
Modulation of Cellular Redox Parameters for Improving Therapeutic Responses in Multiple Myeloma

 ,
,  ,
,  ,
,
Abstract
1. Introduction
1.1. General Consideration on Multiple Myeloma and Oxidative Stress
1.2. Oxidative Stress and Cancer
1.3. Oxidative Stress and Myelomagenesis
2. Oxidative Stress and Proteasome Inhibitors
Oxidative Stress and Chemoresistance to Proteasome Inhibitors
3. Oxidative Stress and Immunomodulatory and Alkylating Drugs
4. Oxidative Stress and Autologous Transplantation in Multiple Myeloma
5. Future Perspectives on the Modulation of Oxidative Stress in Patients with Multiple Myeloma
5.1. Use of Substances of Natural Origin in the Modulation of Oxidative Stress in Multiple Myeloma
5.2. Metabolic Modifications and Oxidative Stress in Multiple Myeloma
6. Modulation of Oxidative Stress to Prevent Treatment Side Effects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.; et al. Cronin K SEER Cancer Statistics Review. Natl. Cancer Inst. 2019, 1975–2017. [Google Scholar]
- Casciaro, M.; Di Salvo, E.; Pace, E.; Ventura-Spagnolo, E.; Navarra, M.; Gangemi, S. Chlorinative stress in age-related diseases: A literature review. Immun. Ageing 2017, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cristani, M.; Speciale, A.; Saija, A.; Gangemi, S.; Minciullo, P.; Cimino, F. Circulating Advanced Oxidation Protein Products as Oxidative Stress Biomarkers and Progression Mediators in Pathological Conditions Related to Inflammation and Immune Dysregulation. Curr. Med. Chem. 2016, 23, 3862–3882. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Xiong, S.; Chng, W.-J.; Zhou, J. Crosstalk between endoplasmic reticulum stress and oxidative stress: A dynamic duo in multiple myeloma. Cell. Mol. Life Sci. 2021, 78, 3883–3906. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Ox-idative stress: Harms and benefits for human health. Oxid. Med. Cell Longev. 2017, 2017, 1–13. [Google Scholar]
- O’Hagan, H.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Hendershot, L.M. Oxidative folding: Cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 2009, 11, 2317–2331. [Google Scholar] [CrossRef]
- Imbesi, S.; Musolino, C.; Allegra, A.; Saija, A.; Morabito, F.; Calapai, G.; Gangemi, S. Oxidative stress in oncohematologic diseases: An update. Expert Rev. Hematol. 2013, 6, 317–325. [Google Scholar] [CrossRef]
- Cieslar, P.; Mášová, L.; Scheiner, T.; Ryšavá, J.; Křížová, P.; Danzigová, Z.; Špička, I.; Tesař, V. Oxidative stress and platelet function in multiple myeloma and renal insufficiency: Clinical relations of different tests. Thromb. Res. 2002, 105, 277–283. [Google Scholar] [CrossRef]
- Sharma, A.; Tripathi, M.; Satyam, A.; Kumar, L. Study of antioxidant levels in patients with multiple myeloma. Leuk. Lymphoma 2009, 50, 809–815. [Google Scholar] [CrossRef]
- Gangemi, S.; Allegra, A.; Alonci, A.; Cristani, M.; Russo, S.; Speciale, A.; Penna, G.; Spatari, G.; Cannavò, A.; Bellomo, G.; et al. Increase of novel biomarkers for oxidative stress in patients with plasma cell disorders and in multiple myeloma patients with bone lesions. Agents Actions 2012, 61, 1063–1067. [Google Scholar] [CrossRef]
- Mehdi, W.A.; Zainulabdeen, J.A.; Mehde, A.A. Investigation of the Antioxidant Status in Multiple Myeloma Patients: Effects of Therapy. Asian Pac. J. Cancer Prev. 2013, 14, 3663–3667. [Google Scholar] [CrossRef][Green Version]
- Smirnova, O.V.; Titova, N.M.; Elmanova, N.G. The Relationship Between the Pro-Oxidant and Antioxidant System Status of Patients with Multiple Myeloma and the Disease Stage. Bull. Exp. Biol. Med. 2014, 157, 375–379. [Google Scholar] [CrossRef]
- Faridvand, Y.; Oskuyi, A.E.; Khadem-Ansari, M.-H. Serum 8-isoprostane levels and paraoxonase 1 activity in patients with stage I multiple myeloma. Redox Rep. Commun. Free Radic. Res. 2016, 21, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ellidag, H.Y.; Eren, E.; Aydin, O.; Yıldırım, M.; Sezer, C.; Yilmaz, N. Multiple Myeloma: Relationship to Antioxidant Esterases. Med. Princ. Pract. 2014, 23, 18–23. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Polito, F.; Oteri, R.; Alibrandi, A.; Allegra, A.G.; Oteri, G.; Di Giorgio, R.M.; Musolino, C.; Aguennouz, M. SIRT2 and SIRT3 expression correlates with redox imbalance and advanced clinical stage in patients with multiple myeloma. Clin. Biochem. 2021, 93, 42–49. [Google Scholar] [CrossRef]
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.-T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015, 5, 972–987. [Google Scholar] [CrossRef]
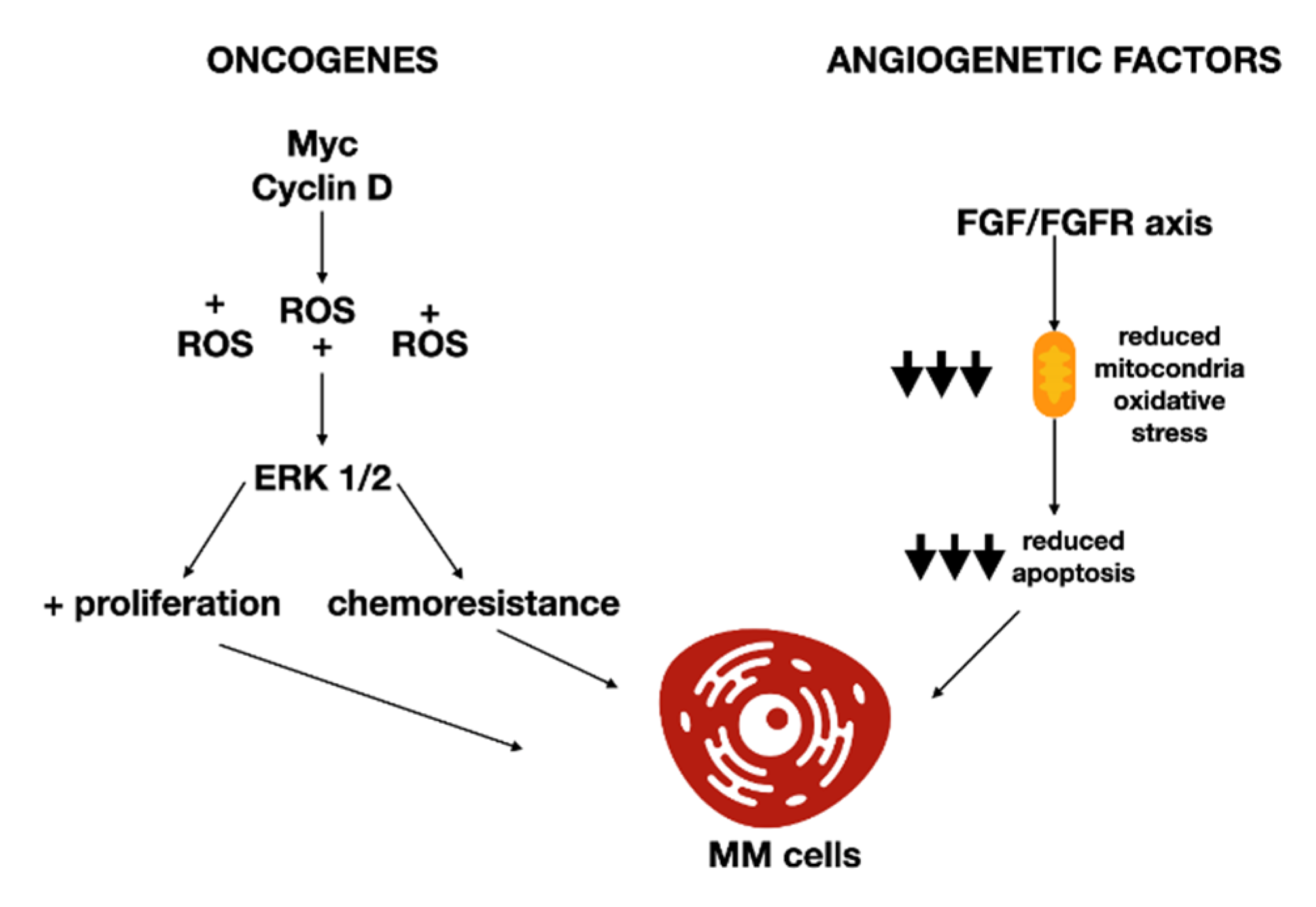
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J.J. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef]
- Massagué, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Bustany, S.; Bourgeais, J.; Tchakarska, G.; Body, S.; Herault, O.; Gouilleux, F.; Sola, B. Cyclin D1 unbalances the redox status controlling cell adhesion, migration, and drug resistance in myeloma cells. Oncotarget 2016, 7, 45214–45224. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF Trapping Inhibits Multiple Myeloma Growth through c-Myc Degradation–Induced Mitochondrial Oxidative Stress. Cancer Res. 2020, 80, 2340–2354. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.J.; Brailey, M. Emerging small molecule approaches to enhance the anti-myeloma benefit of proteasome inhibitors. Cancer Metastasis Rev. 2017, 36, 585–598. [Google Scholar] [CrossRef]
- Harding, T.; Baughn, L.; Kumar, S.; Van Ness, B. The future of myeloma precision medicine: Integrating the compendium of known drug resistance mechanisms with emerging tumor profiling technologies. Leukemia 2019, 33, 863–883. [Google Scholar] [CrossRef]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Gerace, D.; Russo, S.; Innao, V.; Calabrò, L.; Musolino, C. New orally active proteasome inhibitors in multiple myeloma. Leuk. Res. 2014, 38, 1–9. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.H.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, srep05896. [Google Scholar] [CrossRef]
- Parekh, S.; Weniger, M.A.; Wiestner, A. New molecular targets in mantle cell lymphoma. Semin. Cancer Biol. 2011, 21, 335–346. [Google Scholar] [CrossRef]
- Goel, A.; Spitz, D.; Weiner, G. Manipulation of cellular redox parameters for improving therapeutic responses in B-cell lymphoma and multiple myeloma. J. Cell. Biochem. 2011, 113, 419–425. [Google Scholar] [CrossRef]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006, 107, 257–264. [Google Scholar] [CrossRef]
- Ling, Y.-H.; Liebes, L.; Zou, Y.; Perez-Soler, R. Reactive Oxygen Species Generation and Mitochondrial Dysfunction in the Apoptotic Response to Bortezomib, a Novel Proteasome Inhibitor, in Human H460 Non-small Cell Lung Cancer Cells. J. Biol. Chem. 2003, 278, 33714–33723. [Google Scholar] [CrossRef]
- Llobet, D.; Eritja, N.; Encinas, M.; Sorolla, A.; Yeramian, A.; Schoenenberger-Arnaiz, J.A.; Llombart-Cussac, A.; Marti, R.M.; Matias-Guiu, X.; Dolcet, X. Antioxidants block proteasome inhibitor function in endometrial carcinoma cells. Anti-Cancer Drugs 2008, 19, 115–124. [Google Scholar] [CrossRef]
- Yang, J.; Su, Y.; Richmond, A. Antioxidants tiron and N-acetyl-L-cysteine differentially mediate apoptosis in melanoma cells via a reactive oxygen species-independent NF-κB pathway. Free Radic. Biol. Med. 2007, 42, 1369–1380. [Google Scholar] [CrossRef]
- Mannava, S.; Zhuang, D.; Nair, J.R.; Bansal, R.; Wawrzyniak, J.A.; Zucker, S.N.; Fink, E.E.; Moparthy, K.C.; Hu, Q.; Liu, S.; et al. KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood 2012, 119, 1450–1458. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 Amplifies Oxidative Stress via Induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef]
- Arnér, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Horstkotte, J.; Perisic, T.; Schneider, M.; Lange, P.; Schroeder, M.; Kiermayer, C.; Hinkel, R.; Ziegler, T.; Mandal, P.K.; David, R.; et al. Mitochondrial Thioredoxin Reductase Is Essential for Early Postischemic Myocardial Protection. Circulation 2011, 124, 2892–2902. [Google Scholar] [CrossRef]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2016, 30, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism—Emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise Induces Hippocampal BDNF through a PGC-1α/FNDC5 Pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Sweeney, T.R.; Shigenaga, J.K.; Chui, L.G.; Moser, A.; Grunfeld, C.; Feingold, K.R. Tumor necrosis factor and interleukin 1 decrease RXRalpha, PPARalpha, PPARgamma, LXRalpha, and the coactivators SRC-1, PGC-1alpha, and PGC-1beta in liver cells. Metabolism 2007, 56, 267–279. [Google Scholar] [CrossRef]
- Cao, D.; Jin, L.; Zhou, H.; Yu, W.; Hu, Y.; Guo, T. Inhibition of PGC-1α after chemotherapy-mediated insult confines multiple myeloma cell survival by affecting ROS accumulation. Oncol. Rep. 2014, 33, 899–904. [Google Scholar] [CrossRef]
- Gao, L.; Gao, M.; Yang, G.; Tao, Y.; Kong, Y.; Yang, R.; Meng, X.; Ai, G.; Wei, R.; Wu, H.; et al. Synergistic Activity of Carfilzomib and Panobinostat in Multiple Myeloma Cells via Modulation of ROS Generation and ERK1/2. BioMed Res. Int. 2015, 2015, 459052. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.-W.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered redox regulation and S-glutathionylation of BiP contribute to bortezomib resistance in multiple myeloma. Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzeźniakiewicz-Janus, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Espinosa, B.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Linder, S. Oxidative Stress Induced by the Deubiquitinase Inhibitor b-AP15 Is Associated with Mitochondrial Impairment. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From metabolism to molecular therapy. Am. J. Respir. Cell Mol. Biol. 2009, 41, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Goswami, B.; Rajappa, M.; Sharma, M.; Sharma, A. Inflammation: Its role and interplay in the development of cancer, with special focus on gynecological malignancies. Int. J. Gynecol. Cancer 2008, 18, 591–599. [Google Scholar] [CrossRef]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Volti, G.L. Heme oxygenase-1 nuclear translocation regulates bortezomib-induced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Puglisi, F.; Barbato, A.; Vicario, N.; Cambria, D.; Parrinello, N.L.; Romano, A.; Conticello, C.; Forte, S.; et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers 2020, 12, 1999. [Google Scholar] [CrossRef]
- Pei, X.-Y.; Dai, Y.; Grant, S. Synergistic Induction of Oxidative Injury and Apoptosis in Human Multiple Myeloma Cells by the Proteasome Inhibitor Bortezomib and Histone Deacetylase Inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef]
- Innao, V.; Rizzo, V.; Allegra, A.; Musolino, C.; Allegra, A. Promising Anti-Mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma. Cells 2021, 10, 439. [Google Scholar] [CrossRef]
- Fang, Q.; Jiang, S.; Li, C. Evodiamine Selectively Inhibits Multiple Myeloma Cell Growth by Triggering Activation of Intrinsic Apoptosis Pathway. OncoTargets Ther. 2019, 12, 11383–11391. [Google Scholar] [CrossRef]
- Han, H.-J.; Tokino, T.; Nakamura, Y. CSR, a scavenger receptor-like protein with a protective role against cellular damage causedby UV irradiation and oxidative stress. Hum. Mol. Genet. 1998, 7, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- DeWitte-Orr, S.J.; Collins, S.E.; Bauer, C.M.; Bowdish, D.M.; Mossman, K.L. An accessory to the ’Trinity’: SR-As are essential pathogen sensors of extracellular dsRNA, mediating entry and leading to subsequent type I IFN responses. PLoS Pathog. 2010, 6, e1000829. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.O.; Schibler, J.; Fitzgerald, M.P.; Singh, N.; Salem, K.; Zhan, F.; Goel, A. Scavenger receptor class A member 3 (SCARA3) in disease progression and therapy resistance in multiple myeloma. Leuk. Res. 2013, 37, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Zmorzyński, S.; Popek-Marciniec, S.; Szudy-Szczyrek, A.; Wojcierowska-Litwin, M.; Korszeń-Pilecka, I.; Chocholska, S.; Styk, W.; Hus, M.; Filip, A.A. The Association of GSTT1, GSTM1, and TNF-α Polymorphisms With the Risk and Outcome in Multiple Myeloma. Front. Oncol. 2019, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Prelowska, M.K.; Mehlich, D.; Ugurlu, M.T.; Kedzierska, H.; Cwiek, A.; Kosnik, A.; Kaminska, K.; Marusiak, A.A.; Nowis, D. Inhibition of the ʟ-glutamine transporter ASCT2 sensitizes plasma cell myeloma cells to proteasome inhibitors. Cancer Lett. 2021, 507, 13–25. [Google Scholar] [CrossRef]
- Mitra, A.K.; Kumar, H.; Ramakrishnan, V.; Chen, L.; Baughn, L.; Kumar, S.; Rajkumar, S.V.; Van Ness, B.G. In vitro and ex vivo gene expression profiling reveals differential kinetic response of HSPs and UPR genes is associated with PI resistance in multiple myeloma. Blood Cancer J. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; Van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Mountjoy, L.; Sebastian, S.; Jain, T.; Hilal, T.; Gonzalez-Velez, M.; Girardo, M.; Ahmann, G.; Larsen, J.; Bergsagel, L.; Fonseca, R. Prediction of immunomodulatory drugs (IMiDs) sensitivity in myeloma via determination of baseline anti-oxidative stress capacity. Leukemia 2020, 34, 3060–3063. [Google Scholar] [CrossRef]
- Sebastian, S.; Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Panchabhai, S.C.; Van Wier, S.A.; Ahmann, G.J.; Chesi, M.; Bergsagel, P.L.; Stewart, A.K.; et al. Multiple myeloma cells’ capacity to decompose H2O2 determines lenalidomide sensitivity. Blood 2017, 129, 991–1007. [Google Scholar] [CrossRef]
- Zub, K.A.; De Sousa, M.M.L.; Sarno, A.; Sharma, A.; Demirovic, A.; Rao, S.; Young, C.; Aas, P.A.; Ericsson, I.; Sundan, A.; et al. Modulation of Cell Metabolic Pathways and Oxidative Stress Signaling Contribute to Acquired Melphalan Resistance in Multiple Myeloma Cells. PLoS ONE 2015, 10, e0119857. [Google Scholar] [CrossRef]
- Knight, J.A. Free radicals: Their history and current status in aging and disease. Ann. Clin. Lab. Sci. USA 1998, 28, 331–346. [Google Scholar]
- Evens, A.; Mehta, J.; Gordon, L.I. Rust and corrosion in hematopoietic stem cell transplantation: The problem of iron and oxidative stress. Bone Marrow Transplant. 2004, 34, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.; Carreras, E.; Gluckman, E.; Masszi, T. EBMT-ESH Handbook on Haematopoietic Stem Cell Transplantation, 6th ed.; European School of Haematology: Paris, France, 2012; Available online: https://ebmtonline.forumservice.net/ (accessed on 22 December 2021).
- Dos Santos, T.N.; Duarte, F.B.; Filho, P.A.M.; Barbosa, M.C.; Cavalcanti, B.C.; De Vasconcelos, P.R.L.; Dutra, L.L.; Lopes, G.S.; Costa, F.O.; Leitão, J.P.V.; et al. Association of oxidative stress and DNA damage with grafting time in patients with multiple myeloma and lymphoma submitted to autologous hematopoietic stem cell transplantation. Rev. Assoc. Med. Bras. 2016, 62, 39–43. [Google Scholar] [CrossRef]
- Sabuncuog, L.S.; Öztas, Y.; Çetinkaya, D.U.; Özgünes, N.; Özgünes, H. Oxidative protein damage with carbonyl levels and nitrotyrosine expression after chemotherapy in bone marrow transplantation patients. Pharmacology 2012, 89, 283–286. [Google Scholar] [CrossRef]
- Bayraktar, U.D.; Nates, J.L. Intensive care outcomes in adult hematopoietic stem cell transplantation patients. World J. Clin. Oncol. 2016, 109, 98–105. [Google Scholar] [CrossRef]
- Patterson, A.M.; Zhang, S.; Liu, L.; Li, H.; Singh, P.; Liu, Y.; Farag, S.S.; Pelus, L.M. Meloxicam with Filgrastim may Reduce Oxidative Stress in Hematopoietic Progenitor Cells during Mobilization of Autologous Peripheral Blood Stem Cells in Patients with Multiple Myeloma. Stem Cell Rev. Rep. 2021, 17, 2124–2138. [Google Scholar] [CrossRef]
- Röllig, C.; Illmer, T. The efficacy of arsenic trioxide for the treatment of relapsed and refractory multiple myeloma: A systematic review. Cancer Treat. Rev. 2009, 35, 425–430. [Google Scholar] [CrossRef]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef]
- Raninga, P.; Di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K.F. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410–15424. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Das Gupta, S.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Wang, X.; Stafford, W.; Mazurkiewicz, M.; Fryknäs, M.; Brjnic, S.; Zhang, X.; Gullbo, J.; Larsson, R.; Arnér, E.S.J.; D’Arcy, P.; et al. The 19S Deubiquitinase Inhibitor b-AP15 Is Enriched in Cells and Elicits Rapid Commitment to Cell Death. Mol. Pharmacol. 2014, 85, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Tibodeau, J.D.; Isham, C.R.; Bible, K.C. Annatto Constituent Cis-Bixin Has Selective Antimyeloma Effects Mediated by Oxidative Stress and Associated with Inhibition of Thioredoxin and Thioredoxin Reductase. Antioxid. Redox Signal. 2010, 13, 987–997. [Google Scholar] [CrossRef]
- Evens, A.M.; Lecane, P.; Magda, D.; Prachand, S.; Singhal, S.; Nelson, J.; Miller, R.A.; Gartenhaus, R.B.; Gordon, L.I. Motexafin gadolinium generates reactive oxygen species and induces apoptosis in sensitive and highly resistant multiple myeloma cells. Blood 2005, 105, 1265–1273. [Google Scholar] [CrossRef]
- Dvorakova, K.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; McClure, T.; Dorr, R.T. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem. Pharmacol. 2000, 60, 749–758. [Google Scholar] [CrossRef]
- Dvorakova, K.; Waltmire, C.N.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; Dorr, R.T. Induction of mitochondrial changes in myeloma cells by imexon. Blood 2001, 97, 3544–3551. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Z.; Li, M.; Li, X.; Wong, Y.-S.; Ngai, S.-M.; Zheng, W.; Zhang, Y.; Chen, T. Enhancement of Auranofin-Induced Apoptosis in MCF-7 Human Breast Cells by Selenocystine, a Synergistic Inhibitor of Thioredoxin Reductase. PLoS ONE 2013, 8, e53945. [Google Scholar] [CrossRef]
- Baker, A.F.; Landowski, T.; Dorr, R.; Tate, W.R.; Gard, J.M.; Tavenner, B.E.; Dragovich, T.; Coon, A.; Powis, G. The Antitumor Agent Imexon Activates Antioxidant Gene Expression: Evidence for an Oxidative Stress Response. Clin. Cancer Res. 2007, 13, 3388–3394. [Google Scholar] [CrossRef][Green Version]
- Isham, C.R.; Tibodeau, J.D.; Jin, W.; Xu, R.; Timm, M.M.; Bible, K.C. Chaetocin: A promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 2006, 109, 2579–2588. [Google Scholar] [CrossRef]
- Wang, W.; Adachi, M.; Kawamura, R.; Sakamoto, H.; Hayashi, T.; Ishida, T.; Imai, K.; Shinomura, Y. Parthenolide-induced apoptosis in multiple myeloma cells involves reactive oxygen species generation and cell sensitivity depends on catalase activity. Apoptosis 2006, 11, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Kufe, T.; Avigan, D.; Kufe, N. Targeting MUC1-C is synergistic with bortezomib in downregulating TIGAR and inducing ROS-mediated myeloma cell death. Blood 2014, 123, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. The transient receptor potential family of ion channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Rosenberg, E.; Dimenshtein-Voevoda, V.; Sirovsky, Y.; Vladimirsky, J.; Magen, H.; Ostrovsky, O.; Shimoni, A.; Bromberg, Z.; Weiss, L.; et al. Blocking of Transient Receptor Potential Vanilloid 1 (TRPV1) promotes terminal mitophagy in multiple myeloma, disturbing calcium homeostasis and targeting ubiquitin pathway and bortezomib-induced unfolded protein response. J. Hematol. Oncol. 2020, 13, 1–20. [Google Scholar] [CrossRef]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.C.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel Protein Disulfide Isomerase Inhibitor with Anticancer Activity in Multiple Myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2018, 33, 1011–1022. [Google Scholar] [CrossRef]
- Anchoori, R.K.; Tan, M.; Tseng, S.-H.; Peng, S.; Soong, R.-S.; Algethami, A.; Foran, P.; Das, S.; Wang, C.; Wang, T.-L.; et al. Structure-function analyses of candidate small molecule RPN13 inhibitors with antitumor properties. PLoS ONE 2020, 15, e0227727. [Google Scholar] [CrossRef]
- Harizi, H.; Corcuff, J.-B.; Gualde, N. Arachidonic-acid-derived eicosanoids: Roles in biology and immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar] [CrossRef]
- Yen, C.-C.; Hsiao, C.-D.; Chen, W.-M.; Wen, Y.-S.; Lin, Y.-C.; Chang, T.-W.; Yao, F.-Y.; Hung, S.-C.; Wang, J.-Y.; Chiu, J.-H.; et al. Cytotoxic effects of 15d-PGJ2 against osteosarcoma through ROS-mediated AKT and cell cycle inhibition. Oncotarget 2014, 5, 716–725. [Google Scholar] [CrossRef]
- Sperandio, M.; Demasi, A.P.D.; Martinez, E.F.; Saad, S.O.; Pericole, F.V.; Vieira, K.P.; Freitas, N.S.; Araújo, V.; Brown, A.L.; Clemente-Napimoga, J.T.; et al. 15d-PGJ 2 as an endoplasmic reticulum stress manipulator in multiple myeloma in vitro and in vivo. Exp. Mol. Pathol. 2017, 102, 434–445. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: Reassessment of the ARE consensus sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef]
- No, J.H.; Kim, Y.-B.; Song, Y.S. Targeting Nrf2 Signaling to Combat Chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Tuckett, A.Z.; Fennell, M.; Garippa, R.; Zakrzewski, J.L. Repurposing of the CDK inhibitor PHA-767491 as a NRF2 inhibitor drug candidate for cancer therapy via redox modulation. Investig. New Drugs 2018, 36, 590–600. [Google Scholar] [CrossRef]
- Takimoto, R.; Wang, W.; Dicker, D.T.; Rastinejad, F.; Lyssikatos, J.; el-Deiry, W.S. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol. Ther. 2002, 1, 47–55. [Google Scholar] [CrossRef]
- Xu, J.; Timares, L.; Heilpern, C.; Weng, Z.; Li, C.; Xu, H.; Pressey, J.; Elmets, C.A.; Kopelovich, L.; Athar, M. Targeting Wild-Type and Mutant p53 with Small Molecule CP-31398 Blocks the Growth of Rhabdomyosarcoma by Inducing Reactive Oxygen Species–Dependent Apoptosis. Cancer Res. 2010, 70, 6566–6576. [Google Scholar] [CrossRef]
- Arihara, Y.; Takada, K.; Kamihara, Y.; Hayasaka, N.; Nakamura, H.; Murase, K.; Ikeda, H.; Iyama, S.; Sato, T.; Miyanishi, K.; et al. Small molecule CP-31398 induces reactive oxygen species-dependent apoptosis in human multiple myeloma. Oncotarget 2017, 8, 65889–65899. [Google Scholar] [CrossRef]
- Allegra, A.; Speciale, A.; Molonia, M.S.; Guglielmo, L.; Musolino, C.; Ferlazzo, G.; Costa, G.; Saija, A.; Cimino, F. Curcumin ameliorates the in vitro efficacy of carfilzomib in human multiple myeloma U266 cells targeting p53 and NF-κB pathways. Toxicol. Vitr. 2018, 47, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Achkar, I.; Siveen, K.S.; Kuttikrishnan, S.; Prabhu, K.S.; Khan, A.Q.; Ahmed, E.I.; Sahir, F.; Jerobin, J.; Raza, A.; et al. Sanguinarine Induces Apoptosis Pathway in Multiple Myeloma Cell Lines via Inhibition of the JaK2/STAT3 Signaling. Front. Oncol. 2019, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, J.; Bao, J.; Lu, J.; Wang, Y. The Anticancer Properties of Salvia Miltiorrhiza Bunge (Danshen): A Systematic Review. Med. Res. Rev. 2013, 34, 768–794. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xu, S.; Zhang, M.; Wang, W.W.; Zhang, Y.F.; Rehman, K.; Naranmandura, H.; Chen, Z. Anticancer activity in human multiple myeloma U266 cells: Synergy between cryptotanshinone and arsenic trioxide. Metallomics 2013, 5, 871–878. [Google Scholar] [CrossRef]
- Liu, J.-J.; Liu, W.-D.; Yang, H.-Z.; Zhang, Y.; Fang, Z.-G.; Liu, P.-Q.; Lin, D.-J.; Xiao, R.-Z.; Hu, Y.; Wang, C.-Z.; et al. Inactivation of PI3k/Akt signaling pathway and activation of caspase-3 are involved in tanshinone I-induced apoptosis in myeloid leukemia cells in vitro. Ann. Hematol. 2010, 89, 1089–1097. [Google Scholar] [CrossRef]
- Kim, C.; Song, H.-S.; Park, H.; Kim, B. Activation of ER Stress-Dependent miR-216b Has a Critical Role in Salvia miltiorrhiza Ethanol-Extract-Induced Apoptosis in U266 and U937 Cells. Int. J. Mol. Sci. 2018, 19, 1240. [Google Scholar] [CrossRef]
- Marin, E.H.; Paek, H.; Li, M.; Ban, Y.; Karaga, M.K.; Shashidharamurthy, R.; Wang, X. Caffeic acid phenethyl ester exerts apoptotic and oxidative stress on human multiple myeloma cells. Investig. New Drugs 2018, 37, 837–848. [Google Scholar] [CrossRef]
- Li, Q.; Yue, Y.; Chen, L.; Xu, C.; Wang, Y.; Du, L.; Xue, X.; Liu, Q.; Wang, Y.; Fan, F. Resveratrol Sensitizes Carfilzomib-Induced Apoptosis via Promoting Oxidative Stress in Multiple Myeloma Cells. Front. Pharmacol. 2018, 9, 334. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.-G.; Yang, W.M.; Arfuso, F.; Um, J.-Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141. [Google Scholar] [CrossRef]
- Losada, A.; Berlanga, J.J.; Guijarro, J.M.M.; Jiménez-Ruiz, A.; Gago, F.; Avilés, P.; de Haro, C.; Martínez-Leal, J.F. Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharmacol. 2020, 172, 113744. [Google Scholar] [CrossRef]
- Cao, M.-N.; Zhou, Y.-B.; Gao, A.-H.; Cao, J.-Y.; Gao, L.-X.; Sheng, L.; Xu, L.; Su, M.-B.; Cao, X.-C.; Han, M.-M.; et al. Curcusone D, a novel ubiquitin–proteasome pathway inhibitor via ROS-induced DUB inhibition, is synergistic with bortezomib against multiple myeloma cell growth. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2004–2013. [Google Scholar] [CrossRef]
- Choi, T.; Choi, I.Y.; Han, K.; Jeong, S.-M.; Yoo, J.E.; Rhee, S.Y.; Park, Y.-G.; Shin, D.W. Lipid Level, Lipid Variability, and Risk of Multiple Myeloma: A Nationwide Population-Based Study of 3,527,776 Subjects. Cancers 2021, 13, 540. [Google Scholar] [CrossRef] [PubMed]
- de Weille, J.; Fabre, C.; Bakalara, N. Oxysterols in cancer cell proliferation and death. Biochem. Pharmacol. 2013, 86, 154–160. [Google Scholar] [CrossRef]
- Poirot, M.; Silvente-Poirot, S. Cholesterol-5,6-epoxides: Chemistry, biochemistry, metabolic fate and cancer. Biochimie 2013, 95, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Nury, T.; Zarrouk, A.; Yammine, A.; Mackrill, J.J.; Vejux, A.; Lizard, G. Oxiapoptophagy: A type of cell death induced by some oxysterols. J. Cereb. Blood Flow Metab. 2020, 178, 3115–3123. [Google Scholar] [CrossRef] [PubMed]
- Jaouadi, O.; Limam, I.; Abdelkarim, M.; Berred, E.; Chahbi, A.; Caillot, M.; Sola, B.; Ben Aissa-Fennira, F. 5,6-Epoxycholesterol Isomers Induce Oxiapoptophagy in Myeloma Cells. Cancers 2021, 13, 3747. [Google Scholar] [CrossRef] [PubMed]
- Bar-Tana, J.; Ben-Shoshan, S.; Blum, J.; Migron, Y.; Hertz, R.; Pill, J.; Rose-Khan, G.; Witte, E.C. Synthesis and hypolipidemic and antidiabetogenic activities of beta, beta, beta’, beta’-tetrasubstituted, long-chain dioic acids. J. Med. Chem. 1989, 32, 2072–2084. [Google Scholar] [CrossRef]
- Bernardi, P.; Penzo, D.; Wojtczak, L. Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam. Horm. 2002, 65, 97–126. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 2008, 45, 231–241. [Google Scholar] [CrossRef]
- Atiq, R.; Hertz, R.; Eldad, S.; Smeir, E.; Bar-Tana, J. Suppression of B-Raf(V600E) cancers by MAPK hyper-activation. Oncotarget 2016, 7, 18694–18704. [Google Scholar] [CrossRef]
- Samovski, D.; Kalderon, B.; Yehuda-Shnaidman, E.; Bar-Tana, J. Gating of the Mitochondrial Permeability Transition Pore by Long Chain Fatty Acyl Analogs in Vivo. J. Biol. Chem. 2010, 285, 6879–6890. [Google Scholar] [CrossRef]
- Aisen, Y.; Gatt, M.E.; Hertz, R.; Smeir, E.; Bar-Tana, J. Suppression of multiple myeloma by mitochondrial targeting. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Schibler, J.; Tomanek-Chalkley, A.M.; Reedy, J.L.; Zhan, F.; Spitz, U.R.; Schultz, M.K.; Goel, A. Mitochondrial-Targeted Decyl-Triphenylphosphonium Enhances 2-Deoxy-D-Glucose Mediated Oxidative Stress and Clonogenic Killing of Multiple Myeloma Cells. PLoS ONE 2016, 11, e0167323. [Google Scholar] [CrossRef]
- Jhaveri, K.D.; Wanchoo, R. Carfilzomib-induced nephrotoxcity. Kidney Int. 2015, 88, 199–200. [Google Scholar] [CrossRef]
- Abarikwu, S.O.; Adebayo, O.L.; Otuechere, C.A.; Iserhienrhien, B.O.; Badejo, T.A. Selenium and rutin alone or in combination do not have stronger protective effects than their separate effects against cadmium-induced renal damage. Pharm. Biol. 2015, 54, 1–9. [Google Scholar] [CrossRef]
- Al-Harbi, N.O.; Imam, F.; Al-Harbi, M.M.; Al-Shabanah, O.A.; Alotaibi, M.R.; Sobeai, H.M.A.; Afzal, M.; Kazmi, I.; Al Rikabi, A.C. Rutin inhibits carfilzomib-induced oxidative stress and inflammation via the NOS-mediated NF-κB signaling pathway. Inflammopharmacology 2019, 27, 817–827. [Google Scholar] [CrossRef]
- Imam, F.; Al-Harbi, N.O.; Al-Harbia, M.M.; Korashy, H.M.; Ansari, M.A.; Sayed-Ahmed, M.M.; Nagi, M.N.; Iqbal, M.; Anwer, K.; Kazmi, I.; et al. Rutin Attenuates Carfilzomib-Induced Cardiotoxicity Through Inhibition of NF-κB, Hypertrophic Gene Expression and Oxidative Stress. Cardiovasc. Toxicol. 2015, 17, 58–66. [Google Scholar] [CrossRef]
- Imam, F.; Al-Harbi, N.O.; Al-Harbi, M.M.; Ansari, M.A.; Almutairi, M.M.; Alshammari, M.; Almukhlafi, T.S.; Ansari, M.N.; Aljerian, K.; Ahmad, S.F. Apremilast reversed carfilzomib-induced cardiotoxicity through inhibition of oxidative stress, NF-κB and MAPK signaling in rats. Toxicol. Mech. Methods 2016, 26, 700–708. [Google Scholar] [CrossRef]
- Musolino, C.; Oteri, G.; Allegra, A.; Mania, M.; D’Ascola, A.; Avenoso, A.; Innao, V.; Allegra, A.G.; Campo, S. Altered microRNA expression profile in the peripheral lymphoid compartment of multiple myeloma patients with bisphosphonate-induced osteonecrosis of the jaw. Ann. Hematol. 2018, 97, 1259–1269. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Penna, G.; Granata, A.; Siniscalchi, E.N.; Oteri, G.; Loddo, S.; Teti, D.; Cicciù, D.; De Ponte, F.S.; et al. Bisphosphonates Induce Apoptosis of Circulating Endothelial Cells in Multiple Myeloma Patients and in Subjects with Bisphosphonate-Induced Osteonecrosis of the Jaws. Acta Haematol. 2010, 124, 79–85. [Google Scholar] [CrossRef]
- Koçer, G.; Nazıroğlu, M.; Çelik, Ö.; Önal, L.; Özçelik, D.; Koçer, M.; Sönmez, T.T. Basic Fibroblast Growth Factor Attenuates Bisphosphonate-Induced Oxidative Injury but Decreases Zinc and Copper Levels in Oral Epithelium of Rat. Biol. Trace Element Res. 2013, 153, 251–256. [Google Scholar] [CrossRef]
- Karabulut, A.B.; Gül, M.; Karabulut, E.; Kiran, T.; Ocak, S.; Otlu, O. Oxidant and Antioxidant Activity in Rabbit Livers Treated With Zoledronic Acid. Transplant. Proc. 2010, 42, 3820–3822. [Google Scholar] [CrossRef]
- Taniguchi, N.; Osaki, M.; Onuma, K.; Ishikawa, M.; Ryoke, K.; Kodani, I.; Okada, F. Bisphosphonate-induced reactive oxygen species inhibit proliferation and migration of oral fibroblasts: A pathogenesis of bisphosphonate-related osteonecrosis of the jaw. J. Periodontol. 2019, 91, 947–955. [Google Scholar] [CrossRef]
- Bagan, J.; Sáez, G.T.; Tormos, M.C.; Gavalda-Esteve, C.; Bagan, L.; Leopoldo-Rodado, M.; Calvo, J.; Camps, C. Oxidative stress in bisphosphonate-related osteonecrosis of the jaws. J. Oral Pathol. Med. 2014, 43, 371–377. [Google Scholar] [CrossRef]
- Driscoll, J.; Finley, D. A controlled breakdown: Antigen processing and the turnover of viral proteins. Cell 1992, 68, 823–825. [Google Scholar] [CrossRef]
- Brown, M.G.; Driscoll, J.; Monaco, J.J. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature 1991, 353, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Kloetzel, P.M. Immunoproteasomes at the interface of innate and adaptive immune responses: Two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Kararoudi, M.N.; Nagai, Y.; Elmas, E.; Pereira, M.D.S.F.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Parameters | Material | Status | Ref. |
|---|---|---|---|
| Vitamin C, vitamin E, glutathione peroxidase, superoxide dismutase catalase | Serum | Reduced | [11,12,13,14,15] |
| Advanced oxidation protein products, malondialdehyde, markers of fatty acid peroxidation | Serum | Augmented | [11,12,13,14,15] |
| Paraoxonase, arylesterase | Serum | Reduced | [16,17] |
| Nicotinamide adenine dinucleotide | Peripheral blood mononuclear cells | Reduced | [18] |
| Hydrogen peroxide | Peripheral blood mononuclear cells | Augmented | [18] |
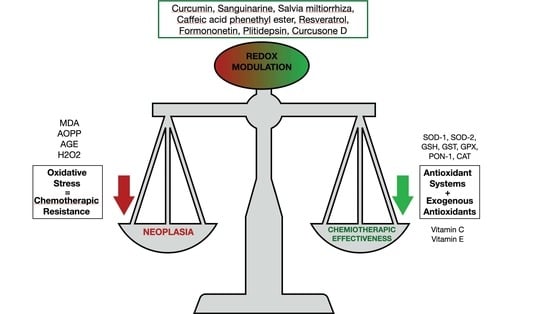
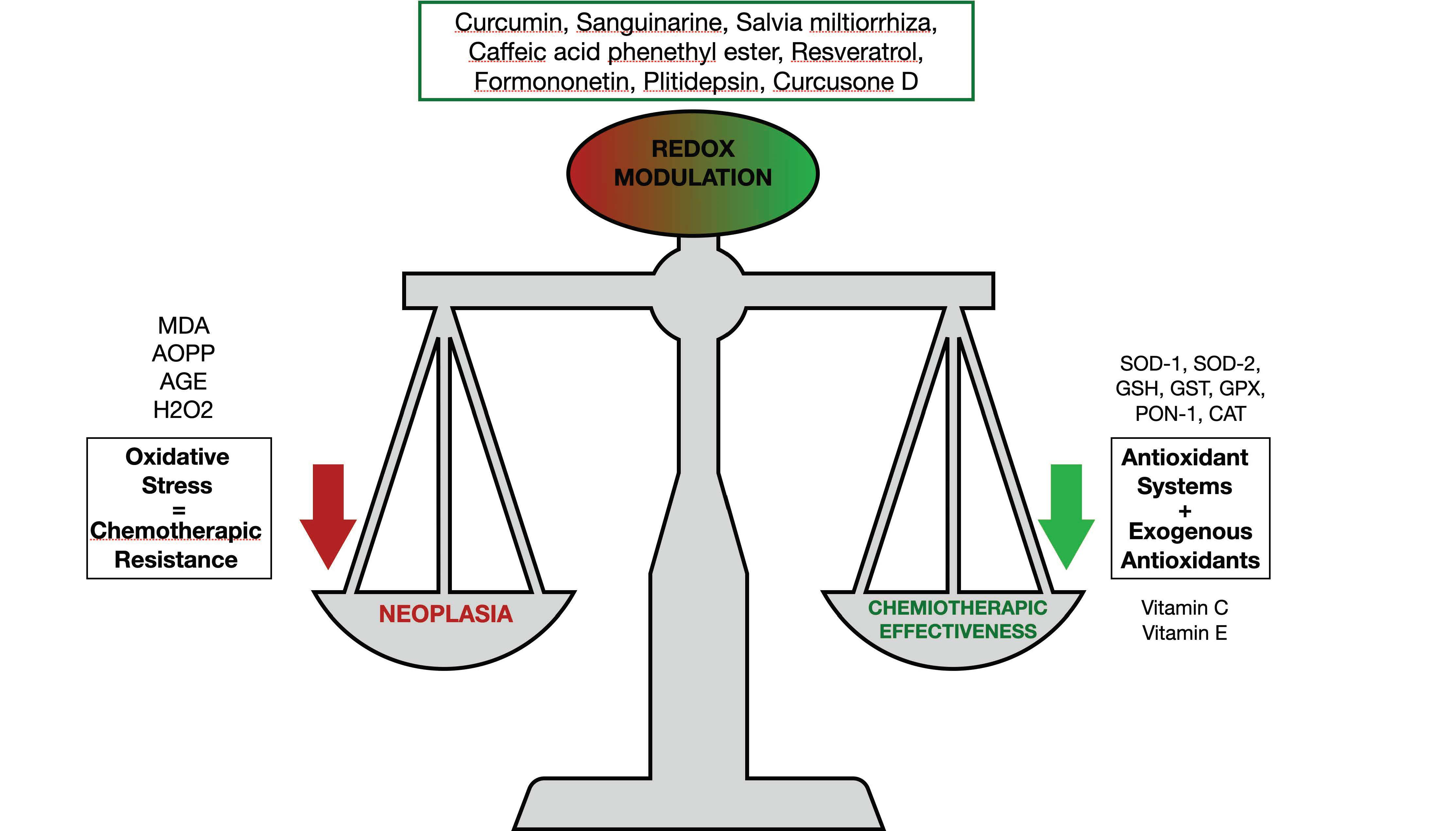
| Molecules | Effects on Oxidative Stress | Mechanisms of Action | Ref. |
|---|---|---|---|
| Curcumin and Carfilzomib | Augmented ROS concentrations | They stimulated the p53/p21 axis and G0/G1 cell cycle arrest, and curcumin augmented the CFZ proapoptotic effect | [109] |
| Sanguinarine and Bortezomib | Mitochondrial membrane potential loss | They increased apoptosis and caused inhibition of the STAT3 pathway | [110] |
| Salvia miltiorrhiza | Augmented ROS production and ER stress | Augmented apoptosis and increased miRNA-216b expression | [114] |
| Caffeic acid phenethyl ester | Stimulation of oxidative stress-response genes (heme oxigenase-1). Reduced intracellular antioxidants | Reduced MM cells proliferation | [115] |
| Resveratrol and Carfilzomib | Augmented ROS production. Delivery of second mitochondria-derived activator of caspase | Augmented apoptosis | [116] |
| Formononetin | Augmented generation of ROS for a GSH/GSSG disequilibrium | Decrease in kinases such as c-Src, JAK1, and JAK2 | [117] |
| Plitidepsin | Augmented oxidative and ER stress, JNK1, and eIF2α phosphorylation | Increased apoptosis | [118] |
| Curcusone D and Bortezomib | Augmented ROS generation | Ubiquitin–proteasome pathway inhibition | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Petrarca, C.; Di Gioacchino, M.; Casciaro, M.; Musolino, C.; Gangemi, S. Modulation of Cellular Redox Parameters for Improving Therapeutic Responses in Multiple Myeloma. Antioxidants 2022, 11, 455. https://doi.org/10.3390/antiox11030455
Allegra A, Petrarca C, Di Gioacchino M, Casciaro M, Musolino C, Gangemi S. Modulation of Cellular Redox Parameters for Improving Therapeutic Responses in Multiple Myeloma. Antioxidants. 2022; 11(3):455. https://doi.org/10.3390/antiox11030455
Chicago/Turabian StyleAllegra, Alessandro, Claudia Petrarca, Mario Di Gioacchino, Marco Casciaro, Caterina Musolino, and Sebastiano Gangemi. 2022. "Modulation of Cellular Redox Parameters for Improving Therapeutic Responses in Multiple Myeloma" Antioxidants 11, no. 3: 455. https://doi.org/10.3390/antiox11030455
APA StyleAllegra, A., Petrarca, C., Di Gioacchino, M., Casciaro, M., Musolino, C., & Gangemi, S. (2022). Modulation of Cellular Redox Parameters for Improving Therapeutic Responses in Multiple Myeloma. Antioxidants, 11(3), 455. https://doi.org/10.3390/antiox11030455