
The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Structural Features and Properties of NRF2
2.1. NRF2 Overexpression in Heart Tissue
2.2. Inhibition of NRF2 Expression in Heart Tissue
3. Role of NRF2 in Obesity-Induced Cardiac Alterations and Adipogenesis
4. NRF2 and Cardiovascular Risk Factors
4.1. NRF2 and Hyperglycemia
4.2. NRF2 and Hyperlipidemia
4.3. Role of NRF2 in Endothelial Dysfunction
4.4. NRF2 and Atherosclerosis
4.5. NRF2 in Hypertension
5. Role of Oxidative Stress and Inflammatory Response in CVD
6. Therapeutic Strategies Implicated in the NRF2 Activation
7. Conclusions
Funding
Conflicts of Interest
References
- Gutiérrez-Cuevas, J.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Monroy-Ramírez, H.C.; Galicia-Moreno, M.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. Molecular Mechanisms of Obesity-Linked Cardiac Dysfunction: An Up-Date on Current Knowledge. Cells 2021, 10, 629. [Google Scholar] [CrossRef]
- Gutiérrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Pathophysiological Molecular Mechanisms of Obesity: A Link between MAFLD and NASH with Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 11629. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Li, H.; Xia, N. The Interplay between Adipose Tissue and Vasculature: Role of Oxidative Stress in Obesity. Front Cardiovasc. Med. 2021, 8, 650214. [Google Scholar] [CrossRef]
- Dua, S.; Bhuker, M.; Sharma, P.; Dhall, M.; Kapoor, S. Body mass index relates to blood pressure among adults. N. Am. J. Med. Sci. 2014, 6, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Piñeiro, J.A.; Gonzalez-Rovira, A.; Sánchez-Gomar, I.; Moreno, J.A.; Durán-Ruiz, M.C. Nrf2 and Heme Oxygenase-1 Involvement in Atherosclerosis Related Oxidative Stress. Antioxidants 2021, 10, 1463. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Tian, W.; De La Vega, M.R.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P. Keap1/Nrf2 Signaling Pathway. Antioxidants 2021, 10, 828. [Google Scholar] [CrossRef]
- Matzinger, M.; Fischhuber, K.; Heiss, E.H. Activation of Nrf2 signaling by natural products-can it alleviate diabetes? Biotechnol. Adv. 2018, 36, 1738–1767. [Google Scholar] [CrossRef]
- Barancik, M.; Gresova, L.; Bartekova, M.; Dovinova, I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol. Res. 2016, 65 (Suppl. S1), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Erkens, R.; Kramer, C.M.; Lückstädt, W.; Panknin, C.; Krause, L.; Weidenbach, M.; Dirzka, J.; Krenz, T.; Mergia, E.; Suvorava, T.; et al. Left ventricular diastolic dysfunction in Nrf2 knock out mice is associated with cardiac hypertrophy, decreased expression of SERCA2a, and preserved endothelial function. Free Radic. Biol. Med. 2015, 89, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef]
- Mikhailov, A.T.; Torrado, M. Myocardial transcription factors in diastolic dysfunction: Clues for model systems and disease. Heart Fail. Rev. 2016, 21, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88 Pt B, 221–232. [Google Scholar] [CrossRef]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 in Cardiovascular Function and Disease. Oxid. Med. Cell Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Vashi, R.; Patel, B.M. NRF2 in Cardiovascular Diseases: A Ray of Hope. J. Cardiovasc. Transl. Res. 2021, 14, 573–586. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Zang, H.; Mathew, R.O.; Cui, T. The Dark Side of Nrf2 in the Heart. Front. Physiol. 2020, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic. Biol. Med. 2015, 88 Pt B, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.A.; Lee, S.; Kwak, M.K. Nfe2l2/NRF2 Activity Is Linked to Mitochondria and AMP-Activated Protein Kinase Signaling in Cancers Through miR-181c/Mitochondria-Encoded Cytochrome c Oxidase Regulation. Antioxid. Redox Signal. 2017, 27, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Dutta, N.; Banerjee, P.; Gajbhiye, R.L.; Sareng, H.R.; Kapse, P.; Pal, S.; Burdelya, L.; Mandal, N.C.; Ravichandiran, V.; et al. Induction of monoamine oxidase A-mediated oxidative stress and impairment of NRF2-antioxidant defence response by polyphenol-rich fraction of Bergenia ligulata sensitizes prostate cancer cells in vitro and in vivo. Free Radic. Biol. Med. 2021, 172, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Yang, X.; Omiecinski, C.J. Intronic DNA elements regulate Nrf2 chemical responsiveness of the human microsomal epoxide hydrolase gene (EPHX1) through a far upstream alternative promoter. Biochim. Biophys. Acta 2014, 1839, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yueh, M.F.; Tukey, R.H. Nrf2-Keap1 signaling pathway regulates human UGT1A1 expression in vitro and in transgenic UGT1 mice. J. Biol. Chem. 2007, 282, 8749–8758. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Aboonabi, A.; Singh, I. Chemopreventive role of anthocyanins in atherosclerosis via activation of Nrf2-ARE as an indicator and modulator of redox. Biomed. Pharmacother. 2015, 72, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Paek, J.; Lo, J.Y.; Narasimhan, S.D.; Nguyen, T.N.; Glover-Cutter, K.; Robida-Stubbs, S.; Suzuki, T.; Yamamoto, M.; Blackwell, T.K.; Curran, S.P. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012, 16, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Zipper, L.M.; Mulcahy, R.T. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem. Biophys. Res. Commun. 2000, 278, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Narasimhan, M.; Tamowski, S.; Darley-Usmar, V.; Rajasekaran, N.S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 2017, 12, 937–945. [Google Scholar] [CrossRef]
- Quiles, J.M.; Narasimhan, M.; Mosbruger, T.; Shanmugam, G.; Crossman, D.; Rajasekaran, N.S. Identification of transcriptome signature for myocardial reductive stress. Redox Biol. 2017, 13, 568–580. [Google Scholar] [CrossRef]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.R.; Narasimhan, M.; Shanmugam, G.; Hong, J.; Devarajan, A.; Palaniappan, S.; Zhang, J.; Halade, G.V.; Darley-Usmar, V.M.; Hoidal, J.R.; et al. Abrogation of Nrf2 impairs antioxidant signaling and promotes atrial hypertrophy in response to high-intensity exercise stress. J. Transl. Med. 2016, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Strom, J.; Chen, Q.M. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 2017, 327, 52–58. [Google Scholar] [CrossRef]
- Taherkhani, S.; Suzuki, K.; Ruhee, R.T. A Brief Overview of Oxidative Stress in Adipose Tissue with a Therapeutic Approach to Taking Antioxidant Supplements. Antioxidants 2021, 10, 594. [Google Scholar] [CrossRef]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.X.; Ganesan, A.N.; Selvanayagam, J.B. Epicardial fat and atrial fibrillation: Current evidence, potential mechanisms, clinical implications, and future directions. Eur. Heart J. 2017, 38, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Clerico, A.; Zaninotto, M.; Passino, C.; Plebani, M. Obese phenotype and natriuretic peptides in patients with heart failure with preserved ejection fraction. Clin. Chem. Lab. Med. 2018, 56, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Peres Diaz, L.S.; Schuman, M.L.; Aisicovich, M.; Toblli, J.E.; Pirola, C.J.; Landa, M.S.; Garcia, S.I. Angiotensin II requires an intact cardiac thyrotropin-releasing hormone (TRH) system to induce cardiac hypertrophy in mouse. J. Mol. Cell Cardiol. 2018, 124, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Elrashidy, R.A. Dysregulation of nuclear factor erythroid 2-related factor 2 signaling and activation of fibrogenic pathways in hearts of high fat diet-fed rats. Mol. Biol. Rep. 2020, 47, 2821–2834. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Stourman, N.V.; Sekhar, K.R.; Rinehart, C.; Yan, X.; Meredith, M.J.; Arteaga, C.L.; Freeman, M.L. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005, 38, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef]
- Chen, D.; Li, Z.; Bao, P.; Chen, M.; Zhang, M.; Yan, F.; Xu, Y.; Ji, C.; Hu, X.; Sanchis, D.; et al. Nrf2 deficiency aggravates Angiotensin II-induced cardiac injury by increasing hypertrophy and enhancing IL-6/STAT3-dependent inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Okino, N.; Sato, T.; Sugatani, J.; Miwa, M. Induction of detoxifying enzymes in rodent white adipose tissue by aryl hydrocarbon receptor agonists and antioxidants. Drug Metab. Dispos. 2006, 34, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Leung, L.; Xue, P.; Wang, W.; Hou, Y.; Liu, D.; Yehuda-Shnaidman, E.; Lee, C.; Lau, J.; Kurtz, T.W.; et al. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010, 285, 9292–9300. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S.; et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein beta during adipogenesis. Free Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dasuri, K.; Fernandez-Kim, S.O.; Bruce-Keller, A.J.; Keller, J.N. Adipose-specific ablation of Nrf2 transiently delayed high-fat diet-induced obesity by altering glucose, lipid and energy metabolism of male mice. Am. J. Transl. Res. 2016, 8, 5309–5319. [Google Scholar]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 2013, 62, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Lai, M.; Zhu, X.; Ren, S.; Yin, Y.; Wang, Z.; Liu, Z.; Zuo, Z.; Hou, Y.; Pi, J.; et al. Rifampicin impairs adipogenesis by suppressing NRF2-ARE activity in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2021, 413, 115393. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, X.; Jia, H.; Wang, H.; Shui, G.; Qin, Y.; Shu, X.; Wang, Y.; Dong, J.; Liu, G.; et al. Nuclear Factor E2-Related Factor 2 Mediates Oxidative Stress-Induced Lipid Accumulation in Adipocytes by Increasing Adipogenesis and Decreasing Lipolysis. Antioxid. Redox Signal. 2020, 32, 173–192. [Google Scholar] [CrossRef]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Sampath, C.; Rashid, M.R.; Sang, S.; Ahmedna, M. Green tea epigallocatechin 3-gallate alleviates hyperglycemia and reduces advanced glycation end products via nrf2 pathway in mice with high fat diet-induced obesity. Biomed. Pharmacother. 2017, 87, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Kang, B.; Suh, H.J.; Choi, H.S. Parthenolide, a feverfew-derived phytochemical, ameliorates obesity and obesity-induced inflammatory responses via the Nrf2/Keap1 pathway. Pharmacol. Res. 2019, 145, 104259. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.K.; Reddy, V.T.; Konopleva, M.; Andreeff, M.; Chan, L. The triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic-acid methyl ester has potent anti-diabetic effects in diet-induced diabetic mice and Lepr(db/db) mice. J. Biol. Chem. 2010, 285, 40581–40592. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Kim, C.Y.; Hwang, J.; Suh, H.J.; Choi, H.S. Brassinin, a phytoalexin in cruciferous vegetables, suppresses obesity-induced inflammatory responses through the Nrf2-HO-1 signaling pathway in an adipocyte-macrophage co-culture system. Phytother. Res. 2019, 33, 1426–1437. [Google Scholar] [CrossRef]
- Liu, Z.; Dou, W.; Ni, Z.; Wen, Q.; Zhang, R.; Qin, M.; Wang, X.; Tang, H.; Cao, Y.; Wang, J.; et al. Deletion of Nrf2 leads to hepatic insulin resistance via the activation of NF-kappaB in mice fed a high-fat diet. Mol. Med. Rep. 2016, 14, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.K.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2012, 264, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Huang, J.; Shen, C.; Liu, Y.; He, S.; Sun, J.; Yu, B. NRF2 deficiency increases obesity susceptibility in a mouse menopausal model. PLoS ONE 2020, 15, e0228559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachur, S.; Lavie, C.J.; de Schutter, A.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular diseases. Minerva Med. 2017, 108, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Baumel-Alterzon, S.; Katz, L.S.; Brill, G.; Garcia-Ocana, A.; Scott, D.K. Nrf2: The Master and Captain of Beta Cell Fate. Trends Endocrinol. Metab. 2021, 32, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Wang, C.C.; Lai, T.Y.; Tsu, H.N.; Wang, C.H.; Liang, H.Y.; Kuo, W.W. Antioxidant effects of diallyl trisulfide on high glucose-induced apoptosis are mediated by the PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int. J. Cardiol. 2013, 168, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.S.; Pei, Y.H.; Peng, Y.P.; Chen, J.; Jiang, S.S.; Gong, J.B. Oscillating high glucose enhances oxidative stress and apoptosis in human coronary artery endothelial cells. J. Endocrinol. Investig. 2014, 37, 645–651. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kan, H.; Cai, L.; Ma, Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 47–58. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Wen, S.Y.; Cheng, S.Y.; Wang, C.H.; Yang, Y.C.; Viswanadha, V.P.; Huang, C.Y.; Kuo, W.W. Nrf2 Activation as a Protective Feedback to Limit Cell Death in High Glucose-Exposed Cardiomyocytes. J. Cell Biochem. 2017, 118, 1659–1669. [Google Scholar] [CrossRef]
- Shukla, K.; Pal, P.B.; Sonowal, H.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Inhibitor Protects against Hyperglycemic Stress by Activating Nrf2-Dependent Antioxidant Proteins. J. Diabetes Res. 2017, 2017, 6785852. [Google Scholar] [CrossRef]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Chen, X.; Zhong, Y.; Wen, X.; Cai, Y.; Li, J.; Fan, Z.; Feng, J. Activation of TGR5 Partially Alleviates High Glucose-Induced Cardiomyocyte Injury by Inhibition of Inflammatory Responses and Oxidative Stress. Oxid. Med. Cell Longev. 2019, 2019, 6372786. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Donepudi, A.C.; More, V.R.; Kulkarni, S.R.; Li, L.; Guo, L.; Yan, B.; Chatterjee, T.; Weintraub, N.; Slitt, A.L. Deficiency in Nrf2 transcription factor decreases adipose tissue mass and hepatic lipid accumulation in leptin-deficient mice. Obesity 2015, 23, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320. [Google Scholar] [PubMed] [Green Version]
- Braud, L.; Pini, M.; Stec, D.F.; Manin, S.; Derumeaux, G.; Stec, D.E.; Foresti, R.; Motterlini, R. Increased Sirt1 secreted from visceral white adipose tissue is associated with improved glucose tolerance in obese Nrf2-deficient mice. Redox Biol. 2021, 38, 101805. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Cuevas, J.; Sandoval-Rodriguez, A.; Monroy-Ramirez, H.C.; Vazquez-Del Mercado, M.; Santos-Garcia, A.; Armendariz-Borunda, J. Prolonged-release pirfenidone prevents obesity-induced cardiac steatosis and fibrosis in a mouse NASH model. Cardiovasc. Drugs Ther. 2021, 35, 927–938. [Google Scholar] [CrossRef]
- Ganesan, K.; Ramkumar, K.M.; Xu, B. Vitexin restores pancreatic beta-cell function and insulin signaling through Nrf2 and NF-kappaB signaling pathways. Eur. J. Pharmacol. 2020, 888, 173606. [Google Scholar] [CrossRef] [PubMed]
- Fratantonio, D.; Speciale, A.; Ferrari, D.; Cristani, M.; Saija, A.; Cimino, F. Palmitate-induced endothelial dysfunction is attenuated by cyanidin-3-O-glucoside through modulation of Nrf2/Bach1 and NF-kappaB pathways. Toxicol. Lett. 2015, 239, 152–160. [Google Scholar] [CrossRef]
- Cao, K.; Lv, W.; Liu, X.; Fan, Y.; Wang, K.; Feng, Z.; Liu, J.; Zang, W.; Xing, L.; Liu, J. Herba Houttuyniae Extract Benefits Hyperlipidemic Mice via Activation of the AMPK/PGC-1alpha/Nrf2 Cascade. Nutrients 2020, 12, 164. [Google Scholar] [CrossRef] [Green Version]
- He, H.J.; Wang, G.Y.; Gao, Y.; Ling, W.H.; Yu, Z.W.; Jin, T.R. Curcumin attenuates Nrf2 signaling defect, oxidative stress in muscle and glucose intolerance in high fat diet-fed mice. World J. Diabetes 2012, 3, 94–104. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371 Pt 3, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, S.; Jiang, X.; Wang, Y.H.; Li, F.; Wang, Y.G.; Zheng, Y.; Cai, L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev. Endocr. Metab. Disord. 2015, 16, 35–45. [Google Scholar] [CrossRef]
- Wang, Y.; You, Y.; Tian, Y.; Sun, H.; Li, X.; Wang, X.; Wang, Y.; Liu, J. Pediococcus pentosaceus PP04 Ameliorates High-Fat Diet-Induced Hyperlipidemia by Regulating Lipid Metabolism in C57BL/6N Mice. J. Agric. Food. Chem. 2020, 68, 15154–15163. [Google Scholar] [CrossRef] [PubMed]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Galley, J.C.; Straub, A.C. Redox Control of Vascular Function. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e178–e184. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Papaharalambus, C.A.; Griendling, K.K. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc. Med. 2007, 17, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.Y.; Gao, L.; Cai, H. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012, 55, 2069–2079. [Google Scholar] [CrossRef] [Green Version]
- da Costa, R.M.; Fais, R.S.; Dechandt, C.R.P.; Louzada-Junior, P.; Alberici, L.C.; Lobato, N.S.; Tostes, R.C. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br. J. Pharmacol. 2017, 174, 3527–3541. [Google Scholar] [CrossRef] [Green Version]
- Neves, K.B.; Nguyen Dinh Cat, A.; Alves-Lopes, R.; Harvey, K.Y.; Costa, R.M.D.; Lobato, N.S.; Montezano, A.C.; Oliveira, A.M.; Touyz, R.M.; Tostes, R.C. Chemerin receptor blockade improves vascular function in diabetic obese mice via redox-sensitive and Akt-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1851–H1860. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qi, J.; Wu, Q.; Jiang, H.; Wang, J.; Chen, W.; Mao, A.; Zhu, M. High glucose inhibits vascular endothelial Keap1/Nrf2/ARE signal pathway via downregulation of monomethyltransferase SET8 expression. Acta Biochim. Biophys. Sin. 2020, 52, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Abu-Saleh, N.; Yaseen, H.; Kinaneh, S.; Khamaisi, M.; Abassi, Z. Combination of hyperglycaemia and hyperlipidaemia induces endothelial dysfunction: Role of the endothelin and nitric oxide systems. J. Cell Mol. Med. 2021, 25, 1884–1895. [Google Scholar] [CrossRef]
- Jimenez, R.; Toral, M.; Gomez-Guzman, M.; Romero, M.; Sanchez, M.; Mahmoud, A.M.; Duarte, J. The Role of Nrf2 Signaling in PPARbeta/delta-Mediated Vascular Protection against Hyperglycemia-Induced Oxidative Stress. Oxid. Med. Cell Longev. 2018, 2018, 5852706. [Google Scholar] [CrossRef] [Green Version]
- Gupte, A.A.; Lyon, C.J.; Hsueh, W.A. Nuclear factor (erythroid-derived 2)-like-2 factor (Nrf2), a key regulator of the antioxidant response to protect against atherosclerosis and nonalcoholic steatohepatitis. Curr. Diabetes Rep. 2013, 13, 362–371. [Google Scholar] [CrossRef]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Pu, C.; Zhou, P.; Wang, P.; Liang, D.; Wang, Q.; Hu, Y.; Li, B.; Hao, X. Cinnamaldehyde prevents endothelial dysfunction induced by high glucose by activating Nrf2. Cell Physiol. Biochem. 2015, 36, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, X.; Niu, C.; Huang, X.; An, N.; Sun, J.; Huang, S.; Ye, W.; Li, S.; Shen, Y.; et al. Baicalin alleviates hyperglycemia-induced endothelial impairment 1 via Nrf2. J. Endocrinol. 2018, 240, 81–98. [Google Scholar] [CrossRef]
- Fratantonio, D.; Speciale, A.; Canali, R.; Natarelli, L.; Ferrari, D.; Saija, A.; Virgili, F.; Cimino, F. Low nanomolar caffeic acid attenuates high glucose-induced endothelial dysfunction in primary human umbilical-vein endothelial cells by affecting NF-kappaB and Nrf2 pathways. Biofactors 2017, 43, 54–62. [Google Scholar] [CrossRef]
- Ortega, F.J.; Mercader, J.M.; Catalan, V.; Moreno-Navarrete, J.M.; Pueyo, N.; Sabater, M.; Gomez-Ambrosi, J.; Anglada, R.; Fernandez-Formoso, J.A.; Ricart, W.; et al. Targeting the circulating microRNA signature of obesity. Clin. Chem. 2013, 59, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.Q.; Ren, K.; Liu, S.H.; Li, W.M.; Huang, C.J.; Yang, X.H. MicroRNA-140-5p aggravates hypertension and oxidative stress of atherosclerosis via targeting Nrf2 and Sirt2. Int. J. Mol. Med. 2019, 43, 839–849. [Google Scholar] [CrossRef] [Green Version]
- da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a Potential Mediator of Cardiovascular Risk in Metabolic Diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzon Dos Santos, J.; Quadros, A.S.; Weschenfelder, C.; Garofallo, S.B.; Marcadenti, A. Oxidative Stress Biomarkers, Nut-Related Antioxidants, and Cardiovascular Disease. Nutrients 2020, 12, 682. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Torzewski, M.; Degreif, A.; Rossmann, H.; Canisius, A.; Lackner, K.J. Impact of glutathione peroxidase-1 deficiency on macrophage foam cell formation and proliferation: Implications for atherogenesis. PLoS ONE 2013, 8, e72063. [Google Scholar] [CrossRef] [Green Version]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef]
- Yet, S.F.; Layne, M.D.; Liu, X.; Chen, Y.H.; Ith, B.; Sibinga, N.E.; Perrella, M.A. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003, 17, 1759–1761. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.R.; Gupte, A.A.; Ji, R.; Ramirez, M.R.; Minze, L.J.; Liu, J.Z.; Arredondo, M.; Ren, Y.; Deng, T.; Wang, J.; et al. Myeloid deletion of nuclear factor erythroid 2-related factor 2 increases atherosclerosis and liver injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2839–2846. [Google Scholar] [CrossRef] [Green Version]
- Ruotsalainen, A.K.; Inkala, M.; Partanen, M.E.; Lappalainen, J.P.; Kansanen, E.; Makinen, P.I.; Heinonen, S.E.; Laitinen, H.M.; Heikkila, J.; Vatanen, T.; et al. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc. Res. 2013, 98, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Ashino, T.; Yamamoto, M.; Yoshida, T.; Numazawa, S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Ashino, T.; Yamamoto, M.; Numazawa, S. Nrf2/Keap1 system regulates vascular smooth muscle cell apoptosis for vascular homeostasis: Role in neointimal formation after vascular injury. Sci. Rep. 2016, 6, 26291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckers, H.J.; Boehm, M.; True, A.L.; Yet, S.F.; San, H.; Park, J.L.; Clinton Webb, R.; Lee, M.E.; Nabel, G.J.; Nabel, E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001, 7, 693–698. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeoung, N.H.; Oh, C.J.; Choi, Y.K.; Lee, H.J.; Kim, H.J.; Kim, J.Y.; Hwang, J.H.; Tadi, S.; Yim, Y.H.; et al. Activation of NAD(P)H:quinone oxidoreductase 1 prevents arterial restenosis by suppressing vascular smooth muscle cell proliferation. Circ. Res. 2009, 104, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Li, X.X.; Wang, L.; Wang, M.; Zhang, Y.; Li, P.L. Contribution of Nrf2 to Atherogenic Phenotype Switching of Coronary Arterial Smooth Muscle Cells Lacking CD38 Gene. Cell Physiol. Biochem. 2015, 37, 432–444. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Song, F.; Li, Y.; Li, J.; Cui, Y.; Hong, Y.; Han, W.; Wu, W.; Lakhani, I.; Li, G.; et al. Acacetin exerts antioxidant potential against atherosclerosis through Nrf2 pathway in apoE(-/-) Mice. J. Cell Mol. Med. 2021, 25, 521–534. [Google Scholar] [CrossRef]
- Yang, X.J.; Liu, F.; Feng, N.; Ding, X.S.; Chen, Y.; Zhu, S.X.; Yang, L.C.; Feng, X.F. Berberine Attenuates Cholesterol Accumulation in Macrophage Foam Cells by Suppressing AP-1 Activity and Activation of the Nrf2/HO-1 Pathway. J. Cardiovasc. Pharmacol. 2020, 75, 45–53. [Google Scholar] [CrossRef]
- Gu, L.; Ye, P.; Li, H.; Wang, Y.; Xu, Y.; Tian, Q.; Lei, G.; Zhao, C.; Gao, Z.; Zhao, W.; et al. Lunasin attenuates oxidant-induced endothelial injury and inhibits atherosclerotic plaque progression in ApoE(-/-) mice by up-regulating heme oxygenase-1 via PI3K/Akt/Nrf2/ARE pathway. FASEB J. 2019, 33, 4836–4850. [Google Scholar] [CrossRef]
- Jia, H.; Cheng, J.; Zhou, Q.; Peng, J.; Pan, Y.; Han, H. Fibroblast growth factor 21 attenuates inflammation and oxidative stress in atherosclerotic rat via enhancing the Nrf1-ARE signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 1308–1317. [Google Scholar] [PubMed]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Chen, J.; Gong, F.; Chen, M.F.; Li, C.; Hong, P.; Sun, S.; Zhou, C.; Qian, Z.J. In Vitro Vascular-Protective Effects of a Tilapia By-Product Oligopeptide on Angiotensin II-Induced Hypertensive Endothelial Injury in HUVEC by Nrf2/NF-kappaB Pathways. Mar. Drugs 2019, 17, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Tian, T.; Wang, Y.; Li, Z.; Xing, K.; Tian, G. SIRT6 protects vascular endothelial cells from angiotensin II-induced apoptosis and oxidative stress by promoting the activation of Nrf2/ARE signaling. Eur. J. Pharmacol. 2019, 859, 172516. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Li, J.; Liu, L.; Fan, L.; Li, X.Y.; Wang, Y.T.; Abraham, N.G.; Cao, J. Effects of heme oxygenase-1 upregulation on blood pressure and cardiac function in an animal model of hypertensive myocardial infarction. Int. J. Mol. Sci. 2013, 14, 2684–2706. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Yet, S.F.; Perrella, M.A. Role of heme oxygenase-1 in the regulation of blood pressure and cardiac function. Exp. Biol. Med. 2003, 228, 447–453. [Google Scholar] [CrossRef]
- Datla, S.R.; Dusting, G.J.; Mori, T.A.; Taylor, C.J.; Croft, K.D.; Jiang, F. Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertension 2007, 50, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Silva-Palacios, A.; Konigsberg, M.; Zazueta, C. Nrf2 signaling and redox homeostasis in the aging heart: A potential target to prevent cardiovascular diseases? Ageing Res. Rev. 2016, 26, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.A.; Neves, K.B.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 2015, 66, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, C.; Xing, Y.; Janicki, J.S.; Yamamoto, M.; Wang, X.L.; Tang, D.Q.; Cui, T. Up-regulation of p27(kip1) contributes to Nrf2-mediated protection against angiotensin II-induced cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.H.; Rizzi, E.S.; Alves-Lopes, R.; Pinheiro, L.C.; Tostes, R.C.; Tanus-Santos, J.E. Antioxidant and antihypertensive responses to oral nitrite involves activation of the Nrf2 pathway. Free Radic. Biol. Med. 2019, 141, 261–268. [Google Scholar] [CrossRef]
- Montenegro, M.F.; Amaral, J.H.; Pinheiro, L.C.; Sakamoto, E.K.; Ferreira, G.C.; Reis, R.I.; Marcal, D.M.; Pereira, R.P.; Tanus-Santos, J.E. Sodium nitrite downregulates vascular NADPH oxidase and exerts antihypertensive effects in hypertension. Free Radic. Biol. Med. 2011, 51, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Yang, T.; Liu, M.; Peleli, M.; Zollbrecht, C.; Weitzberg, E.; Lundberg, J.O.; Persson, A.E.; Carlstrom, M. NADPH oxidase in the renal microvasculature is a primary target for blood pressure-lowering effects by inorganic nitrate and nitrite. Hypertension 2015, 65, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.H.; Ferreira, G.C.; Pinheiro, L.C.; Montenegro, M.F.; Tanus-Santos, J.E. Consistent antioxidant and antihypertensive effects of oral sodium nitrite in DOCA-salt hypertension. Redox Biol. 2015, 5, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Teng, X.; Jin, S.; Dong, J.; Guo, Q.; Tian, D.; Wu, Y. Hydrogen sulfide improves endothelial dysfunction by inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2019, 37, 1633–1643. [Google Scholar] [CrossRef]
- Meephat, S.; Prasatthong, P.; Potue, P.; Bunbupha, S.; Pakdeechote, P.; Maneesai, P. Diosmetin Ameliorates Vascular Dysfunction and Remodeling by Modulation of Nrf2/HO-1 and p-JNK/p-NF-kappaB Expression in Hypertensive Rats. Antioxidants 2021, 10, 1487. [Google Scholar] [CrossRef]
- Huo, C.J.; Yu, X.J.; Sun, Y.J.; Li, H.B.; Su, Q.; Bai, J.; Li, Y.; Liu, K.L.; Qi, J.; Zhou, S.W.; et al. Irisin lowers blood pressure by activating the Nrf2 signaling pathway in the hypothalamic paraventricular nucleus of spontaneously hypertensive rats. Toxicol. Appl. Pharmacol. 2020, 394, 114953. [Google Scholar] [CrossRef]
- Bai, J.; Yu, X.J.; Liu, K.L.; Wang, F.F.; Jing, G.X.; Li, H.B.; Zhang, Y.; Huo, C.J.; Li, X.; Gao, H.L.; et al. Central administration of tert-butylhydroquinone attenuates hypertension via regulating Nrf2 signaling in the hypothalamic paraventricular nucleus of hypertensive rats. Toxicol. Appl. Pharmacol. 2017, 333, 100–109. [Google Scholar] [CrossRef]
- Zhuang, L.N.; Hu, W.X.; Zhang, M.L.; Xin, S.M.; Jia, W.P.; Zhao, J.; Pei, G. Beta-arrestin-1 protein represses diet-induced obesity. J. Biol. Chem. 2011, 286, 28396–28402. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Jiao, P.L.; Sun, J.C.; Wang, W.; Ye, P.; Wang, Y.K.; Leng, Y.Q.; Wang, W.Z. beta-Arrestin1 Reduces Oxidative Stress via Nrf2 Activation in the Rostral Ventrolateral Medulla in Hypertension. Front. Neurosci. 2021, 15, 657825. [Google Scholar] [CrossRef]
- Gao, L.; Zimmerman, M.C.; Biswal, S.; Zucker, I.H. Selective Nrf2 Gene Deletion in the Rostral Ventrolateral Medulla Evokes Hypertension and Sympathoexcitation in Mice. Hypertension 2017, 69, 1198–1206. [Google Scholar] [CrossRef]
- Lin, Y.J.; Lin, I.C.; Yu, H.R.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Early Postweaning Treatment with Dimethyl Fumarate Prevents Prenatal Dexamethasone- and Postnatal High-Fat Diet-Induced Programmed Hypertension in Male Rat Offspring. Oxid. Med. Cell Longev. 2018, 2018, 5343462. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Yu, H.R.; Lin, I.C.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int. J. Mol. Sci. 2019, 20, 3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooqui, Z.; Mohammad, R.S.; Lokhandwala, M.F.; Banday, A.A. Nrf2 inhibition induces oxidative stress, renal inflammation and hypertension in mice. Clin. Exp. Hypertens. 2021, 43, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Van der Heiden, K.; le Luong, A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Kloska, D.; Kopacz, A.; Piechota-Polanczyk, A.; Nowak, W.N.; Dulak, J.; Jozkowicz, A.; Grochot-Przeczek, A. Nrf2 in aging-Focus on the cardiovascular system. Vascul. Pharmacol. 2019, 112, 42–53. [Google Scholar] [CrossRef]
- Ungvari, Z.; Bailey-Downs, L.; Sosnowska, D.; Gautam, T.; Koncz, P.; Losonczy, G.; Ballabh, P.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Vascular oxidative stress in aging: A homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H363–H372. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Gao, J.; Zheng, Y.; Wang, X.; Chen, C.; Cao, K.; Xu, J.; Li, Y.; Lu, W.; Liu, J.; et al. Zeaxanthin induces Nrf2-mediated phase II enzymes in protection of cell death. Cell Death Dis. 2014, 5, e1218. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Yang, Y.; Li, T.; Ma, Z.; Hu, W.; Deng, C.; Fan, C.; Lv, J.; Sun, Y.; Yi, W. An overview of the mechanisms and novel roles of Nrf2 in cardiovascular diseases. Expert Opin. Ther. Targets 2016, 20, 1413–1424. [Google Scholar] [CrossRef]
- Greco, T.; Shafer, J.; Fiskum, G. Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic. Biol. Med. 2011, 51, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Li, B.; Lai, Y.; Li, H.; Windust, A.; Hofseth, L.J.; Nagarkatti, M.; Nagarkatti, P.; Wang, X.L.; Tang, D.; et al. Identifying panaxynol, a natural activator of nuclear factor erythroid-2 related factor 2 (Nrf2) from American ginseng as a suppressor of inflamed macrophage-induced cardiomyocyte hypertrophy. J. Ethnopharmacol. 2015, 168, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Rui, W.; Guan, L.; Zhang, F.; Zhang, W.; Ding, W. PM2.5-induced oxidative stress increases adhesion molecules expression in human endothelial cells through the ERK/AKT/NF-kappaB-dependent pathway. J. Appl. Toxicol. 2016, 36, 48–59. [Google Scholar] [CrossRef]
- Zeng, C.; Zhong, P.; Zhao, Y.; Kanchana, K.; Zhang, Y.; Khan, Z.A.; Chakrabarti, S.; Wu, L.; Wang, J.; Liang, G. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-kappaB both in vitro and in vivo. J. Mol. Cell Cardiol. 2015, 79, 1–12. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Boliang, W.; Xiaoxi, T.; Guoqiang, F.; Jianbo, X.; Gang, W. Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomed. Pharmacother. 2020, 122, 109547. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Senthil, K.K.J.; Gokila, V.M.; Wang, S.Y. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget 2017, 8, 96568–96587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Li, H.Y.; Si, C.Y.; Liu, Y.Z.; Teng, S. Asiatic acid inhibits cardiac fibrosis throughNrf2/HO-1 and TGF-beta1/Smads signaling pathways in spontaneous hypertension rats. Int. Immunopharmacol. 2019, 74, 105712. [Google Scholar] [CrossRef]
- Zhong, P.; Wu, L.; Qian, Y.; Fang, Q.; Liang, D.; Wang, J.; Zeng, C.; Wang, Y.; Liang, G. Blockage of ROS and NF-kappaB-mediated inflammation by a new chalcone L6H9 protects cardiomyocytes from hyperglycemia-induced injuries. Biochim. Biophys. Acta 2015, 1852, 1230–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuvaraj, S.; Ramprasath, T.; Saravanan, B.; Vasudevan, V.; Sasikumar, S.; Selvam, G.S. Chrysin attenuates high-fat-diet-induced myocardial oxidative stress via upregulating eNOS and Nrf2 target genes in rats. Mol. Cell Biochem. 2021, 476, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Q.; Shi, L.; Qin, L.; Zhang, Q.; Mi, M. Equol Attenuates Atherosclerosis in Apolipoprotein E-Deficient Mice by Inhibiting Endoplasmic Reticulum Stress via Activation of Nrf2 in Endothelial Cells. PLoS ONE 2016, 11, e0167020. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, Y.; Zhong, P.; Peng, K.; Xu, Z.; Chen, X.; Lu, K.; Chen, G.; Li, X.; Liang, G. Inhibition of inflammation and oxidative stress by an imidazopyridine derivative X22 prevents heart injury from obesity. J. Cell Mol. Med. 2016, 20, 1427–1442. [Google Scholar] [CrossRef]
- Chen, R.R.; Fan, X.H.; Chen, G.; Zeng, G.W.; Xue, Y.G.; Liu, X.T.; Wang, C.Y. Corrigendum to “Irisin attenuates angiotensin II-induced cardiac fibrosis via Nrf2 mediated inhibition of ROS/ TGFbeta1/Smad2/3 signaling axis”. Chem. Biol. Interact. 2020, 325, 109130. [Google Scholar] [CrossRef]
- Jiang, X.; Li, Y.; Wang, W.; Han, X.; Han, J.; Chen, M.; Zhang, J.; Wang, C.; Li, S.; Luo, J.; et al. Nuclear Factor Erythroid 2 Related Factor 2 Activator JC-5411 Inhibits Atherosclerosis Through Suppression of Inflammation and Regulation of Lipid Metabolism. Front. Pharmacol. 2020, 11, 532568. [Google Scholar] [CrossRef]
- Du, J.; Zhu, M.; Li, H.; Liang, G.; Li, Y.; Feng, S. Metformin attenuates cardiac remodeling in mice through the Nrf2/Keap1 signaling pathway. Exp. Ther. Med. 2020, 20, 838–845. [Google Scholar] [CrossRef]
- Chen, P.Y.; Shih, N.L.; Hao, W.R.; Chen, C.C.; Liu, J.C.; Sung, L.C. Inhibitory Effects of Momordicine I on High-Glucose-Induced Cell Proliferation and Collagen Synthesis in Rat Cardiac Fibroblasts. Oxid. Med. Cell Longev. 2018, 2018, 3939714. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; He, W.; Zhang, C.; Wu, J.; Li, Z.; Wang, M.; Feng, S.; Liang, G. Pentamethylquercetin Attenuates Cardiac Remodeling via Activation of the Sestrins/Keap1/Nrf2 Pathway in MSG-Induced Obese Mice. Biomed. Res. Int. 2020, 2020, 3243906. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Duan, Z.; Xu, J.; Liang, J.; Zhang, S.; Zhang, H.; Zhang, X.; Wang, Y. Pterostilbene reduces endothelial cell injury in vascular arterial walls by regulating the Nrf2-mediated AMPK/STAT3 pathway in an atherosclerosis rat model. Exp. Ther. Med. 2020, 19, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, Z.; Zhang, L.; Yao, X.; Zhong, X.; Cheng, G.; Wang, L.; Wan, Q. Spiraeoside protects human cardiomyocytes against high glucose-induced injury, oxidative stress, and apoptosis by activation of PI3K/Akt/Nrf2 pathway. J. Biochem. Mol. Toxicol. 2020, 34, e22548. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.T.; Loh, S.H.; Lee, C.Y.; Lee, S.P.; Chen, Y.L.; Cheng, T.H.; Tsai, C.S. Tanshinone IIA Inhibits High Glucose-Induced Collagen Synthesis via Nuclear Factor Erythroid 2-Related Factor 2 in Cardiac Fibroblasts. Cell Physiol. Biochem. 2018, 51, 2250–2261. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Huang, X.; Xie, Y.; Qu, Y.; Long, H.; Gu, N.; Jiang, W. Z-Ligustilide protects vascular endothelial cells from oxidative stress and rescues high fat diet-induced atherosclerosis by activating multiple NRF2 downstream genes. Atherosclerosis 2019, 284, 110–120. [Google Scholar] [CrossRef] [PubMed]
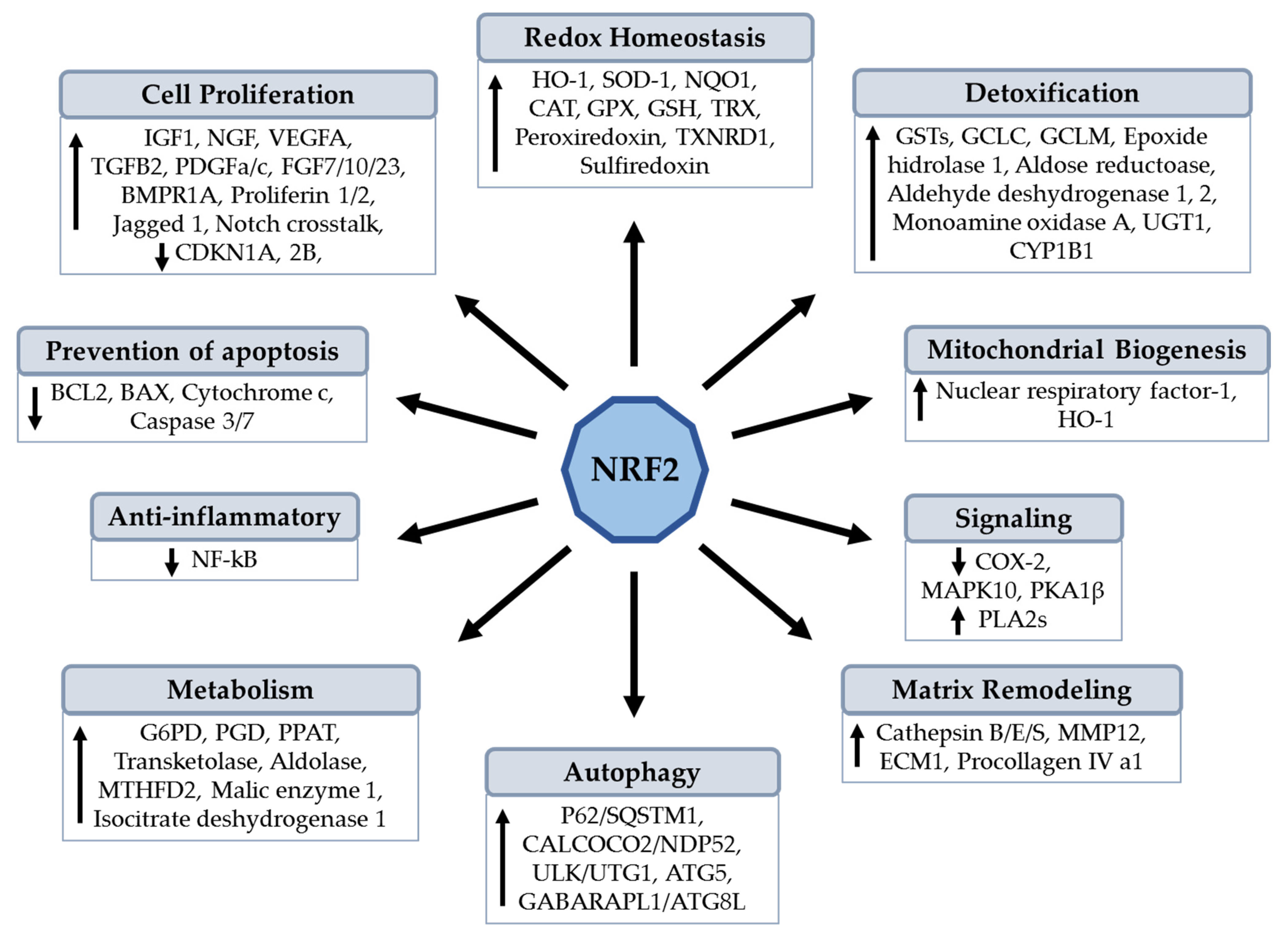
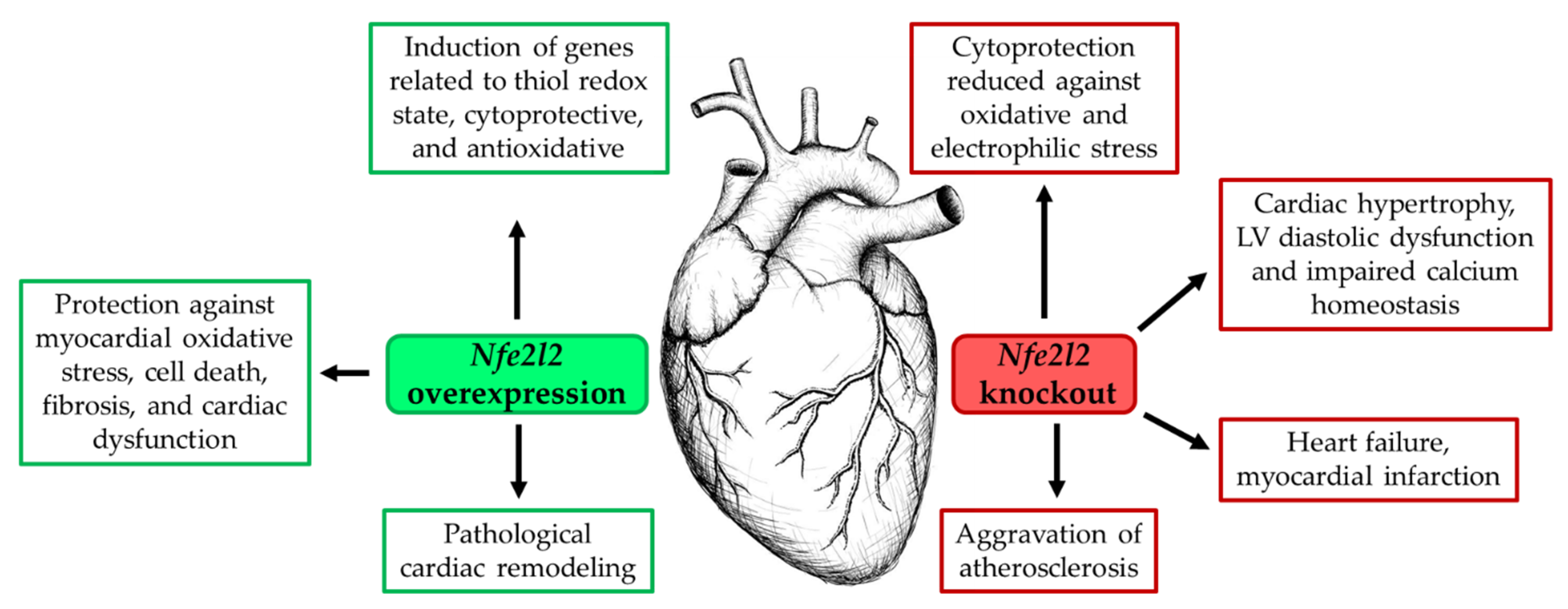
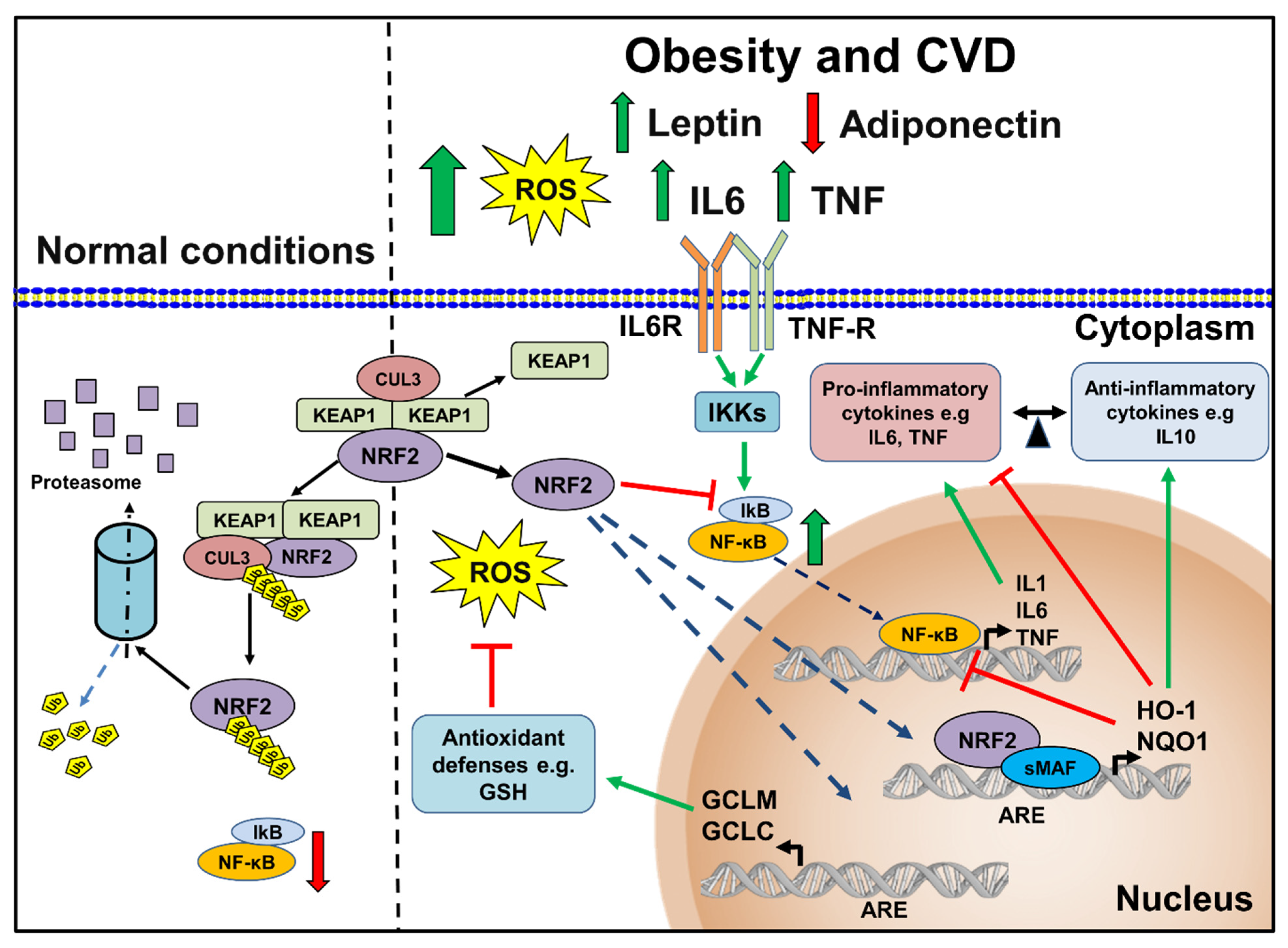

{kind=link}
{kind=link}
{kind=link}
| Mouse and Cell Line Models | Experimental Conditons | Main Effects Reported |
|---|---|---|
| Nfe2l2-KO male mice | Obesity induced by an HFD for 12 weeks | Reduced adipose tissue mass, Impaired adipocyte differentiation [58] |
| Nfe2l2-KO male mice | Mice were fed an HFD for 180 days | Mice were partially protected from HFD-induced obesity and developed a less insulin-resistant phenotype [60] |
| Nfe2l2-KO male mice | Obesity induced by an HFD for 8 weeks | Mice showed an accelerated onset of obesity and NASH via the induction of hepatic IR. In addition, mice had increased in total and hepatic weight [72] |
| Nfe2l2-KO male mice | Obesity induced by a high-fat Western diet for 12 weeks | Mice were resistant to high-fat, Western-diet-induced glucose intolerance. Absence of NRF2 activity did not prevent diet-induced obesity [73] |
| Nfe2l2-KO mice | Obesity induced by an HFD for 12 weeks | Mice showed significant oxidative stress in the WAT. Body weight and WAT weight were significantly lower in Nfe2l2-KO mice, including triglycerides content in the liver and muscle [66] |
| Male mice with adipose-specific ablation of Nfe2l2 | Obesity induced by an HFD for 14 weeks | Body weight and body fat content of Nfe2l2-KO mice showed comparable results with Nfe2l2 control mice, but exhibited reduced blood glucose, reduced number but increased size of adipocytes [61] |
| Nfe2l2-KO ovariectomized female mice | Ovariectomy was performed once the mice reached 15-16 weeks old | Nfe2l2-KO ovariectomized mice had a greater body weight gain, an increase in blood glucose level, and a reduction in LDL and the level of 5-HT [74] |
| Nfe2l2-KD in primary cultured mouse preadipocytes and 3T3-L1 cells | Mouse primary preadipocytes were isolated from WAT. Lentiviral transduction of 3T3-L1 cells with particles for shRNA targeting NRF2 | Hampered adipogenic differentiation induced by hormonal cocktails [59] |
| Male mice with cell-specific deletion of Nfe2l2 in adipocytes (ANKO) or hepatocytes (HeNKO) | Obesity induced by an HFD for 24 weeks | Mice showed similar increases in body weight and body fat content. ANKO mice showed high fasting glucose levels and high levels of cholesterol and nonesterified fatty acids. HeNKO mice showed low insulin levels and trended toward improved insulin sensitivity without having any difference in liver triglyceride accumulation [63] |
| Adipocyte-specific Nfe2l2-knockout male and female mice on a leptin-deficient ob/ob background | Monitored during an 11-week period in mice 4–15 weeks of age | Mice showed reduced WAT mass but severe metabolic syndrome with aggravated insulin resistance, hyperglycemia, and hypertriglyceridemia [64] |
| Keap1-KD male mice, which have increased NRF2 activity | Obesity induced by a high-fat Western diet for 12 weeks | Mice exhibited prolonged elevation of circulating glucose during a glucose tolerance test. Enhancement of NRF2 activity did not prevent diet-induced obesity [73] |
| NRF2 overexpression or Keap1-KD in 3T3-L1 cells | Lentiviral transduction of 3T3-L1 cells with particles for shRNAs targeting NRF2 | Accelerates hormone-induced adipocyte differentiation [58] |
| Lep(ob/ob)-Keap1-KD mice, which have increased NRF2 activity | Keap1-KD mice were fed an HFD for 36 days | Lep(ob/ob)-Keap1-KD mice exhibited less lipid accumulation, smaller adipocytes, decreased food intake, and reduced lipogenic gene expression. Obesity and lipid accumulation in white adipose tissue was decreased in Keap1-KD mice. Constitutive NRF2 activation inhibited lipid accumulation in WAT, suppressed adipogenesis, induced insulin resistance and glucose intolerance, and increased hepatic steatosis in Lep(ob/ob) mice [67] |
| Male C57BL/6 J mice | Obesity induced by an HFD for 17 weeks with or without ECGC, an NRF2 activator | Dietary EGCG significantly reduced weight gain, plasma glucose, insulin level, liver and kidney weight. Prevention of HFD-induced AGEs formation [68] |
| Male C57BL/6 J mice | Obesity induced by an HFD for 12 weeks with or without parthenolide, an NRF2 activator | Parthenolide-administered mice showed a significant reduction in body weightand WAT. Parthenolide inhibitedobesity-induced inflammatory responses [69] |
| Drug or Compound | Representative Model | Effects Reported | Pathological Condition |
|---|---|---|---|
| Acacetin | EA.hy926 cells and apolipoprotein E deficiency (ApoE−/−) female mice with Western diet | In cells, decreased ROS. In vivo, attenuated atherosclerosis by increasing reductase levels and aortic roots, decreasing plasma inflammatory factor levels | Atherosclerosis [128] |
| Antrodin C | HUVECs | Prevented high glucose-induced senescence, ameliorated ROS and apoptosis | Hyperglycemia [171] |
| Asiatic acid | Cultured cardiac fibroblasts. Male WKY rats and male SHRs | In vitro, inhibited ANG II-induced cardiac fibrosis. In vivo, attenuated myocardial hypertrophy, reduced collage deposition, MDA, and ROS | Hypertension [172] |
| Brassinin | 3T3-L1 and RAW264.7 cells | Suppressed lipid accumulation, decreased inflammatory cytokines and ROS | Obesity [71] |
| Chalcone L6H9 | H9C2 cells and male C57BL/6 mice with STZ-induced diabetes | In vitro, reduced inflammation, ROS, mitochondrial dysfunction, cell apoptosis, fibrosis, and hypertrophy. In vivo, decreased cardiac cytokines and ROS level, decreasing cardiac apoptosis, hypertrophy, and fibrosis | Hyperglycemia [173] |
| Chrysin | Male Sprague Dawley rats | Attenuated myocardial oxidative stress via upregulating eNOS and NRF2 target genes | Obesity [174] |
| Curcumin | H9C2 cells and male C57BL/6 mice | In vitro, decreased ROS, inflammation, apoptosis, and hypertrophy. In vivo, suppressed oxidative stress, inflammation, apoptosis, fibrosis, hypertrophy, and tissue remodeling | Obesity [167] |
| Cyanidin-3-O-glucosid | HUVECs | Via NRF2/BACH1 and NF-κB pathways, improved intracellular redox status, inhibited NF-κB proinflammatory pathway and adhesion molecules | Endothelial dysfunction induced by palmitic acid [89] |
| Equol | HUVECs and apolipoprotein E knockout (ApoE−/−) male mice fed a HFD | In cells, inhibited apoptosis induced by t-BHP and thapsigargin, attenuated ER stress markers. In vivo, reduced triglycerides, total cholesterol, and LDL-cholesterol and increased HDL-cholesterol | Atherosclerosis [175] |
| Herba houttuyniae | Male C57BL/6 J mice | Via activation of the PRKAA2/PPARG/NRF2 cascade, attenuated lipids, improved cardiac remodeling, and ameliorated cardiac oxidative stress | Hyperlipidemia [90] |
| Imidazopyridine derivative X22 | H9c2 cells and male Wistar rats | Inhibited ROS, inflammation, apoptosis, fibrosis, and hypertrophy. NF-κB also was inhibited | Obesity [176] |
| Irisin | Cardiac fibroblasts and male C57BL/6 mice | Attenuated ANG II-induced cardiac fibrosis via NRF2 mediated inhibition of ROS/TGFB1/SMAD2/3 signaling axis | Cardiac dysfunction [177] |
| JC-5411 (Phenethyl isothiocyanate formulation) | Apolipoprotein E deficient (ApoE−/−) male mice | Reduced atherosclerotic plaque area in both in face aorta and aortic sinus through suppression of inflammation and regulation of lipid metabolism | Atherosclerosis [178] |
| Lunasin | EA.hy926 cells and apolipopro-tein E deficiency (ApoE−/−) male mice fed a HFD | Upregulated HO-1 via the PI3K/AKT1/NRF2/ARE pathway, attenuating H2O2 and apoptosis | Atherosclerosis [130] |
| Metformin | Male C57BL/6 J mice | Ameliorated obesity phenotype and metabolic disorders, reduced the heart weight index, and attenuated cardiac fibrosis | Obesity [179] |
| Momordicine I | Neonatal rat cardiac fibroblasts | Abolished fibroblast proliferation and collagen synthesis | Hyperglycemia [180] |
| Parthenolide | 3T3-L1 and RAW264.7 cells. Male C57BL/6 J mice | In cells, suppressed inflammatory responses by downregulating IL6 and CCL2. In animals, reduced body weight and WAT, downregulating NF-κB and MAPKs | Obesity [69] |
| Pentamethylquercetin | CD1 male and female mice treated with monosodium glutamate | Ameliorated obesity phenotypes, decreased the heart wall thickness, and attenuated cardiac fibrosis | Obesity [181] |
| Pterostilbene | HUAEC and Male Sprague-Dawley rats with endothelial injury of the iliac arteries and feeding with a 2.5% cholesterol diet with 1% glucose | In vitro, decreased oxidative stress injury and apoptosis. In animals, decreased inflammation, atherogenesis, reduced aortic plaque size, reduced macrophage infiltration, and suppressed oxidative stress and apoptosis | Atherosclerosis [182] |
| Spiraeoside | AC16 cells | Inhibited ROS and MDA production, increased activities of SOD1, GPX1, and CAT. Prevented apoptosis | Hyperglycemia [183] |
| Tanshinone IIA | Neonatal rat cardiac fibroblasts | Abolished cell proliferation and collagen synthesis via activation of NRF2 and inhibition of TGFB1 production and SMAD2/3 phosphorylation | Hyperglycemia [184] |
| Z-Ligustilide | EA.hy926 cells and HFD-fed Ldlr-deficient male mice | In vitro, alleviated oxidative stress and cell injury caused by t-BHP. In vivo, restrained atherosclerosis progression, attenuated atherosclerotic plaque formation, alleviated lipid peroxidation, and increased antioxidant enzyme activity in aortas | Atherosclerosis [185] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramírez, H.C.; Sandoval-Rodriguez, A.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. https://doi.org/10.3390/antiox11020235
Gutiérrez-Cuevas J, Galicia-Moreno M, Monroy-Ramírez HC, Sandoval-Rodriguez A, García-Bañuelos J, Santos A, Armendariz-Borunda J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants. 2022; 11(2):235. https://doi.org/10.3390/antiox11020235
Chicago/Turabian StyleGutiérrez-Cuevas, Jorge, Marina Galicia-Moreno, Hugo Christian Monroy-Ramírez, Ana Sandoval-Rodriguez, Jesús García-Bañuelos, Arturo Santos, and Juan Armendariz-Borunda. 2022. "The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors" Antioxidants 11, no. 2: 235. https://doi.org/10.3390/antiox11020235
APA StyleGutiérrez-Cuevas, J., Galicia-Moreno, M., Monroy-Ramírez, H. C., Sandoval-Rodriguez, A., García-Bañuelos, J., Santos, A., & Armendariz-Borunda, J. (2022). The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants, 11(2), 235. https://doi.org/10.3390/antiox11020235