
Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells

 ,
,  , ,
, ,  , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Immunofluorescence
2.4. Cloning of pLVX2 hDAO for Lentiviral Overexpression
2.5. Lentiviral Transduction
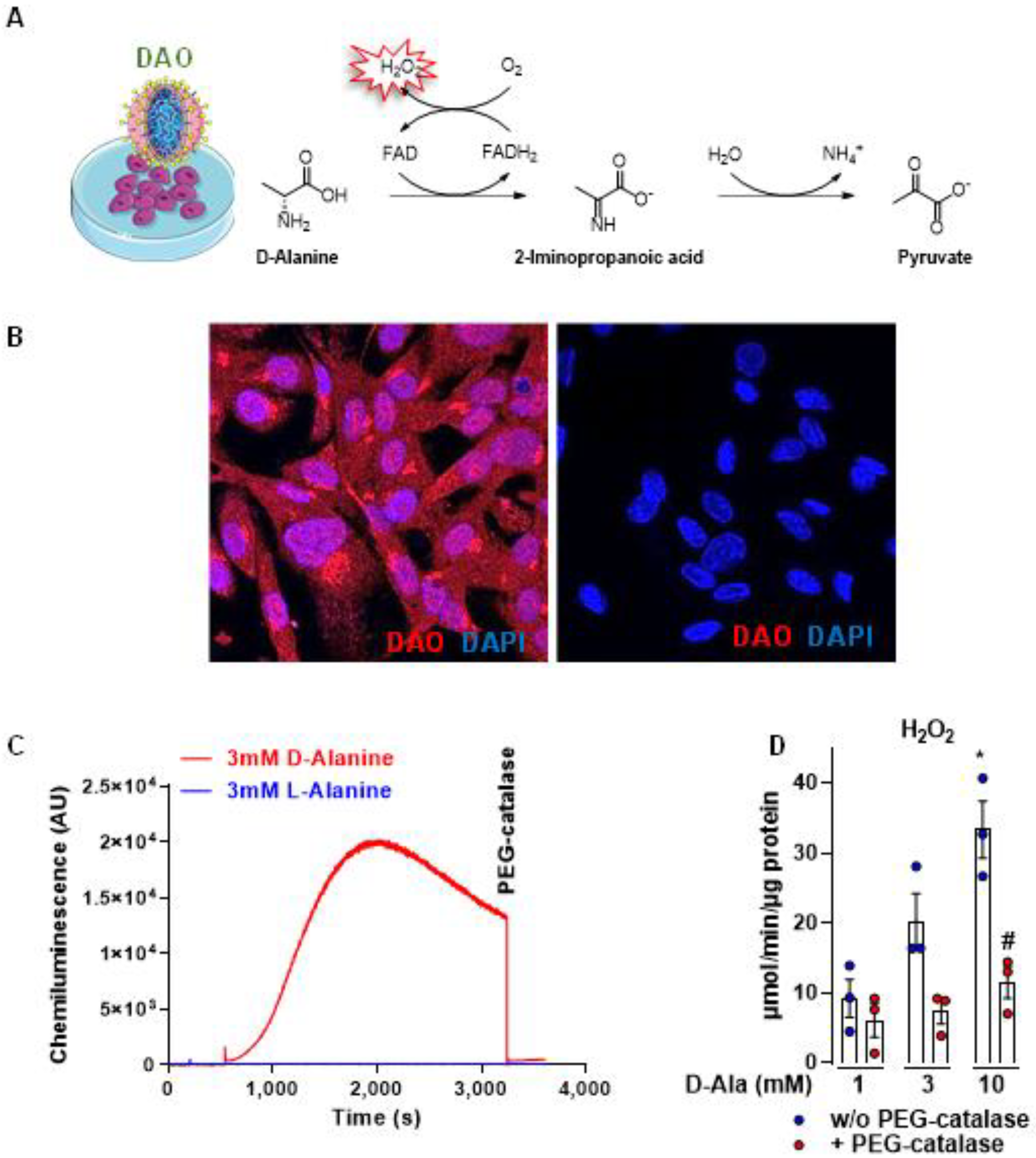
2.6. Determination of ROS Production
2.7. Redox Western Blots
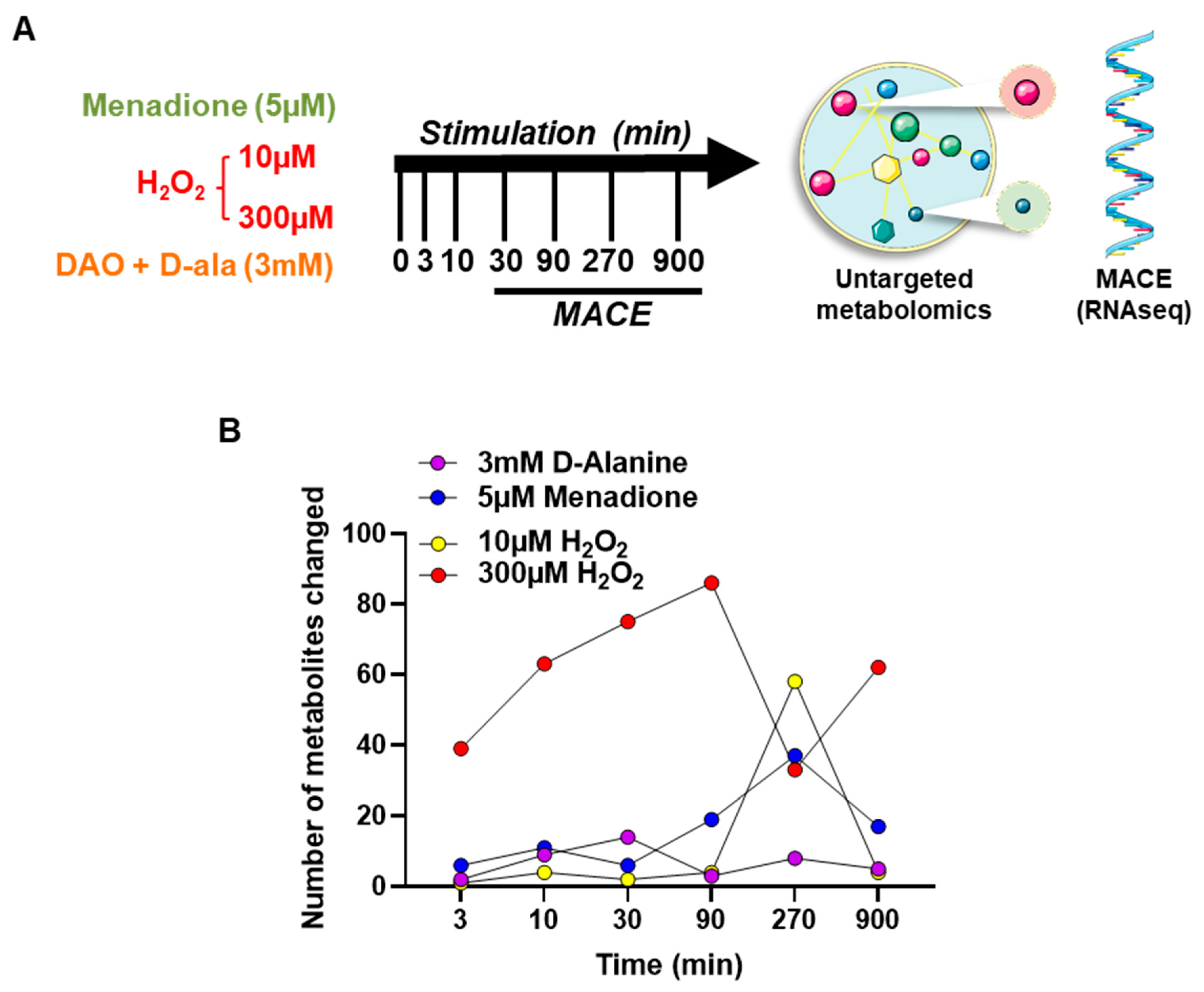
2.8. Metabolomics
2.9. RNA Isolation and RNAseq by Massive Analysis of cDNA Ends (MACE)
2.10. Gene Correlation and Transcription Factor Analysis
2.11. Statistics
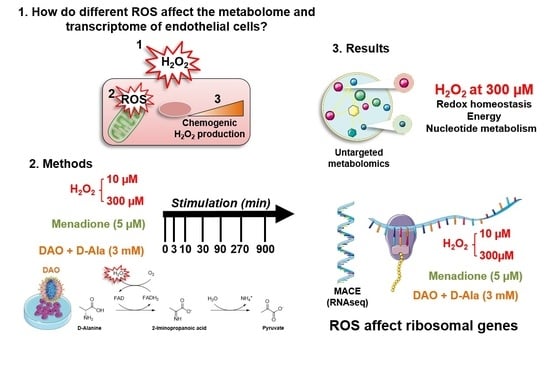
3. Results
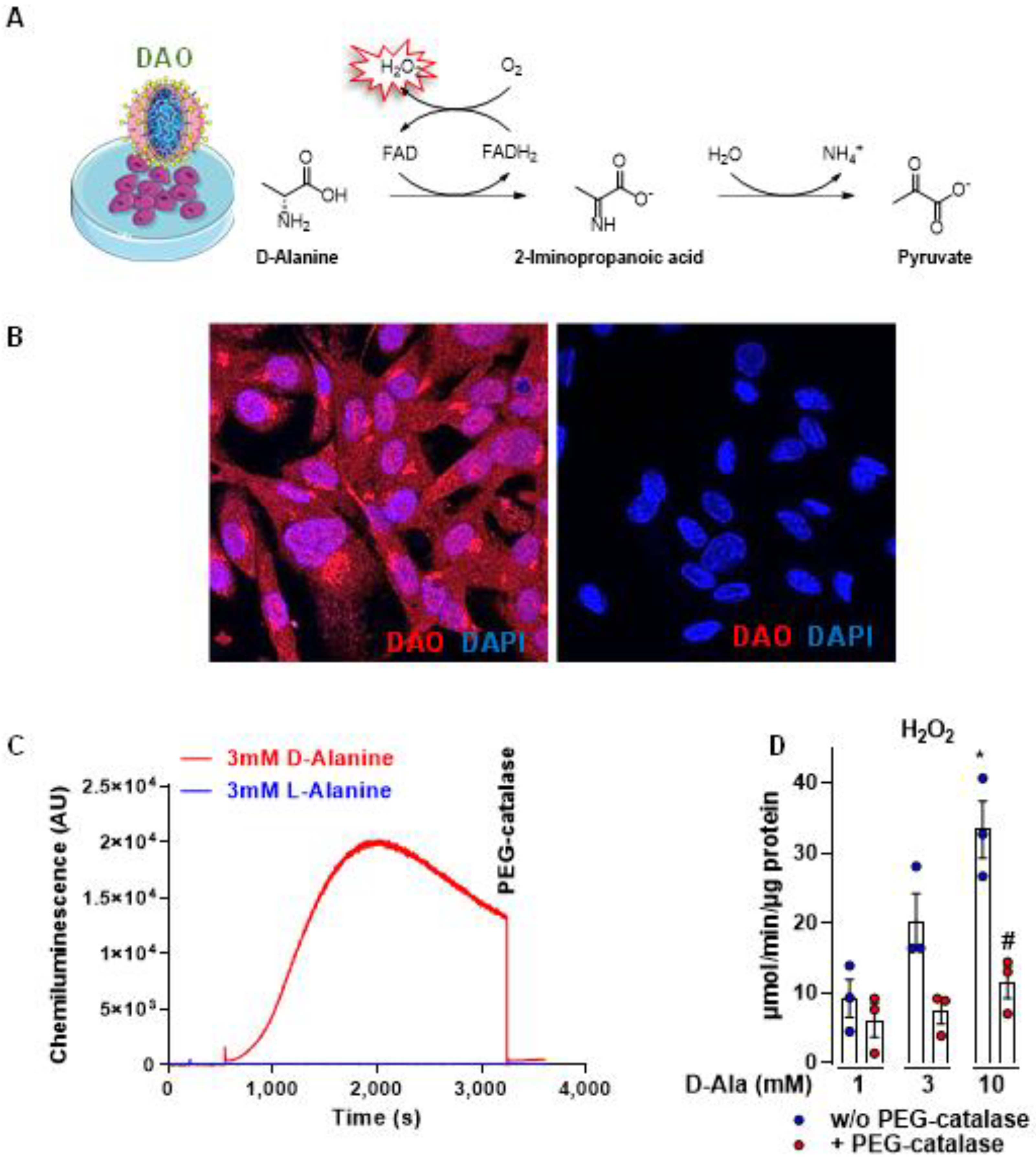
3.1. DAO Is an Efficient Chemogenetic Tool to Produce H2O2 Intracellularly
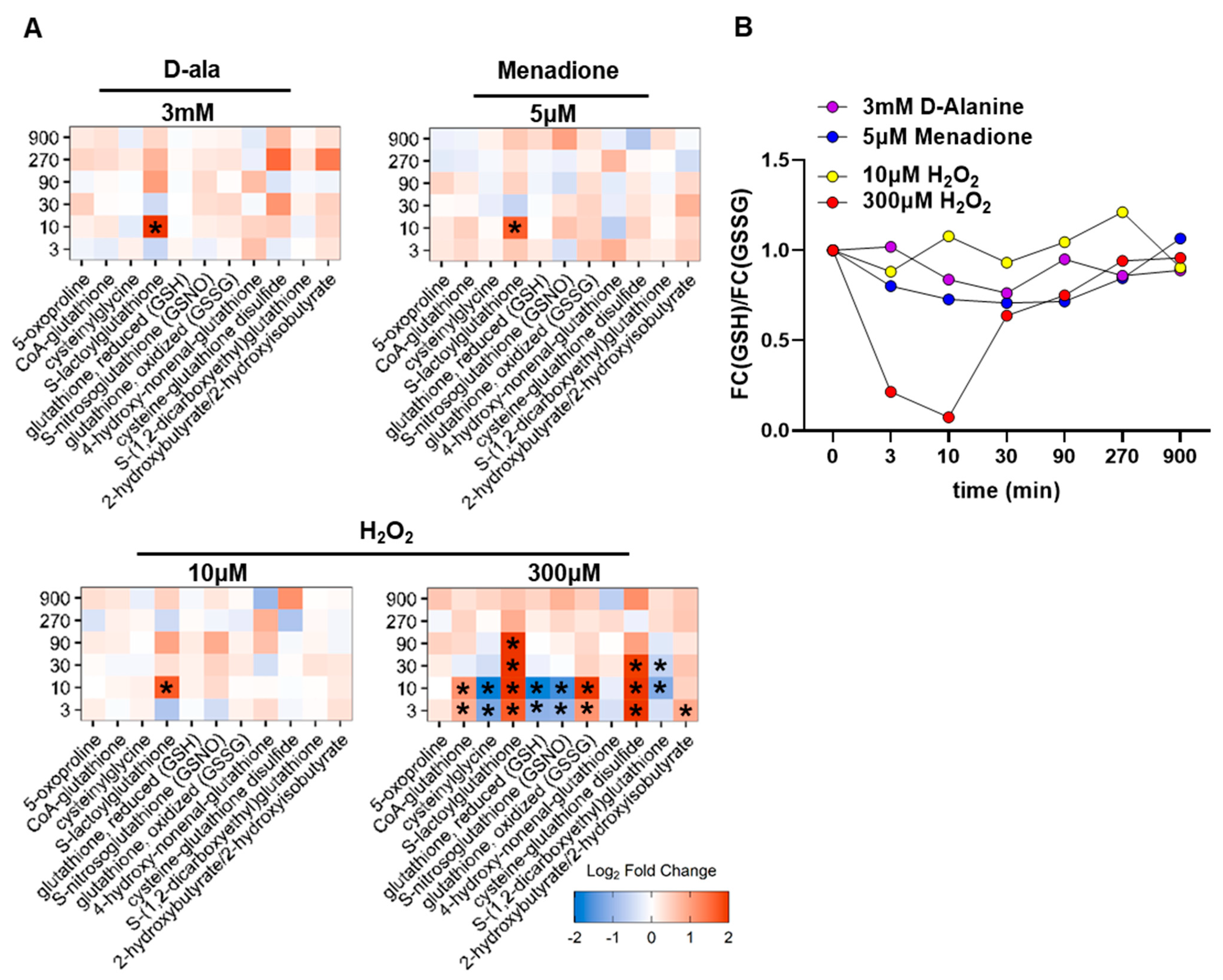
3.2. Different Types of ROS Elicit a Differential Metabolic Response in HUVEC
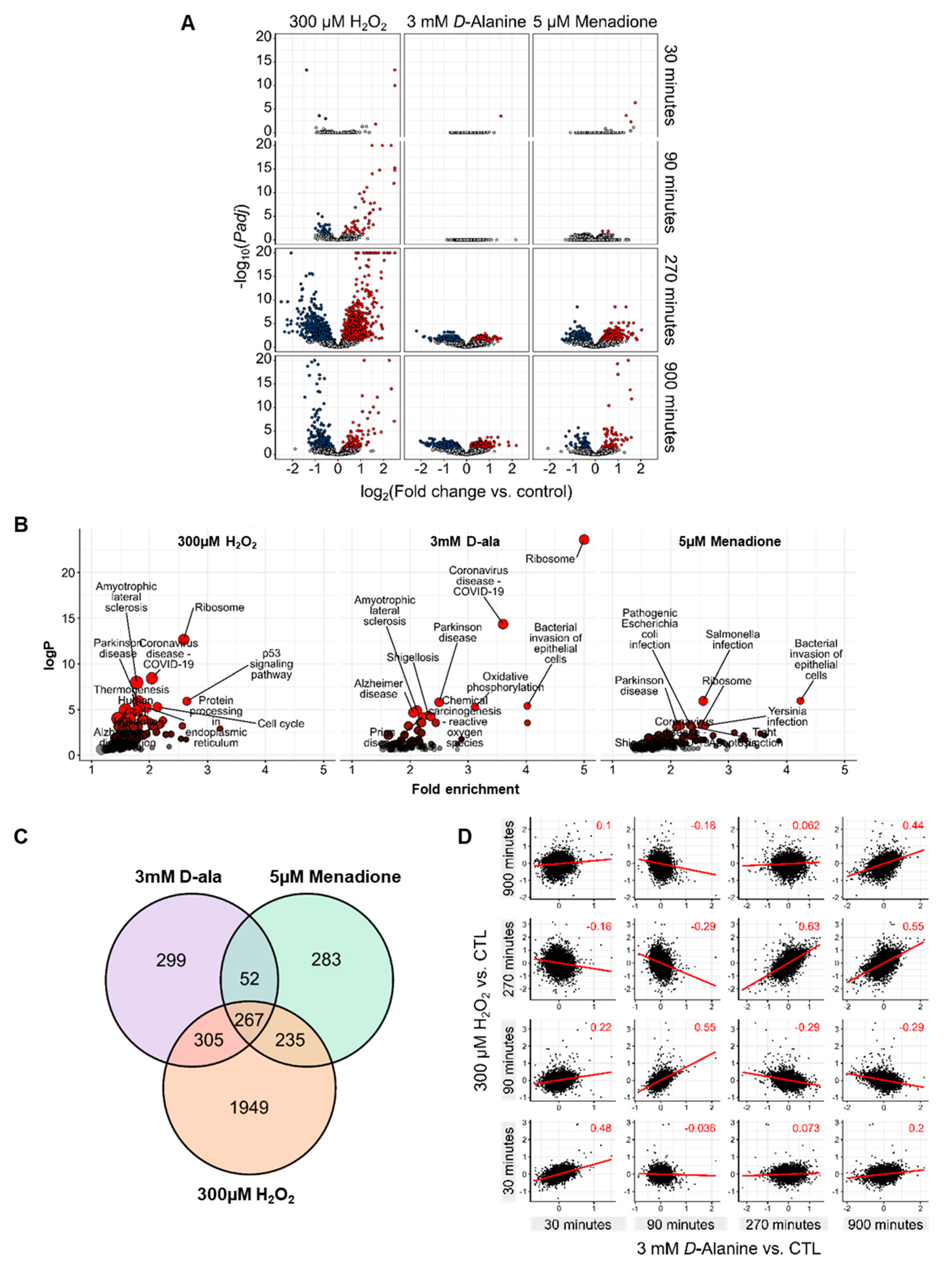
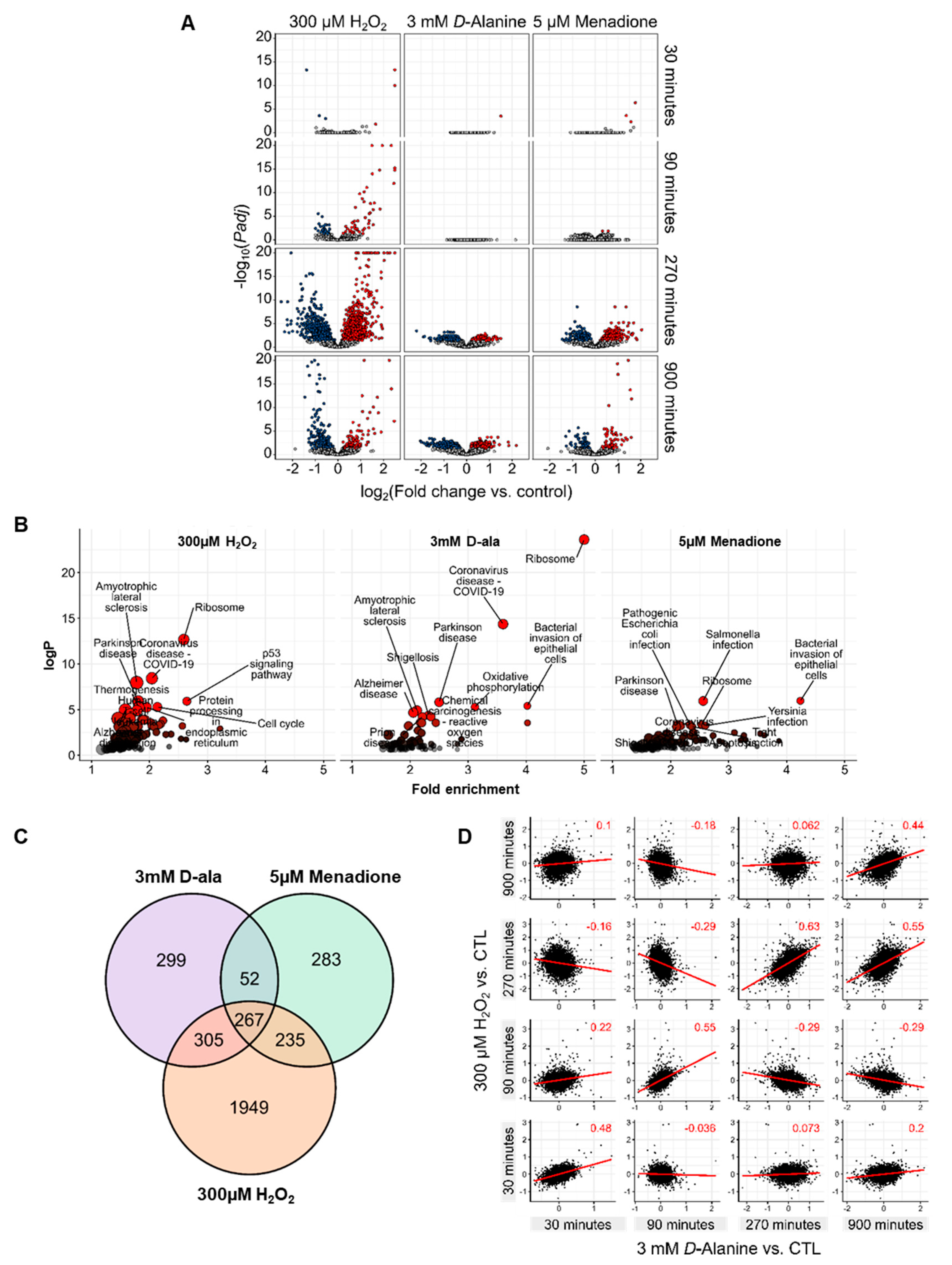
3.3. ROS Lead to Different Gene Expression Responses by HUVEC
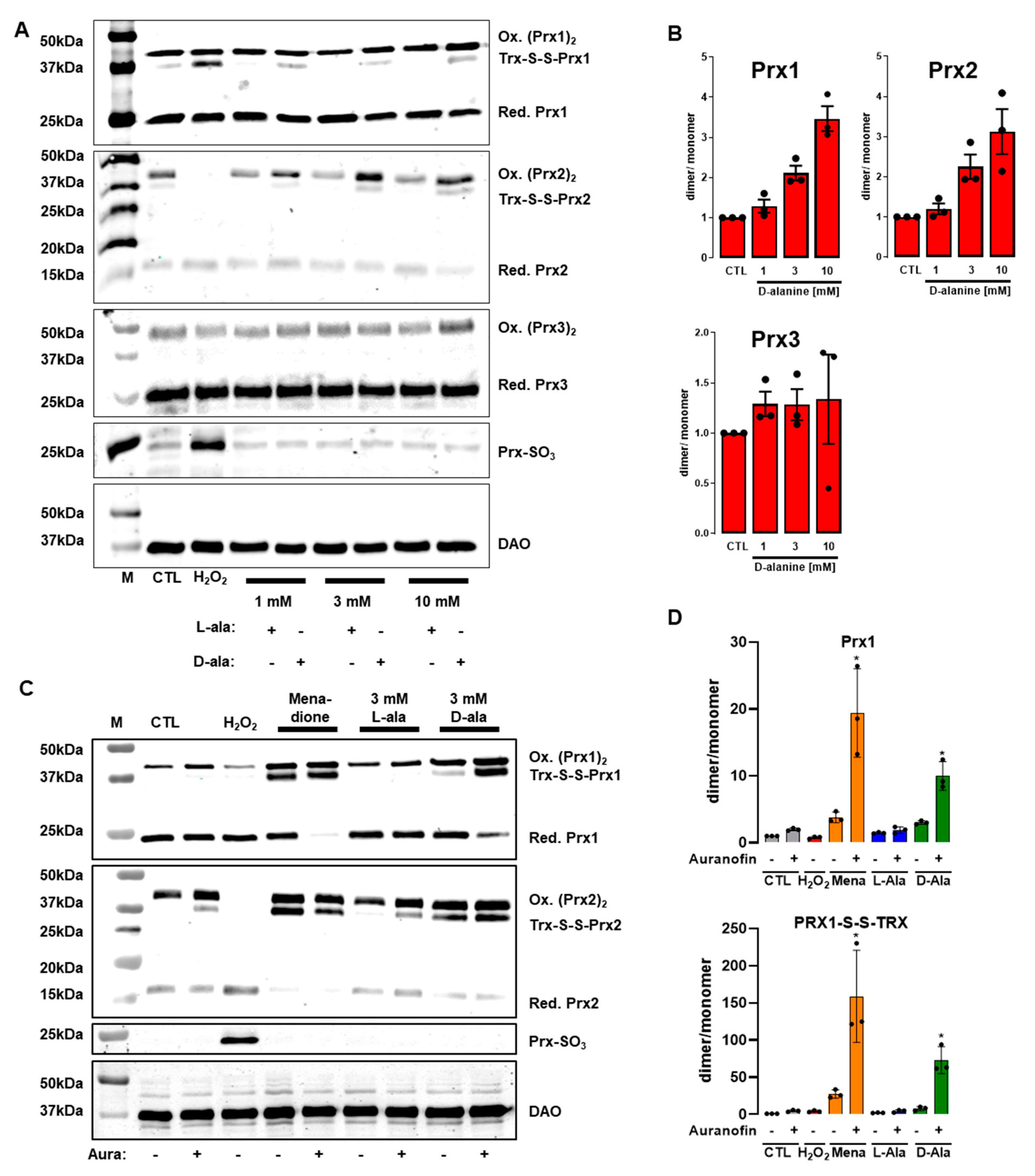
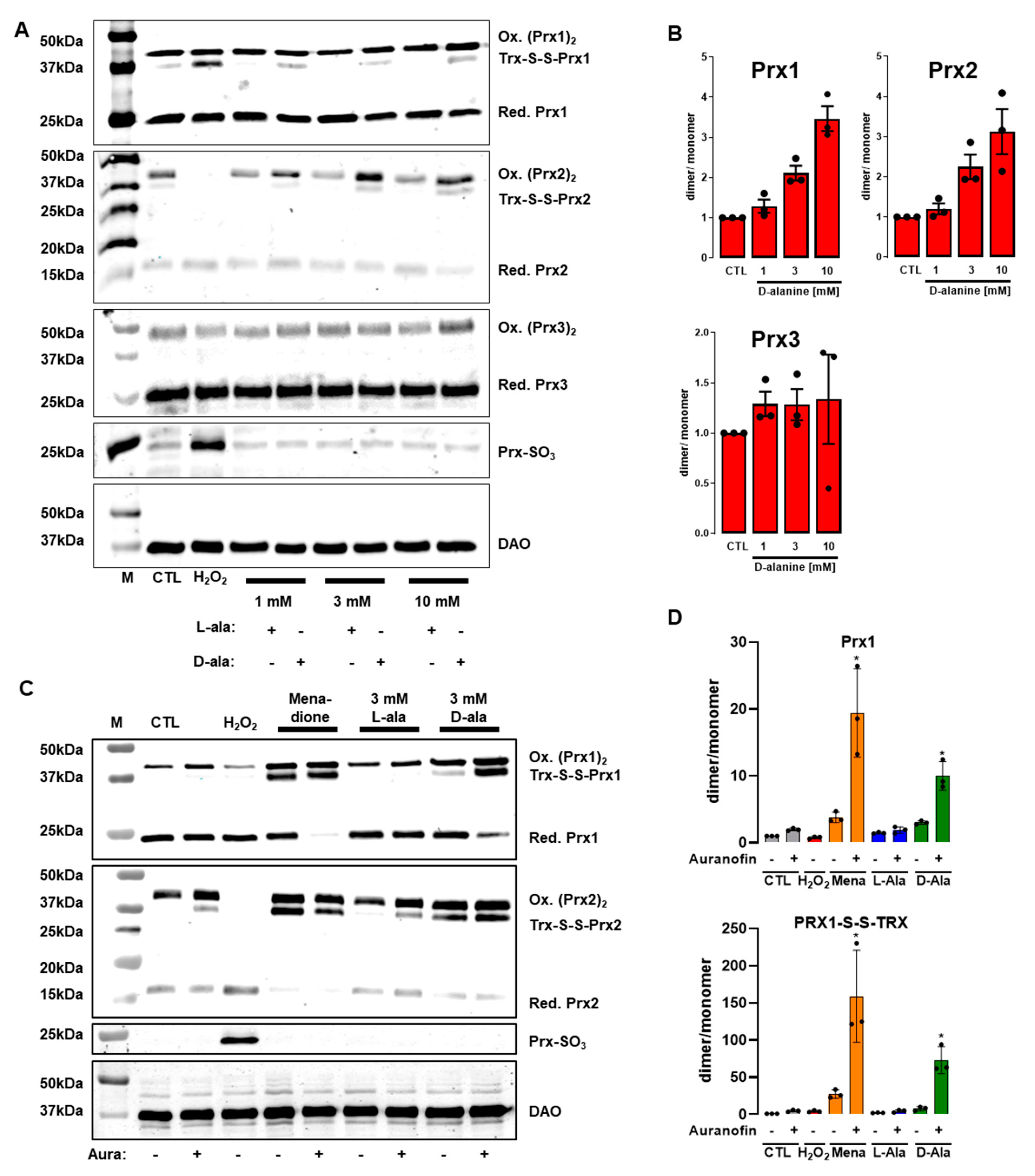
3.4. Only 300 µM H2O2 Overoxidizes Peroxiredoxins in HUVEC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K. Reactive Oxygen Species in the Vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Brown, D.; Griendling, K.K. Regulation of Signal Transduction by Reactive Oxygen Species in the Cardiovascular System. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.; et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF 2α-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Löwe, O.; Rezende, F.; Heidler, J.; Wittig, I.; Helfinger, V.; Brandes, R.P.; Schröder, K. BIAM switch assay coupled to mass spectrometry identifies novel redox targets of NADPH oxidase. Redox Biol. 2019, 21, 101125. [Google Scholar] [CrossRef]
- Pryor, W.A.; Squadrito, G.L. The chemistry of peroxynitrite: A product from the reaction of nitric oxide with superoxide. Am. J. Physiol. 1995, 268, L699–L722. [Google Scholar] [CrossRef]
- Gardner, P.; Fridovich, I. Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 1991, 266, 19328–19333. [Google Scholar] [CrossRef]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2002; pp. 9–23. [Google Scholar]
- Thomas, S.R.; Chen, K.; Keaney, J. Hydrogen Peroxide Activates Endothelial Nitric-oxide Synthase through Coordinated Phosphorylation and Dephosphorylation via a Phosphoinositide 3-Kinase-dependent Signaling Pathway. J. Biol. Chem. 2002, 277, 6017–6024. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; West, N.E.; Pillai, R.; Taggart, D.P.; Channon, K.M. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 2002, 39, 1088–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravi, S.S.S.; Eroglu, E.; Waldeck-Weiermair, M.; Sorrentino, A.; Steinhorn, B.; Belousov, V.; Michel, T. Differential endothelial signaling responses elicited by chemogenetic H2O2 synthesis. Redox Biol. 2020, 36, 101605. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Rezende, F.; Schröder, K. Redox Regulation Beyond ROS: Why ROS Should Not Be Measured as Often. Circ. Res. 2018, 123, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, L.Y.; Oliveira, P.V.; Laurindo, F.R. Peri/Epicellular Thiol Oxidoreductases as Mediators of Extracellular Redox Signaling. Antioxid. Redox Signal. 2020, 33, 280–307. [Google Scholar] [CrossRef]
- Matlashov, M.E.; Belousov, V.V.; Enikolopov, G. How Much H2O2 Is Produced by Recombinant D-Amino Acid Oxidase in Mammalian Cells? Antioxid. Redox Signal. 2014, 20, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Pollegioni, L.; Sacchi, S.; Murtas, G. Human D-Amino Acid Oxidase: Structure, Function, and Regulation. Front. Mol. Biosci. 2018, 5, 107. [Google Scholar] [CrossRef] [Green Version]
- Steinhorn, B.; Sorrentino, A.; Badole, S.; Bogdanova, Y.; Belousov, V.; Michel, T. Chemogenetic generation of hydrogen peroxide in the heart induces severe cardiac dysfunction. Nat. Commun. 2018, 9, 4044. [Google Scholar] [CrossRef]
- Valon, L.; Marín-Llauradó, A.; Wyatt, T.; Charras, G.; Trepat, X. Optogenetic control of cellular forces and mechanotransduction. Nat. Commun. 2017, 8, 14396. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Rinsma, M.; Janssen, J.M.; Liu, J.; Maggio, I.; Gonçalves, M.A. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016, 44, 6482–6492. [Google Scholar] [CrossRef] [Green Version]
- Rezende, F.; Löwe, O.; Helfinger, V.; Prior, K.-K.; Walter, M.; Zukunft, S.; Fleming, I.; Weissmann, N.; Brandes, R.P.; Schröder, K. Unchanged NADPH Oxidase Activity in Nox1-Nox2-Nox4 Triple Knockout Mice: What Do NADPH-Stimulated Chemiluminescence Assays Really Detect? Antioxid. Redox Signal. 2016, 24, 392–399. [Google Scholar] [CrossRef]
- Prior, K.-K.; Leisegang, M.S.; Josipovic, I.; Löwe, O.; Shah, A.M.; Weissmann, N.; Schröder, K.; Brandes, R.P. CRISPR/Cas9-mediated knockout of p22phox leads to loss of Nox1 and Nox4, but not Nox5 activity. Redox Biol. 2016, 9, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Al-Khelaifi, F.; Diboun, I.; Donati, F.; Botrè, F.; Alsayrafi, M.; Georgakopoulos, C.; Suhre, K.; Yousri, N.A.; Elrayess, M.A. A pilot study comparing the metabolic profiles of elite-level athletes from different sporting disciplines. Sports Med. Open 2018, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, Nontargeted Ultrahigh Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry Platform for the Identification and Relative Quantification of the Small-Molecule Complement of Biological Systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Bridgewater, B.R. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High-Throughput Profiling Metabolomics. Metabolomics 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dehaven, C.D.; Evans, A.M.; Dai, H.; Lawton, K.A. Software Techniques for Enabling High-Throughput Analysis of Metabolomic Datasets. Metabolomics; Roessner, U., Ed.; Intech, 2012. Available online: https://www.intechopen.com/chapters/28007 (accessed on 15 February 2022).
- Howe, K.L.; Contreras-Moreira, B.; De Silva, N.; Maslen, G.; Akanni, W.; Allen, J.; Alvarez-Jarreta, J.; Barba, M.; Bolser, D.M.; Cambell, L.; et al. Ensembl Genomes 2020—enabling non-vertebrate genomic research. Nucleic Acids Res. 2020, 48, D689–D695. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.D.; Pastene, D.O.; Breedijk, A.; Rodriguez, A.; Hofmann, B.B.; Sticht, C.; Von Ochsenstein, E.; Allgayer, H.; Born, J.V.D.; Bakker, S.; et al. Methylglyoxal down-regulates the expression of cell cycle associated genes and activates the p53 pathway in human umbilical vein endothelial cells. Sci. Rep. 2019, 9, 1152. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Nikotina, A.D.; Semenyuk, P.I.; Evstafyeva, D.B.; Mikhaylova, E.R.; Muronetz, V.I.; Shevtsov, M.A.; Tolkacheva, A.V.; Dobrodumov, A.V.; Shavarda, A.L.; et al. Small molecules preventing GAPDH aggregation are therapeutically applicable in cell and rat models of oxidative stress. Free Radic. Biol. Med. 2016, 92, 29–38. [Google Scholar] [CrossRef]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor-Flores, D.; Talwar, D.; Pedre, B.; Dick, T.P. Real-time monitoring of peroxiredoxin oligomerization dynamics in living cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16313–16323. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Compartmentalization of Redox Signaling Through NADPH Oxidase—Derived ROS. Antioxid. Redox Signal. 2009, 11, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; A Struys, E.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Ralser, M.; Wamelink, M.M.C.; Latkolik, S.; Jansen, E.E.W.; Lehrach, H.; Jakobs, C. Metabolic reconfiguration precedes transcriptional regulation in the antioxidant response. Nat. Biotechnol. 2009, 27, 604–605. [Google Scholar] [CrossRef]
- Colussi, C.; Albertini, M.C.; Coppola, S.; Rovidati, S.; Galli, F.; Ghibelli, L. H2O2-induced block of glycolysis as an active ADP-ribosylation reaction protecting cells from apoptosis. FASEB J. 2000, 14, 2266–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Santoro, M.M. ROS signaling and redox biology in endothelial cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- De Bari, L.; Scirè, A.; Minnelli, C.; Cianfruglia, L.; Kalapos, M.P.; Armeni, T. Interplay among Oxidative Stress, Methylglyoxal Pathway and S-Glutathionylation. Antioxidants 2020, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Beyfuss, K.; Hood, D.A. A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Rep. 2018, 23, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Shcherbik, N.; Pestov, D.G. The Impact of Oxidative Stress on Ribosomes: From Injury to Regulation. Cells 2019, 8, 1379. [Google Scholar] [CrossRef] [Green Version]
- Gerashchenko, M.V.; Lobanov, A.V.; Gladyshev, V.N. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2012, 109, 17394–17399. [Google Scholar] [CrossRef] [Green Version]
- Walluscheck, D.; Poehlmann, A.; Hartig, R.; Lendeckel, U.; Schönfeld, P.; Hotz-Wagenblatt, A.; Reissig, K.; Bajbouj, K.; Roessner, A.; Schneider-Stock, R. ATF2 knockdown reinforces oxidative stress-induced apoptosis in TE7 cancer cells. J. Cell. Mol. Med. 2013, 17, 976–988. [Google Scholar] [CrossRef]
- Muthalagu, N.; Murphy, D.J. Is oxidative stress MYC’s Achilles heel? Cell Death Differ. 2018, 25, 1189–1190. [Google Scholar] [CrossRef]
- Kraemer, S.M.; Goldstrohm, D.A.; Berger, A.; Hankey, S.; Rovinsky, S.A.; Moye-Rowley, W.S.; Stargell, L.A. TFIIA Plays a Role in the Response to Oxidative Stress. Eukaryot. Cell 2006, 5, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, L.M.; Ferrer-Sueta, G.; Denicola, A. Peroxiredoxins as Preferential Targets in H2O2-Induced Signaling. In Hydrogen Peroxide and Cell Signaling; Cadenas, E., Ed.; Elsevier Acad. Press: Amsterdam, The Netherlands, 2013; pp. 41–63. [Google Scholar]
- Flohé, L. The impact of thiol peroxidases on redox regulation. Free Radic. Res. 2016, 50, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Barata, A.G.; Dick, T.P. A role for peroxiredoxins in H2O2- and MEKK-dependent activation of the p38 signaling pathway. Redox Biol. 2020, 28, 101340. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, R.M.; Hughes, S.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.D.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Stöcker, S.; Maurer, M.; Ruppert, T.; Dick, T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018, 14, 148–155. [Google Scholar] [CrossRef]
- Jin, Z.-G.; Melaragno, M.G.; Liao, D.-F.; Yan, C.; Haendeler, J.; Suh, Y.-A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A Is a Secreted Growth Factor Induced by Oxidative Stress. Circ. Res. 2000, 87, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Blanc, A.; Pandey, N.R.; Srivastava, A.K. Synchronous activation of ERK 1/2, p38mapk and PKB/Akt signaling by H2O2 in vascular smooth muscle cells: Potential involvement in vascular disease (review). Int. J. Mol. Med. 2003, 11, 229–234. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, N.; Warwick, T.; Noack, K.; Malacarne, P.F.; Cooper, A.J.L.; Weissmann, N.; Schröder, K.; Brandes, R.P.; Rezende, F. Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells. Antioxidants 2022, 11, 434. https://doi.org/10.3390/antiox11020434
Müller N, Warwick T, Noack K, Malacarne PF, Cooper AJL, Weissmann N, Schröder K, Brandes RP, Rezende F. Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells. Antioxidants. 2022; 11(2):434. https://doi.org/10.3390/antiox11020434
Chicago/Turabian StyleMüller, Niklas, Timothy Warwick, Kurt Noack, Pedro Felipe Malacarne, Arthur J. L. Cooper, Norbert Weissmann, Katrin Schröder, Ralf P. Brandes, and Flávia Rezende. 2022. "Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells" Antioxidants 11, no. 2: 434. https://doi.org/10.3390/antiox11020434
APA StyleMüller, N., Warwick, T., Noack, K., Malacarne, P. F., Cooper, A. J. L., Weissmann, N., Schröder, K., Brandes, R. P., & Rezende, F. (2022). Reactive Oxygen Species Differentially Modulate the Metabolic and Transcriptomic Response of Endothelial Cells. Antioxidants, 11(2), 434. https://doi.org/10.3390/antiox11020434