusterin in the eye: An old dog with new tricks at the ocular surface. Exp. Eye Res. 2016, 147, 57–71. [Google Scholar] [CrossRef] [PubMed]
- de Campos, T.D.P.; da Cruz Rodrigues, K.C.; Pereira, R.M.; Anaruma, C.P.; dos Santos Canciglieri, R.; de Melo, D.G.; da Silva, A.S.R.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R.; et al. The protective roles of clusterin in ocular diseases caused by obesity and diabetes mellitus type 2. Mol. Biol. Rep. 2021, 48, 4637–4645. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.H.; Jun, H.O.; Yu, Y.S.; Min, B.H.; Park, K.H.; Kim, K.W. Protective effect of clusterin from oxidative stress-induced apoptosis in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 561–566. [Google Scholar] [CrossRef]
- Ren, L.; Han, F.; Xuan, L.; Lv, Y.; Gong, L.; Yan, Y.; Wan, Z.; Guo, L.; Liu, H.; Xu, B.; et al. Clusterin ameliorates endothelial dysfunction in diabetes by suppressing mitochondrial fragmentation. Free Radic. Biol. Med. 2019, 145, 357–373. [Google Scholar] [CrossRef]
- Gharibyan, A.L.; Islam, T.; Pettersson, N.; Golchin, S.A.; Lundgren, J.; Johansson, G.; Genot, M.; Schultz, N.; Wennström, M.; Olofsson, A. Apolipoprotein E interferes with IAPP aggregation and protects pericytes from IAPP-Induced Toxicity. Biomolecules 2020, 10, 134. [Google Scholar] [CrossRef]
- Shea, T.B.; Rogers, E.; Ashline, D.; Ortiz, D.; Sheu, M.S. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic. Biol. Med. 2002, 33, 1115–1120. [Google Scholar] [CrossRef]
- Cecilia, O.-M.; José Alberto, C.-G.; José, N.-P.; Ernesto Germán, C.-M.; Ana Karen, L.-C.; Luis Miguel, R.-P.; Ricardo Raúl, R.-R.; Adolfo Daniel, R.-C. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J. Diabetes Res. 2019, 2019, 8562408. [Google Scholar] [CrossRef] [PubMed]
- Yumnamcha, T.; Guerra, M.; Singh, L.P.; Ibrahim, A.S. Metabolic dysregulation and neurovascular dysfunction in diabetic retinopathy. Antioxidants 2020, 9, 1244. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef]
- Xu, J.; Chen, L.J.; Yu, J.; Wang, H.J.; Zhang, F.; Liu, Q.; Wu, J. Involvement of Advanced Glycation End Products in the Pathogenesis of Diabetic Retinopathy. Cell. Physiol. Biochem. 2018, 48, 705–717. [Google Scholar] [CrossRef]
- Du, X.; Stockklauser-Färber, K.; Rösen, P. Generation of reactive oxygen intermediates, activation of NF-κB, and induction of apoptosis in human endothelial cells by glucose: Role of nitric oxide synthase? Free Radic. Biol. Med. 1999, 27, 752–763. [Google Scholar] [CrossRef]
- Vielma, A.H.; Retamal, M.A.; Schmachtenberg, O. Nitric oxide signaling in the retina: What have we learned in two decades? Brain Res. 2012, 1430, 112–125. [Google Scholar] [CrossRef]
- Opatrilova, R.; Kubatka, P.; Caprnda, M.; Büsselberg, D.; Krasnik, V.; Vesely, P.; Saxena, S.; Ruia, S.; Mozos, I.; Rodrigo, L.; et al. Nitric oxide in the pathophysiology of retinopathy: Evidences from preclinical and clinical researches. Acta Ophthalmol. 2018, 96, 222–231. [Google Scholar] [CrossRef]
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 2007, 26, 205–238. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Odenbach, S. Role of interleukin-1β in the pathogenesis of diabetic retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Nawaz, M.I.; Kangave, D.; Mairaj Siddiquei, M.; Geboes, K. Angiogenic and Vasculogenic Factors in the Vitreous from Patients with Proliferative Diabetic Retinopathy. J. Diabetes Res. 2013, 2013, 539658. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Struyf, S.; Opdenakker, G.; van Damme, J.; Geboes, K. Expression of stem cell factor/c-kit signaling pathway components in diabetic fibrovascular epiretinal membranes. Mol. Vis. 2010, 16, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Awata, T.; Neda, T.; Iizuka, H.; Kurihara, S.; Ohkubo, T.; Takata, N.; Osaki, M.; Watanabe, M.; Nakashima, Y.; Sawa, T.; et al. Endothelial Nitric Oxide Synthase Gene Is Associated with Diabetic Macular Edema in Type 2 Diabetes. Diabetes Care 2004, 27, 2184–2190. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.L.; Sessa, W.C. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 2014, 127, 2120. [Google Scholar] [CrossRef][Green Version]
- Yuasa, I.; Ma, N.; Matsubara, H.; Fukui, Y.; Uji, Y. Inducible nitric oxide synthase mediates retinal DNA damage in Goto-Kakizaki rat retina. Jpn. J. Ophthalmol. 2008, 52, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Carmo, A.; Cunha-Vaz, J.G.; Carvalho, A.P.; Lopes, M.C. Nitric oxide synthase activity in retinas from non-insulin-dependent diabetic Goto-Kakizaki rats: Correlation with blood-retinal barrier permeability. Nitric Oxide-Biol. Chem. 2000, 4, 590–596. [Google Scholar] [CrossRef]
- Mishra, A.; Newman, E.A. Inhibition of inducible nitric oxide synthase reverses the loss of functional hyperemia in diabetic retinopathy. Glia 2010, 58, 1996–2004. [Google Scholar] [CrossRef]
- Ellis, E.A.; Guberski, D.L.; Hutson, B.; Grant, M.B. Time course of NADH oxidase, inducible nitric oxide synthase and peroxynitrite in diabetic retinopathy in the BBZ/Wor rat. Nitric Oxide-Biol. Chem. 2002, 6, 295–304. [Google Scholar] [CrossRef]
- Nakagami, Y. Nrf2 Is an Attractive Therapeutic Target for Retinal Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7469326. [Google Scholar] [CrossRef]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription factor Nrf2-mediated antioxidant defense system in the development of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Gardner, T.W.; Sundstrom, J.M. A proposal for early and personalized treatment of diabetic retinopathy based on clinical pathophysiology and molecular phenotyping. Vis. Res. 2017, 139, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 activation is a potential therapeutic approach to attenuate diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Apte, R.S. Age-Related Macular Degeneration. N. Engl. J. Med. 2021, 385, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, Y.; Miller, J.; Kim, I. Age-Related Macular Degeneration: Advances in Management and Diagnosis. J. Clin. Med. 2015, 4, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef]
- Supuran, C.T. Agents for the prevention and treatment of age-related macular degeneration and macular edema: A literature and patent review. Expert Opin. Ther. Pat. 2019, 29, 761–767. [Google Scholar] [CrossRef]
- Hernández-Zimbrón, L.F.; Zamora-Alvarado, R.; Ochoa-De la Paz, L.; Velez-Montoya, R.; Zenteno, E.; Gulias-Cañizo, R.; Quiroz-Mercado, H.; Gonzalez-Salinas, R. Age-Related Macular Degeneration: New Paradigms for Treatment and Management of AMD. Oxid. Med. Cell. Longev. 2018, 2018, 8374647. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and nitrosative stress in age-related macular degeneration: A review of their role in different stages of disease. Antioxidants 2021, 10, 653. [Google Scholar] [CrossRef]
- Ozawa, Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 2020, 37, 101779. [Google Scholar] [CrossRef] [PubMed]
- Čolak, E.; Žorić, L. Antioxidants and Age-Related Macular Degeneration. In Handbook of Nutrition, Diet, and the Eye; Preedy, V.R., Watson, R.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 85–106. ISBN 9781607410072. [Google Scholar]
- Rivera, J.C.; Dabouz, R.; Noueihed, B.; Omri, S.; Tahiri, H.; Chemtob, S. Ischemic Retinopathies: Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 3940241. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Valapala, M.; Shang, P.; Hose, S.; Grebe, R.; Lutty, G.A.; Zigler, J.S.; Kaarniranta, K.; Handa, J.T. Lysosomes: Regulators of autophagy in the retinal pigmented epithelium. Exp. Eye Res. 2016, 144, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Kim, Y.A.; Yoo, Y.H. Loss of Pigment Epithelial Cells Is Prevented by Autophagy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; Volume 11, pp. 105–117. ISBN 9780128094273. [Google Scholar]
- Zhang, Z.-Y.; Bao, X.-L.; Cong, Y.-Y.; Fan, B.; Li, G.-Y. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef]
- Krohne, T.U.; Stratmann, N.K.; Kopitz, J.; Holz, F.G. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp. Eye Res. 2010, 90, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and Exosomes in the Aged Retinal Pigment Epithelium: Possible Relevance to Drusen Formation and Age-Related Macular Degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells—Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Marmorstein, L.Y.; Munier, F.L.; Arsenijevic, Y.; Schorderet, D.F.; Mclaughlin, P.J.; Chung, D.; Traboulsi, E.; Marmorstein, A.D. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 13067–13072. [Google Scholar] [CrossRef]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Léveillard, T.; Philp, N.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Egbejimi, A.; Dharmat, R.; Xu, P.; Zhao, Z.; Long, B.; Miao, H.; Chen, R.; Wensel, T.G.; Cai, J.; et al. Phagocytosed photoreceptor outer segments activate mTORC1 in the retinal pigment epithelium. Sci. Signal. 2018, 11, eaag3315. [Google Scholar] [CrossRef] [PubMed]
- Richer, S.; Ulanski, L.; Popenko, N.A.; Pratt, S.G.; Hitchmoth, D.; Chous, P.; Patel, S.; Sockanathan, S.; Sardi, B. Age-related Macular Degeneration Beyond the Age-related Eye Disease Study II. Adv. Ophthalmol. Optom. 2016, 1, 335–369. [Google Scholar] [CrossRef]
- SanGiovanni, J.P. The Relationship of Dietary ω-3 Long-Chain Polyunsaturated Fatty Acid Intake with Incident Age-Related Macular Degeneration. Arch. Ophthalmol. 2008, 126, 1274–1279. [Google Scholar] [CrossRef]
- Seddon, J.M. Cigarette Smoking, Fish Consumption, Omega-3 Fatty Acid Intake, and Associations with Age-Related Macular Degeneration. Arch. Ophthalmol. 2006, 124, 995–1001. [Google Scholar] [CrossRef]
- Koss, M.J.; Hoffmann, J.; Nguyen, N.; Pfister, M.; Mischak, H.; Mullen, W.; Husi, H.; Rejdak, R.; Koch, F.; Jankowski, J.; et al. Proteomics of Vitreous Humor of Patients with Exudative Age-Related Macular Degeneration. PLoS ONE 2014, 9, e96895. [Google Scholar] [CrossRef]
- Nobl, M.; Reich, M.; Dacheva, I.; Siwy, J.; Mullen, W.; Schanstra, J.P.; Choi, C.Y.; Kopitz, J.; Kretz, F.T.A.A.; Auffarth, G.U.; et al. Proteomics of vitreous in neovascular age-related macular degeneration. Exp. Eye Res. 2016, 146, 107–117. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ferrington, D.A.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef]
- Tseng, C.F.; Lin, C.C.; Huang, H.Y.; Liu, H.C.; Mao, S.J.T. Antioxidant role of human haptoglobin. Proteomics 2004, 4, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lu, H.; Dutt, K.; Smith, A.; Hunt Margaret, D.; Hunt, R.C. Expression of the protective proteins hemopexin and haptoglobin cells of the neural retina. Exp. Eye Res. 1998, 67, 83–93. [Google Scholar] [CrossRef]
- Kliffen, M.; de Jong, P.T.; Luider, T.M. Protein analysis of human maculae in relation to age-related maculopathy. Lab. Investig. 1995, 73, 267–272. [Google Scholar]
- Čolak, E.; Žorić, L.; Radosavljević, A.; Ignjatović, S. The Association of Serum Iron-Binding Proteins and the Antioxidant Parameter Levels in Age-Related Macular Degeneration. Curr. Eye Res. 2018, 43, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Seider, N.; Beiran, I.; Miller-Lotan, R.; Dori, D.; Karp, J.; Miller, B.; Levy, A.P. Haptoglobin phenotype in age-related macular degeneration patients. Am. J. Ophthalmol. 2003, 136, 911–914. [Google Scholar] [CrossRef]
- Holekamp, N.M.; Bouck, N.; Volpert, O. Pigment epithelium-derived factor is deficient in the vitreous of patients with choroidal neovascularization due to age-related macular degeneration. Am. J. Ophthalmol. 2002, 134, 220–227. [Google Scholar] [CrossRef]
- Duh, E.J.; Yang, H.S.; Haller, J.A.; De Juan, E.; Humayun, M.S.; Gehlbach, P.; Melia, M.; Pieramici, D.; Harlan, J.B.; Campochiaro, P.A.; et al. Vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor: Implications for ocular angiogenesis. Am. J. Ophthalmol. 2004, 137, 668–674. [Google Scholar] [CrossRef]
- Rinsky, B.; Beykin, G.; Grunin, M.; Amer, R.; Khateb, S.; Tiosano, L.; Almeida, D.; Hagbi-Levi, S.; Elbaz-Hayoun, S.; Chowers, I. Analysis of the Aqueous Humor Proteome in Patients with Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2021, 62, 18. [Google Scholar] [CrossRef]
- Kang, G.-Y.; Bang, J.Y.; Choi, A.J.; Yoon, J.; Lee, W.-C.; Choi, S.; Yoon, S.; Kim, H.C.; Baek, J.-H.; Park, H.S.; et al. Exosomal Proteins in the Aqueous Humor as Novel Biomarkers in Patients with Neovascular Age-related Macular Degeneration. J. Proteome Res. 2014, 13, 581–595. [Google Scholar] [CrossRef]
- Yuan, X.; Gu, X.; Crabb, J.S.; Yue, X.; Shadrach, K.; Hollyfield, J.G.; Crabb, J.W. Quantitative proteomics: Comparison of the macular bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol. Cell. Proteom. 2010, 9, 1031–1046. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Miyagi, M.; Shadrach, K.G.; Rayborn, M.E.; Crabb, J.W.; Hollyfield, J.G. Clusterin is present in drusen in age-related macular degeneration. Exp. Eye Res. 2002, 74, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant lipid and protein components of drusen. PLoS ONE 2010, 5, e10329. [Google Scholar] [CrossRef] [PubMed]
- An, E.; Lu, X.; Flippin, J.; Devaney, J.M.; Halligan, B.; Hoffman, E.; Csaky, K.; Hathout, Y. Secreted Proteome Profiling in Human RPE Cell Cultures Derived from Donors with Age Related Macular Degeneration and Age Matched Healthy Donors. J. Proteome Res. 2006, 5, 2599–2610. [Google Scholar] [CrossRef]
- Hatters, D.M.; Wilson, M.R.; Easterbrook-Smith, S.B.; Howlett, G.J. Suppression of apolipoprotein C-II amyloid formation by the extracellular chaperone, clusterin. Eur. J. Biochem. 2002, 269, 2789–2794. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef]
- Kumita, J.R.; Poon, S.; Caddy, G.L.; Hagan, C.L.; Dumoulin, M.; Yerbury, J.J.; Stewart, E.M.; Robinson, C.V.; Wilson, M.R.; Dobson, C.M. The Extracellular Chaperone Clusterin Potently Inhibits Human Lysozyme Amyloid Formation by Interacting with Prefibrillar Species. J. Mol. Biol. 2007, 369, 157–167. [Google Scholar] [CrossRef][Green Version]
- Ratnayaka, J.A.; Serpell, L.C.; Lotery, A.J. Dementia of the eye: The role of amyloid beta in retinal degeneration. Eye 2015, 29, 1013–1026. [Google Scholar] [CrossRef]
- Luibl, V.; Isas, J.M.; Kayed, R.; Glabe, C.G.; Langen, R.; Chen, J. Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomers. J. Clin. Investig. 2006, 116, 378–385. [Google Scholar] [CrossRef]
- Gupta, V.B.V.; Gupta, V.B.V.; Chitranshi, N.; Gangoda, S.; Vander Wall, R.; Abbasi, M.; Golzan, M.; Dheer, Y.; Shah, T.; Avolio, A.; et al. One protein, multiple pathologies: Multifaceted involvement of amyloid β in neurodegenerative disorders of the brain and retina. Cell. Mol. Life Sci. 2016, 73, 4279–4297. [Google Scholar] [CrossRef]
- Feng, L.; Liao, X.; Zhang, Y.; Wang, F. Protective effects on age-related macular degeneration by activated autophagy induced by amyloid-β in retinal pigment epithelial cells. Discov. Med. 2019, 27, 153–160. [Google Scholar]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative Stress, Hypoxia, and Autophagy in the Neovascular Processes of Age-Related Macular Degeneration. BioMed Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Kojima, A.; Shimada, N.; Nakahama, K.; Safranova, O.; Iwata, N.; Saido, T.C.; Mochizuki, M.; et al. Altered Function of Factor I Caused by Amyloid β: Implication for Pathogenesis of Age-Related Macular Degeneration from Drusen. J. Immunol. 2008, 181, 712–720. [Google Scholar] [CrossRef]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s Aβ-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; Gagliardi, G.; Garanto, A.; de Breuk, A.; Lechanteur, Y.T.E.; Katti, S.; van den Heuvel, L.P.; Volokhina, E.B.; den Hollander, A.I. Implications of genetic variation in the complement system in age-related macular degeneration. Prog. Retin. Eye Res. 2021, 84, 100952. [Google Scholar] [CrossRef]
- Kawa, M.P.; Machalinska, A.; Roginska, D.; Machalinski, B. Complement System in Pathogenesis of AMD: Dual Player in Degeneration and Protection of Retinal Tissue. J. Immunol. Res. 2014, 2014, 483960. [Google Scholar] [CrossRef]
- Toomey, C.B.; Johnson, L.V.; Bowes Rickman, C. Complement factor H in AMD: Bridging genetic associations and pathobiology. Prog. Retin. Eye Res. 2018, 62, 38–57. [Google Scholar] [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef]
- Romero-Vazquez, S.; Llorens, V.; Soler-Boronat, A.; Figueras-Roca, M.; Adan, A.; Molins, B. Interlink between Inflammation and Oxidative Stress in Age-Related Macular Degeneration: Role of Complement Factor H. Biomedicines 2021, 9, 763. [Google Scholar] [CrossRef]
- Paraoan, L.; Sharif, U.; Carlsson, E.; Supharattanasitthi, W.; Mahmud, N.M.; Kamalden, T.A.; Hiscott, P.; Jackson, M.; Grierson, I. Secretory proteostasis of the retinal pigmented epithelium: Impairment links to age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100859. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L.; Bell, B.A.; Rayborn, M.E.; Samuels, I.S.; King, A.; Hollyfield, J.G.; Xie, C.; Cai, H. Absence of DJ-1 causes age-related retinal abnormalities in association with increased oxidative stress. Free Radic. Biol. Med. 2017, 104, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L.; Bell, B.A.; Rayborn, M.E.; Yang, X.; Kaul, C.; Grossman, G.H.; Samuels, I.S.; Hollyfield, J.G.; Xie, C.; Cai, H.; et al. Loss of DJ-1 elicits retinal abnormalities, visual dysfunction, and increased oxidative stress in mice. Exp. Eye Res. 2015, 139, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Bonilha, V.L. Oxidative Stress Regulation and DJ-1 Function in the Retinal Pigment Epithelium: Implications for AMD. In Retinal Degenerative Diseases; Springer: Berlin/Heidelberg, Germany, 2018; pp. 3–9. [Google Scholar]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef]
- Milani, P.; Ambrosi, G.; Gammoh, O.; Blandini, F.; Cereda, C. SOD1 and DJ-1 Converge at Nrf2 Pathway: A Clue for Antioxidant Therapeutic Potential in Neurodegeneration. Oxid. Med. Cell. Longev. 2013, 2013, 836760. [Google Scholar] [CrossRef]
- Zhu, Y.; Aredo, B.; Chen, B.; Zhao, C.X.; He, Y.; Ufret-Vincenty, R.L. Mice with a Combined Deficiency of Superoxide Dismutase 1 (Sod1), DJ-1 (Park7), and Parkin (Prkn) Develop Spontaneous Retinal Degeneration with Aging. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3740–3751. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-Related Retinopathy in NRF2-Deficient Mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.; Chen, S.; Wang, Y.; Cao, L.; Zhang, Y.; Kang, W.; Li, H.; Gui, Y.; Chen, S.; et al. DJ-1 modulates the expression of Cu/Zn-superoxide dismutase-1 through the Erk1/2-Elk1 pathway in neuroprotection. Ann. Neurol. 2011, 70, 591–599. [Google Scholar] [CrossRef]
- Yamashita, S.; Mori, A.; Kimura, E.; Mita, S.; Maeda, Y.; Hirano, T.; Uchino, M. DJ-1 forms complexes with mutant SOD1 and ameliorates its toxicity. J. Neurochem. 2010, 113, 860–870. [Google Scholar] [CrossRef]
- Notomi, S.; Ishihara, K.; Efstathiou, N.E.; Lee, J.J.; Hisatomi, T.; Tachibana, T.; Konstantinou, E.K.; Ueta, T.; Murakami, Y.; Maidana, D.E.; et al. Genetic LAMP2 deficiency accelerates the age-associated formation of basal laminar deposits in the retina. Proc. Natl. Acad. Sci. USA 2019, 116, 23724–23734. [Google Scholar] [CrossRef]
- Lee, S.M.; Koh, H.-J.; Park, D.-C.; Song, B.J.; Huh, T.-L.; Park, J.-W. Cytosolic NADP+-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Cano, M.; Datta, S.; Wang, L.; Liu, T.; Flores-Bellver, M.; Sachdeva, M.; Sinha, D.; Handa, J.T. Nrf2 deficiency decreases NADPH from impaired IDH shuttle and pentose phosphate pathway in retinal pigmented epithelial cells to magnify oxidative stress-induced mitochondrial dysfunction. Aging Cell 2021, 20, e13444. [Google Scholar] [CrossRef] [PubMed]
- Mitry, D.; Charteris, D.G.; Fleck, B.W.; Campbell, H.; Singh, J. The epidemiology of rhegmatogenous retinal detachment: Geographical variation and clinical associations. Br. J. Ophthalmol. 2010, 94, 678–684. [Google Scholar] [CrossRef]
- Nielsen, B.R.; Alberti, M.; Bjerrum, S.S.; la Cour, M. The incidence of rhegmatogenous retinal detachment is increasing. Acta Ophthalmol. 2020, 98, 603–606. [Google Scholar] [CrossRef]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Holz, F.G.; Finger, R.P. Incidence of Rhegmatogenous Retinal Detachment in Europe—A Systematic Review and Meta-Analysis. Ophthalmologica 2019, 242, 81–86. [Google Scholar] [CrossRef]
- Constable, I.J.; Nagpal, M. Proliferative Vitreoretinopathy. In Retina; Elsevier: Amsterdam, The Netherlands, 2013; Volume 3, pp. 1806–1825. ISBN 9781455707379. [Google Scholar]
- Pastor, J.C.; Rojas, J.; Pastor-Idoate, S.; Di Lauro, S.; Gonzalez-Buendia, L.; Delgado-Tirado, S. Proliferative vitreoretinopathy: A new concept of disease pathogenesis and practical consequences. Prog. Retin. Eye Res. 2016, 51, 125–155. [Google Scholar] [CrossRef]
- Idrees, S.; Sridhar, J.; Kuriyan, A.E. Proliferative vitreoretinopathy: A review. Int. Ophthalmol. Clin. 2019, 59, 221–240. [Google Scholar] [CrossRef]
- Nemet, A.; Moshiri, A.; Yiu, G.; Loewenstein, A.; Moisseiev, E. A Review of Innovations in Rhegmatogenous Retinal Detachment Surgical Techniques. J. Ophthalmol. 2017, 2017, 4310643. [Google Scholar] [CrossRef]
- Haugstad, M.; Moosmayer, S.; Bragadόttir, R. Primary rhegmatogenous retinal detachment–surgical methods and anatomical outcome. Acta Ophthalmol. 2017, 95, 247–251. [Google Scholar] [CrossRef]
- Feltgen, N.; Walter, P. Rhegmatogenous Retinal Detachment. Dtsch. Arztebl. Int. 2014, 111, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Sultan, Z.N.; Agorogiannis, E.I.; Iannetta, D.; Steel, D.; Sandinha, T. Rhegmatogenous retinal detachment: A review of current practice in diagnosis and management. BMJ Open Ophthalmol. 2020, 5, e000474. [Google Scholar] [CrossRef]
- Lumi, X.; Hawlina, M.; Glavač, D.; Facskó, A.; Moe, M.C.; Kaarniranta, K.; Petrovski, G. Ageing of the vitreous: From acute onset floaters and flashes to retinal detachment. Ageing Res. Rev. 2015, 21, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, F.; Aylward, B. Rhegmatogenous retinal detachment: A reappraisal of its pathophysiology and treatment. Ophthalmic Res. 2013, 51, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Ankamah, E.; Sebag, J.; Ng, E.; Nolan, J.M. Vitreous Antioxidants, Degeneration, and Vitreo-Retinopathy: Exploring the Links. Antioxidants 2019, 9, 7. [Google Scholar] [CrossRef]
- Cederlund, M.; Ghosh, F.; Arnér, K.; Andréasson, S.; Åkerström, B. Vitreous levels of oxidative stress biomarkers and the radical-scavenger α1-microglobulin/A1M in human rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Maeno, A.; Suzuki, Y.; Adachi, K.; Takahashi, S.; Yokoi, Y.; Nakazawa, M. Characterization of the biological antioxidant potential in the vitreous fluid from patients with rhegmatogenous retinal detachment. Acta Ophthalmol. 2016, 94, e515–e516. [Google Scholar] [CrossRef]
- Pietras-Baczewska, A.; Nowomiejska, K.; Brzozowska, A.; Toro, M.D.; Załuska, W.; Sztanke, M.; Sztanke, K.; Rejdak, R. Antioxidant status in the vitreous of eyes with rhegmatogenous retinal detachment with and without proliferative vitreoretinopathy, macular hole and epiretinal membrane. Life 2021, 11, 453. [Google Scholar] [CrossRef]
- Takahashi, S.; Maeno, A.; Nakazawa, M.; Suzuki, Y.; Adachi, K. Oxidative Stress in the Vitreous Fluid with Rhegmatogenous Retinal Detachment. J. Clin. Exp. Ophthalmol. 2017, 6, 682. [Google Scholar] [CrossRef]
- Kwon, O.W.; Song, J.H.; Roh, M.I.; Song, J.H. Retinal Detachment and Proliferative Vitreoretinopathy. In Retinal Pharmacotherapy; Elsevier: Amsterdam, The Netherlands, 2010; pp. 154–162. [Google Scholar]
- Lo, A.C.Y.; Woo, T.T.Y.; Wong, R.L.M.; Wong, D. Apoptosis and other cell death mechanisms after retinal detachment: Implications for photoreceptor rescue. Ophthalmologica 2011, 226, 10–17. [Google Scholar] [CrossRef]
- Tamiya, S.; Liu, L.H.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, P.; Yandiev, Y.; Hui, Y.-N.; Wang, Y. Pathogenesis of Proliferative Vitreoretinopathy. In Retina; Ryan, S.J., Hinton, D.R., Wiedemann, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 3, pp. 1640–1646. ISBN 9781455707379. [Google Scholar]
- Garweg, J.G.; Tappeiner, C.; Halberstadt, M. Pathophysiology of Proliferative Vitreoretinopathy in Retinal Detachment. Surv. Ophthalmol. 2013, 58, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.A.; Sotani, Y.; Ibrahim, D.G.; Kiuchi, Y. Role of macrophage migration inhibitory factor (MIF) in the effects of oxidative stress on human retinal pigment epithelial cells. Cell Biochem. Funct. 2017, 35, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.-H.; Lee, J.-J.; Wu, P.-C.; Kuo, H.-K.; Kuo, Y.-H.; Huang, H.-M. Oxidative stress enhanced the transforming growth factor-β2-induced epithelial-mesenchymal transition through chemokine ligand 1 on ARPE-19 cell. Sci. Rep. 2020, 10, 4000. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.I.; Murakami, Y.; Thanos, A.; Vavvas, D.G.; Miller, J.W. Edaravone, an ROS scavenger, ameliorates photoreceptor cell death after experimental retinal detachment. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3825–3831. [Google Scholar] [CrossRef]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic Acid (TUDCA) Protects Photoreceptors from Cell Death after Experimental Retinal Detachment. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef]
- Lei, H.; Velez, G.; Cui, J.; Samad, A.; Maberley, D.; Matsubara, J.; Kazlauskas, A. N-acetylcysteine suppresses retinal detachment in an experimental model of proliferative vitreoretinopathy. Am. J. Pathol. 2010, 177, 132–140. [Google Scholar] [CrossRef]
- Yu, J.; Liu, F.; Cui, S.J.; Liu, Y.; Song, Z.Y.; Cao, H.; Chen, F.E.; Wang, W.J.; Sun, T.; Wang, F. Vitreous proteomic analysis of proliferative vitreoretinopathy. Proteomics 2008, 8, 3667–3678. [Google Scholar] [CrossRef]
- Yu, J.; Peng, R.; Chen, H.; Cui, C.; Ba, J. Elucidation of the pathogenic mechanism of rhegmatogenous retinal detachment with proliferative vitreoretinopathy by proteomic analysis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8146–8153. [Google Scholar] [CrossRef]
- Yu, J.; Peng, R.; Chen, H.; Cui, C.; Ba, J.; Wang, F. Kininogen 1 and insulin-like growth factor binding protein 6: Candidate serum biomarkers of proliferative vitreoretinopathy. Clin. Exp. Optom. 2014, 97, 72–79. [Google Scholar] [CrossRef]
- Santos, F.; Gaspar, L.; Ciordia, S.; Rocha, A.; Castro e Sousa, J.; Paradela, A.; Passarinha, L.; Tomaz, C. iTRAQ Quantitative Proteomic Analysis of Vitreous from Patients with Retinal Detachment. Int. J. Mol. Sci. 2018, 19, 1157. [Google Scholar] [CrossRef] [PubMed]
- Öhman, T.; Gawriyski, L.; Miettinen, S.; Varjosalo, M.; Loukovaara, S. Molecular pathogenesis of rhegmatogenous retinal detachment. Sci. Rep. 2021, 11, 966. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, L.M.; Santos, F.M.; Albuquerque, T.; Castro-de-Sousa, J.P.; Passarinha, L.A.; Tomaz, C.T. Proteome analysis of vitreous humor in retinal detachment using two different flow-charts for protein fractionation. J. Chromatogr. B 2017, 1061–1062, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Gao, M.; Zhou, Y.; Liu, H.; Sun, X. DIA Comparative Proteomic Analysis of Retro-oil Fluid and Vitreous Fluid From Retinal Detachment Patients. Front. Mol. Biosci. 2021, 8, 763002. [Google Scholar] [CrossRef]
- Diederen, R.M.H.; La Heij, E.C.; Deutz, N.E.P.; Kessels, A.G.H.; van Eijk, H.M.H.; Hendrikse, F. Increased nitric oxide (NO) pathway metabolites in the vitreous fluid of patients with rhegmatogenous retinal detachment or diabetic traction retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 683–688. [Google Scholar] [CrossRef]
- Klöcker, N.; Cellerino, A.; Bähr, M. Free Radical Scavenging and Inhibition of Nitric Oxide Synthase Potentiates the Neurotrophic Effects of Brain-Derived Neurotrophic Factor on Axotomized Retinal Ganglion Cells In Vivo. J. Neurosci. 1998, 18, 1038–1046. [Google Scholar] [CrossRef]
- Nagai, N.; Ito, Y.; Shibata, T.; Kubo, E.; Sasaki, H. A positive feedback loop between nitric oxide and amyloid β (1-42) accelerates mitochondrial damage in human lens epithelial cells. Toxicology 2017, 381, 19–30. [Google Scholar] [CrossRef]
- Akama, K.T.; Van Eldik, L.J. β-Amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1β- and tumor necrosis factor-α (TNFα)-dependent, and involves a TNFα receptor-associated factor- and NFκB-inducing kinase-dependent signaling mechanism. J. Biol. Chem. 2000, 275, 7918–7924. [Google Scholar] [CrossRef]
- Combs, C.K.; Colleen Karlo, J.; Kao, S.C.; Landreth, G.E. β-amyloid stimulation of microglia anti monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef]
- Van Lint, P.; Libert, C. Matrix metalloproteinase-8: Cleavage can be decisive. Cytokine Growth Factor Rev. 2006, 17, 217–223. [Google Scholar] [CrossRef]
- Sethi, C.S.; Bailey, T.A.; Luthert, P.J.; Chong, N.H.V. Matrix metalloproteinase biology applied to vitreoretinal disorders. Br. J. Ophthalmol. 2000, 84, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akaike, T.; Nagano, T.; Miyajima, S.; Suga, M.; Ando, M.; Ichimori, K.; Maeda, H. Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: A novel mechanism for procollagenase activation involving nitric oxide. Arch. Biochem. Biophys. 1997, 342, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Scott, R.A.H.; Wallace, G.; Berry, M.; Logan, A.; Blanch, R.J. Inflammatory and fibrogenic factors in proliferative vitreoretinopathy development. Transl. Vis. Sci. Technol. 2020, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Kon, C.H.; Occleston, N.L.; Charteris, D.; Daniels, J.; Aylward, G.W.; Khaw, P.T. A prospective study of matrix metalloproteinases in proliferative vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1524–1529. [Google Scholar]
- Symeonidis, C.; Diza, E.; Papakonstantinou, E.; Souliou, E.; Dimitrakos, S.A.; Karakiulakis, G. Correlation of the Extent and Duration of Rhegmatogenous Retinal Detachment with the Expression of Matrix Metalloproteinases in the Vitreous. Retina 2007, 27, 1279–1285. [Google Scholar] [CrossRef]
- Symeonidis, C.; Diza, E.; Papakonstantinou, E.; Souliou, E.; Karakiulakis, G.; Dimitrakos, S.A. Expression of matrix metalloproteinases in the subretinal fluid correlates with the extent of rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2007, 245, 560–568. [Google Scholar] [CrossRef]
- Symeonidis, C.; Papakonstantinou, E.; Souliou, E.; Karakiulakis, G.; Dimitrakos, S.A.; Diza, E. Correlation of matrix metalloproteinase levels with the grade of proliferative vitreoretinopathy in the subretinal fluid and vitreous during rhegmatogenous retinal detachment. Acta Ophthalmol. 2011, 89, 339–345. [Google Scholar] [CrossRef]
- Phipps, J.A.; Dixon, M.A.; Jobling, A.I.; Wang, A.Y.; Greferath, U.; Vessey, K.A.; Fletcher, E.L. The renin-angiotensin system and the retinal neurovascular unit: A role in vascular regulation and disease. Exp. Eye Res. 2019, 187, 107753. [Google Scholar] [CrossRef]
- Holappa, M.; Vapaatalo, H.; Vaajanen, A. Many Faces of Renin-angiotensin System-Focus on Eye. Open Ophthalmol. J. 2017, 11, 122–142. [Google Scholar] [CrossRef]
- Hoerster, R.; Fauser, S.; Cursiefen, C.; Kirchhof, B.; Heindl, L.M. The influence of systemic renin-angiotensin-inhibition on ocular cytokines related to proliferative vitreoretinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1721–1725. [Google Scholar] [CrossRef]
- Schmaier, A.H. The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J. Clin. Investig. 2002, 109, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.H. The kallikrein-kinin and the renin-angiotensin systems have a multilayered interaction. Am. J. Physiol. Integr. Comp. Physiol. 2003, 285, R1–R13. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Shimazawa, M.; Hara, H. The kallikrein system in retinal damage/protection. Eur. J. Pharmacol. 2015, 749, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wei, Q.; Liu, Q.; Ren, C.; Liu, J.; Zhang, R.; He, M.; Wang, Q.; Du, Y.; Yu, J. Effect of bradykinin on TGF-β1-induced retinal pigment epithelial cell proliferation and extracellular matrix secretion. BMC Ophthalmol. 2016, 16, 199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Du, Y.; He, M.; Ren, C.; Wang, Q.; Liu, J.; Wu, Y.; Yu, J. Effect of bradykinin on cultured retinal pigment epithelial cells. Int. J. Clin. Exp. Pathol. 2016, 9, 2998–3006. [Google Scholar]
- Wu, Z.; Ding, N.; Yu, M.; Wang, K.; Luo, S.; Zou, W.; Zhou, Y.; Yan, B.; Jiang, Q. Identification of Potential Biomarkers for Rhegmatogenous Retinal Detachment Associated with Choroidal Detachment by Vitreous iTRAQ-Based Proteomic Profiling. Int. J. Mol. Sci. 2016, 17, 2052. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.; Larson, A.J.; Frederick, J.M.; Southwick, K.; Thulin, C.D.; Bernstein, P.S. Identification and characterization of a Pi isoform of glutathione S-transferase (GSTP1) as a zeaxanthin-binding protein in the macula of the human eye. J. Biol. Chem. 2004, 279, 49447–49454. [Google Scholar] [CrossRef]
- Ganea, E.; Harding, J.J. Glutathione-related enzymes and the eye. Curr. Eye Res. 2006, 31, 1–11. [Google Scholar] [CrossRef]
- Lee, W.-H.; Joshi, P.; Wen, R. Glutathione S-Transferase Pi Isoform (GSTP1) Expression in Murine Retina Increases with Developmental Maturity. In Advances in Experimental Medicine and Biology; Bowes Rickman, C., LaVail, M.M., Anderson, R.E., Grimm, C., Hollyfield, J., Ash, J., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2014; Volume 801, pp. 23–30. ISBN 978-3-319-17120-3. [Google Scholar]
- Liang, F.Q.; Alssadi, R.; Morehead, P.; Awasthi, Y.C.; Godley, B.F. Enhanced expression of glutathione-S-transferase A1-1 protects against oxidative stress in human retinal pigment epithelial cells. Exp. Eye Res. 2005, 80, 113–119. [Google Scholar] [CrossRef]
- Hamadmad, S.; Shah, M.H.; Kusibati, R.; Kim, B.; Erickson, B.; Heisler-Taylor, T.; Bhattacharya, S.K.; Abdel-Rahman, M.H.; Cebulla, C.M.; Van Law, H.; et al. Significant upregulation of small heat shock protein αA-crystallin in retinal detachment. Exp. Eye Res. 2019, 189, 107811. [Google Scholar] [CrossRef]
- Kayama, M.; Nakazawa, T.; Thanos, A.; Morizane, Y.; Murakami, Y.; Theodoropoulou, S.; Abe, T.; Vavvas, D.; Miller, J.W. Heat shock protein 70 (HSP70) is critical for the photoreceptor stress response after retinal detachment via modulating anti-apoptotic Akt kinase. Am. J. Pathol. 2011, 178, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Urbak, L.; Vorum, H. Heat Shock Proteins in the Human Eye. Int. J. Proteom. 2010, 2010, 479571. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef] [PubMed]
- Villarejo-Zori, B.; Jiménez-Loygorri, J.I.; Zapata-Muñoz, J.; Bell, K.; Boya, P. New insights into the role of autophagy in retinal and eye diseases. Mol. Aspects Med. 2021, 82, 101038. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yao, J.; Jia, L.; Ferguson, T.A.; Weber, S.; Sundstrom, J.M.; Wubben, T.J.; Besirli, C.G.; Zacks, D.N. Autophagy activation and photoreceptor survival in retinal detachment. Exp. Eye Res. 2021, 205, 108492. [Google Scholar] [CrossRef] [PubMed]
- Danser, A.H.J.; Derkx, F.H.M.; Admiraal, P.J.J.; Deinum, J.; de Jong, P.T.V.M.; Schalekamp, M.A.D.H. Angiotensin levels in the eye. Investig. Ophthalmol. Vis. Sci. 1994, 35, 1008–1018. [Google Scholar]
- Nagai, N.; Izumi-Nagai, K.; Oike, Y.; Koto, T.; Satofuka, S.; Ozawa, Y.; Yamashiro, K.; Inoue, M.; Tsubota, K.; Umezawa, K.; et al. Suppression of diabetes-induced retinal inflammation by blocking the angiotensin II type 1 receptor or its downstream nuclear factor-κB pathway. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4342–4350. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef]
- Xie, X.; He, Z.; Chen, N.; Tang, Z.; Wang, Q.; Cai, Y. The Roles of Environmental Factors in Regulation of Oxidative Stress in Plant. BioMed Res. Int. 2019, 2019, 9732325. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Azat Aziz, M.; Shehab Diab, A.; Abdulrazak Mohammed, A. Antioxidant Categories and Mode of Action. In Antioxidants; Shalaby, E., Ed.; IntechOpen: London, UK, 2019; pp. 1–20. [Google Scholar]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Nie, K.; Jiang, H.; Fan, W. Effects of lutein supplementation in age-related macular degeneration. PLoS ONE 2019, 14, e0227048. [Google Scholar] [CrossRef] [PubMed]
- VandenLangenberg, G.M.; Mares-Perlman, J.A.; Klein, R.; Klein, B.E.K.; Brady, W.E.; Palta, M. Associations between Antioxidant and Zinc Intake and the 5-Year Incidence of Early Age-related Maculopathy in the Beaver Dam Eye Study. Am. J. Epidemiol. 1998, 148, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Mitchell, P.; Webb, K.; Leeder, S.R. Dietary antioxidants and age-related maculopathy: The Blue Mountains Eye Study. Ophthalmology 1999, 106, 761–767. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group A Randomized, Placebo-Controlled, Clinical Trial of High-Dose Supplementation with Vitamins C and E, Beta Carotene, and Zinc for Age-Related Macular Degeneration and Vision Loss. Arch. Ophthalmol. 2001, 119, 1417–1436. [CrossRef]
- Smirnoff, N. L-Ascorbic acid biosynthesis. In Vitamins and Hormones; Academic Press: Cambridge, MA, USA, 2001; Volume 61, pp. 241–266. [Google Scholar]
- Teichert, J.; Preiss, R. HPLC-methods for determination of lipoic acid and its reduced form in human plasma. Int. J. Clin. Pharmacol. Ther. Toxicol. 1992, 30, 511–512. [Google Scholar]
- Baillie, J.K.; Bates, M.G.D.; Thompson, A.A.R.; Waring, W.S.; Partridge, R.W.; Schnopp, M.F.; Simpson, A.; Gulliver-Sloan, F.; Maxwell, S.R.J.; Webb, D.J. Endogenous Urate Production Augments Plasma Antioxidant Capacity in Healthy Lowland Subjects Exposed to High Altitude. Chest 2007, 131, 1473–1478. [Google Scholar] [CrossRef]
- El-Sohemy, A.; Baylin, A.; Kabagambe, E.; Ascherio, A.; Spiegelman, D.; Campos, H. Individual carotenoid concentrations in adipose tissue and plasma as biomarkers of dietary intake. Am. J. Clin. Nutr. 2002, 76, 172–179. [Google Scholar] [CrossRef]
- Evelson, P.; Travacio, M.; Repetto, M.; Escobar, J.; Llesuy, S.; Lissi, E.A. Evaluation of Total Reactive Antioxidant Potential (TRAP) of Tissue Homogenates and Their Cytosols. Arch. Biochem. Biophys. 2001, 388, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.; Barbas, C. Vitamin E: Action, metabolism and perspectives. J. Physiol. Biochem. 2001, 57, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Baggio, E.; Gandini, R.; Plancher, A.C.; Passeri, M.; Carmosino, G. Italian multicenter study on the safety and efficacy of coenzyme Q10 as adjunctive therapy in heart failure (interim analysis). Clin. Investig. 1993, 71, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Pathways of Oxidative Damage. Annu. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Tinggi, U. Selenium: Its role as antioxidant in human health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef]
- Powell, S.R. The Antioxidant Properties of Zinc. J. Nutr. 2000, 130, 1447–1454. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Rohowetz, L.; Kraus, J.; Koulen, P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef]
- Trinity, J.D.; Symons, J.D. ClinicalTrials.gov Identifier: NCT04351113. Targeting Oxidative Stress to Prevent Vascular and Skeletal Muscle Dysfunction during Disuse. Available online: https://clinicaltrials.gov/ct2/show/NCT04351113 (accessed on 9 February 2022).
- Wan, P.; Su, W.; Zhang, Y.; Li, Z.; Deng, C.; Zhuo, Y. Trimetazidine protects retinal ganglion cells from acute glaucoma via the Nrf2/Ho-1 pathway. Clin. Sci. 2017, 131, 2363–2375. [Google Scholar] [CrossRef]
- Wang, X.; Yuan, Z. Activation of Nrf2/HO-1 pathway protects retinal ganglion cells from a rat chronic ocular hypertension model of glaucoma. Int. Ophthalmol. 2019, 39, 2303–2312. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, Z.; Liu, C.H.; Gong, Y.; Cakir, B.; Liegl, R.; Sun, Y.; Meng, S.S.; Burnim, S.B.; Arellano, I.; et al. Fibroblast growth factor 21 protects photoreceptor function in type 1 diabetic mice. Diabetes 2018, 67, 974–985. [Google Scholar] [CrossRef]
- Hsu, M.-Y.; Hsiao, Y.-P.; Lin, Y.-T.; Chen, C.; Lee, C.-M.; Liao, W.-C.; Tsou, S.-C.; Lin, H.-W.; Chang, Y.-Y. Quercetin Alleviates the Accumulation of Superoxide in Sodium Iodate-Induced Retinal Autophagy by Regulating Mitochondrial Reactive Oxygen Species Homeostasis through Enhanced Deacetyl-SOD2 via the Nrf2-PGC-1α-Sirt1 Pathway. Antioxidants 2021, 10, 1125. [Google Scholar] [CrossRef]
- Araújo, R.S.; Bitoque, D.B.; Silva, G.A. Development of strategies to modulate gene expression of angiogenesis-related molecules in the retina. Gene 2021, 791, 145724. [Google Scholar] [CrossRef]
- Zhang, X.; Lai, D.; Bao, S.; Hambly, B.D.; Gillies, M.C. Triamcinolone Acetonide Inhibits p38MAPK Activation and Neuronal Apoptosis in Early Diabetic Retinopathy. Curr. Mol. Med. 2013, 13, 946–958. [Google Scholar] [CrossRef]
- Holmlund, J.T.; Venigalla, M.; Zhen, W.; Adkins, D.; Pathare, P.; Agarwala, S.; Kunos, C.; Chen, Y.; Buatti, J.; Mahmood, A.; et al. ClinicalTrials.gov Identifier: NCT01921426. A Phase 1 Dose Escalation Study of GC4419 in Combination with Chemoradiation for Squamous Cell Cancer of the Head & Neck. Available online: https://clinicaltrials.gov/show/NCT01921426 (accessed on 9 February 2022).
- Verges, B. ClinicalTrials.gov Identifier: NCT02826083. Study to Evaluate the Effects of XXS on Oxidative Stress in Patients with Mild or Moderate Hyperlipidemia (XXS SYMPA). Available online: https://clinicaltrials.gov/ct2/show/NCT02826083 (accessed on 9 February 2022).
- Jicha, G.A.; Power, R. ClinicalTrials.gov Identifier: NCT01731093. Safety and Tolerability of Antioxidant (AT-001) for Reducing Brain Oxidative Stress. Available online: https://clinicaltrials.gov/ct2/show/NCT01731093 (accessed on 9 February 2022).
- Crimmins, S. ClinicalTrials.gov Identifier: NCT03056014. Antioxidant Use in Diabetes to Reduce Oxidative Stress. Available online: https://clinicaltrials.gov/ct2/show/NCT03056014 (accessed on 9 February 2022).
- Jiaotong, X.; Ma, L. ClinicalTrials.gov Identifier: NCT01056094. Effects of Lutein Supplementation on Oxidative Stress and Inflammation in Healthy Nonsmokers. Available online: https://clinicaltrials.gov/ct2/show/NCT01056094 (accessed on 9 February 2022).
- Norell, T.; Bamman, M.M. ClinicalTrials.gov Identifier: NCT04732247. Oxytocin for Oxidative Stress and Inflammation. Available online: https://clinicaltrials.gov/ct2/show/NCT04732247 (accessed on 9 February 2022).
- Roman-Pintos, L.M.; Miranda-Diaz, A.G.; Rodriguez-Carrizalez, A.D.; Villegas-Rivera, G. ClinicalTrials.gov Identifier: NCT02129231. Statins for Oxidative Stress and Mitochondrial Function in Diabetic Polyneuropathy. Available online: https://clinicaltrials.gov/ct2/show/NCT02129231 (accessed on 9 February 2022).
- Villegas-Rivera, G.; Román-Pintos, L.M.; Cardona-Muñoz, E.G.; Arias-Carvajal, O.; Rodríguez-Carrizalez, A.D.; Troyo-Sanromán, R.; Pacheco-Moisés, F.P.; Moreno-Ulloa, A.; Miranda-Díaz, A.G. Effects of Ezetimibe/Simvastatin and Rosuvastatin on Oxidative Stress in Diabetic Neuropathy: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Oxid. Med. Cell. Longev. 2015, 2015, 756294. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-G.; Park, J. ClinicalTrials.gov Identifier: NCT01339143. Compare the Effect of DPP-IV Inhibitor or TZD on Glycemic Variability and Oxidative Stress in Patient with 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT01339143 (accessed on 9 February 2022).
- Riche, D.M. ClinicalTrials.gov Identifier: NCT01267227. Effect of Pterostilbene on Cholesterol, Blood Pressure and Oxidative Stress. Available online: https://clinicaltrials.gov/ct2/show/NCT01267227 (accessed on 9 February 2022).
- Rodríguez-Carrizalez, A.D.; López-Contreras, A.K.; Olvera-Montaño, C. ClinicalTrials.gov Identifier: NCT04071977. Combined Antioxidant Therapy on Oxidative Stress in Aqueous and Vitreous Humor of Diabetic Retinopathy Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT04071977 (accessed on 9 February 2022).
- Puoci, F. ClinicalTrials.gov Identifier: NCT04912947. Evaluation of the Antioxidant and Anti-Inflammatory Capacity of Nutraceutical IMMU·SYSTEM Food Supplement (EVAANIS) (EVAANIS). Available online: https://clinicaltrials.gov/ct2/show/NCT04912947 (accessed on 9 February 2022).
- Stevens, J.F. ClinicalTrials.gov Identifier: NCT02432651. Xanthohumol and Prevention of DNA Damage. Available online: https://clinicaltrials.gov/ct2/show/NCT02432651 (accessed on 9 February 2022).
- Tareen, N.; Drew, C. ClinicalTrials.gov Identifier: NCT01251315. The Effects of a Glutathione Precursor (FT061452),on Serum and Intracellular Glutathione Levels. Available online: https://clinicaltrials.gov/ct2/show/NCT01251315 (accessed on 9 February 2022).
- Francisco Javier López Román ClinicalTrials.gov Identifier: NCT03798821. The Antioxidant Efficacy of a Product Probiotic in Research (BIO). Available online: https://clinicaltrials.gov/ct2/show/NCT03798821 (accessed on 9 February 2022).
- Macarro, M.S.; Ávila-gandía, V.; Pérez-piñero, S.; Cánovas, F.; García-muñoz, A.M.; Abellán-ruiz, M.S.; Victoria-montesinos, D.; Luque-rubia, A.J.; Climent, E.; Genovés, S.; et al. Antioxidant effect of a probiotic product on a model of oxidative stress induced by high-intensity and duration physical exercise. Antioxidants 2021, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, A.T. ClinicalTrials.gov Identifier: NCT02051842. Effect of Metadoxine on Oxidative Stress in Non-alcoholic Hepatic Steatosis. Available online: https://clinicaltrials.gov/ct2/show/NCT02051842 (accessed on 9 February 2022).
- Lee, M.-K. ClinicalTrials.gov Identifier: NCT01404676. The Effect of Vildagliptin Based Treatment Versus Sulfonylurea on Glycemic Variability, Oxidative Stress, GLP-1, and Endothelial Function in Patients with Type 2 Diabetes. Available online: https://clinicaltrials.gov/ct2/show/NCT01404676 (accessed on 9 February 2022).
- Gitelman, S.E. ClinicalTrials.gov Identifier: NCT00187564. Pilot Study on the Effect of Oral Controlled-Release Alpha-Lipoic Acid on Oxidative Stress in Adolescents with Type 1 Diabetes Mellitus. Available online: https://clinicaltrials.gov/ct2/show/NCT00187564 (accessed on 9 February 2022).
- Lee, S.-H.; Kim, E.-S.; Yoon, K.-H. ClinicalTrials.gov Identifier: NCT00699322. Effect of Dipeptidyl Peptidase-IV Inhibitor and Sulfonylurea on Glucose Variability and Oxidative Stress. Available online: https://clinicaltrials.gov/ct2/show/NCT00699322 (accessed on 9 February 2022).
- de Aguiar, L.G.K. ClinicalTrials.gov Identifier: NCT01827280. Acute and Short-term Chronic Effects of Galvus (Vildagliptin) in Diabetes Type 2 Obese Women. Available online: https://clinicaltrials.gov/ct2/show/NCT01827280 (accessed on 9 February 2022).
- Schiapaccassa, A.; Maranhão, P.A.; de Souza, M.d.G.C.; Panazzolo, D.G.; Nogueira Neto, J.F.; Bouskela, E.; Kraemer-Aguiar, L.G. 30-days effects of vildagliptin on vascular function, plasma viscosity, inflammation, oxidative stress, and intestinal peptides on drug-naïve women with diabetes and obesity: A randomized head-to-head metformin-controlled study. Diabetol. Metab. Syndr. 2019, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P. ClinicalTrials.gov Identifier: NCT02372630. The Effect of LINAGLIPTIN on Inflammation, Oxidative Stress and Insulin Resistance in Obese Type 2 Diabetes Subjects (1971). Available online: https://clinicaltrials.gov/ct2/show/NCT02372630 (accessed on 9 February 2022).
- Ochoa, J.J. ClinicalTrials.gov Identifier: NCT01940627. Influence of a Short Term Supplementation with Ubiquinol on Diverse Aspects Related to the Physical Activity. Available online: https://clinicaltrials.gov/ct2/show/NCT01940627 (accessed on 9 February 2022).
- Enghusen, H. ClinicalTrials.gov Identifier: NCT02256254. SIMOX—Induction of Oxidative Stress (SIMOX). Available online: https://clinicaltrials.gov/ct2/show/study/NCT02256254 (accessed on 9 February 2022).
- Gung, C.; Luo, C.-F. ClinicalTrials.gov Identifier: NCT01334424. Oxidative Stress in Robot-Assisted Laparoscopic Radical Prostatectomy. Available online: https://clinicaltrials.gov/ct2/show/NCT01334424 (accessed on 9 February 2022).




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).