
Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain

 , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Creation of iPLA2γ/PNPLA8 Knockout Mice
2.3. Isolation of Mitochondria
2.4. High-Resolution Respirometry
2.5. Intramitochondrial Detection of Superoxide Formation
2.6. Extramitochondrial Detection of H2O2 Release
2.7. LC–MS-Based Lipidomic Profiling
2.8. Agent/Drug Application to Mice
2.9. Tissue Homogenates
2.10. Quantification of Interleukin-6 Levels
2.11. Quantification of Protein Carbonyls
2.12. Statistical Analysis
3. Results
3.1. Fatty Acid-Induced Increase in Respiration following Redox Activation of iPLA2γ
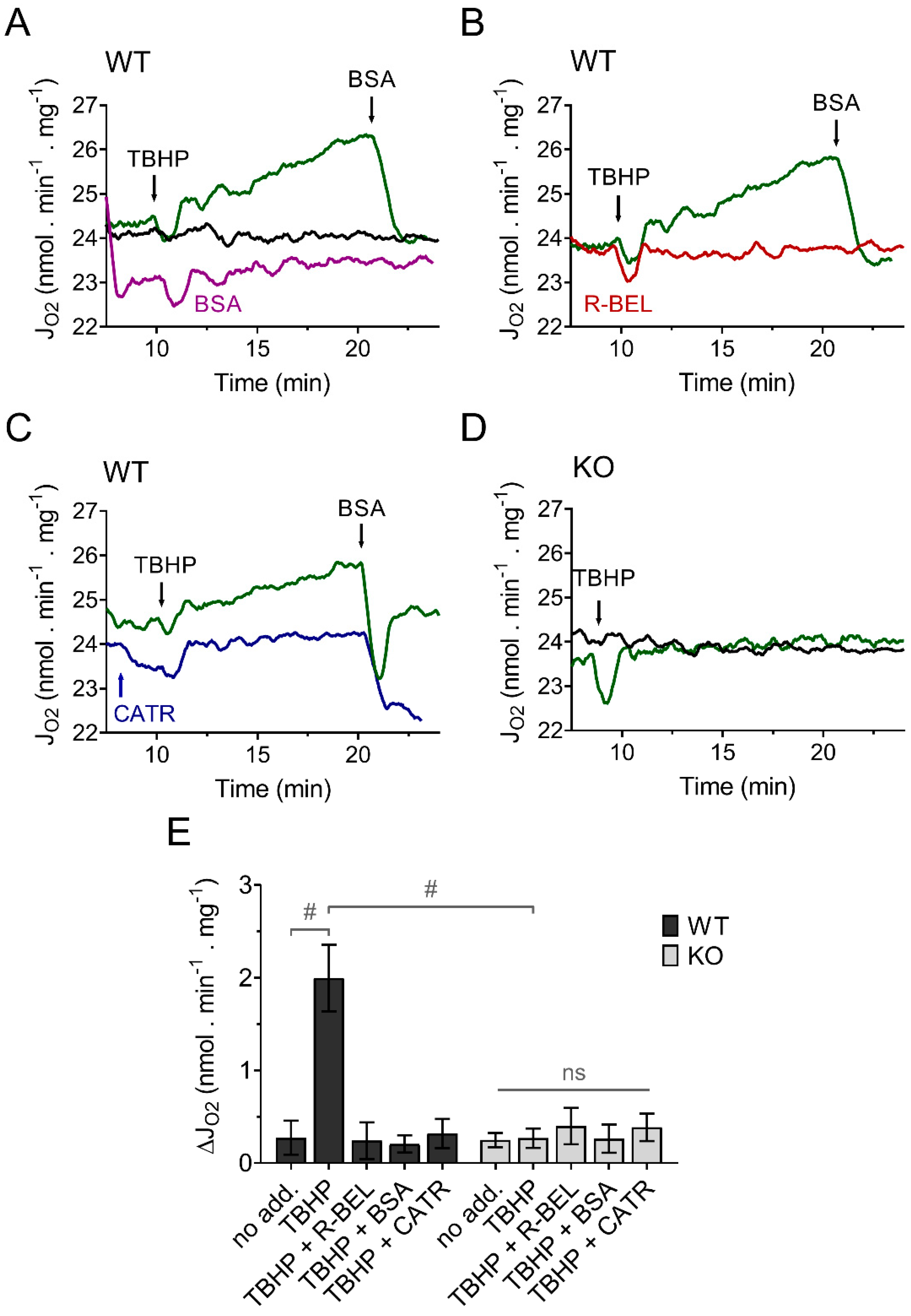
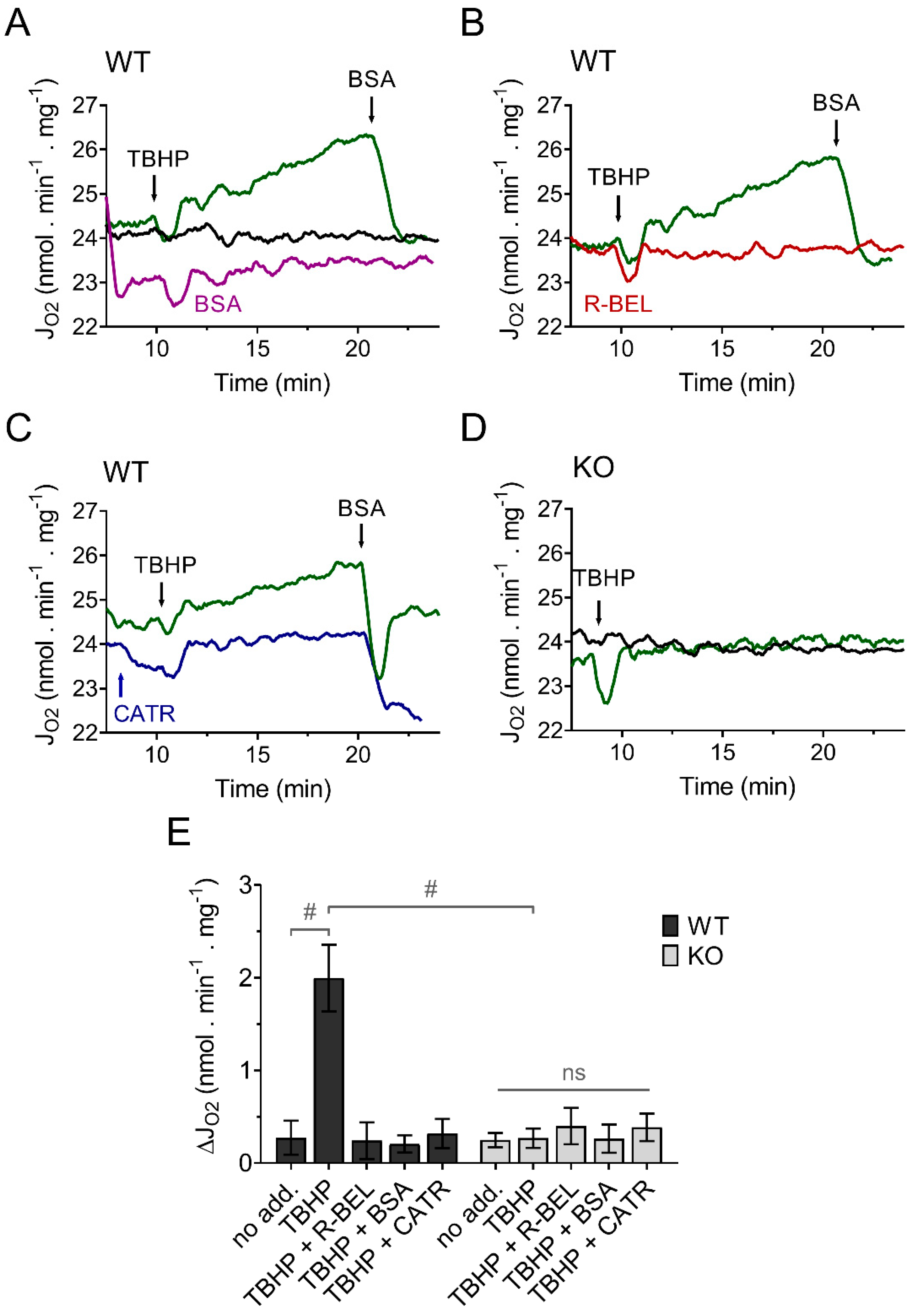
3.1.1. Uncovering the Redox Activation of iPLA2γ in Isolated Brain Mitochondria
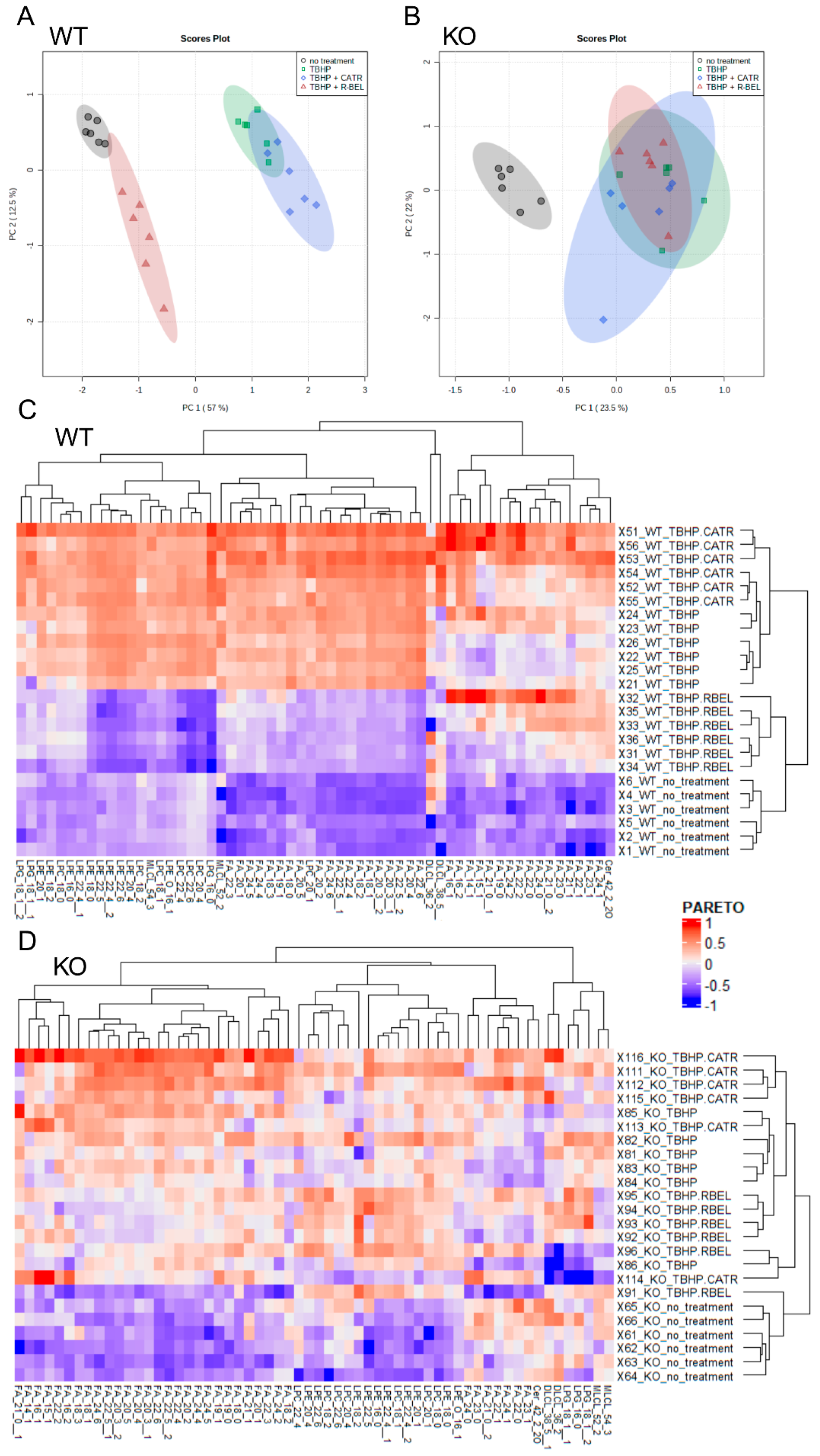
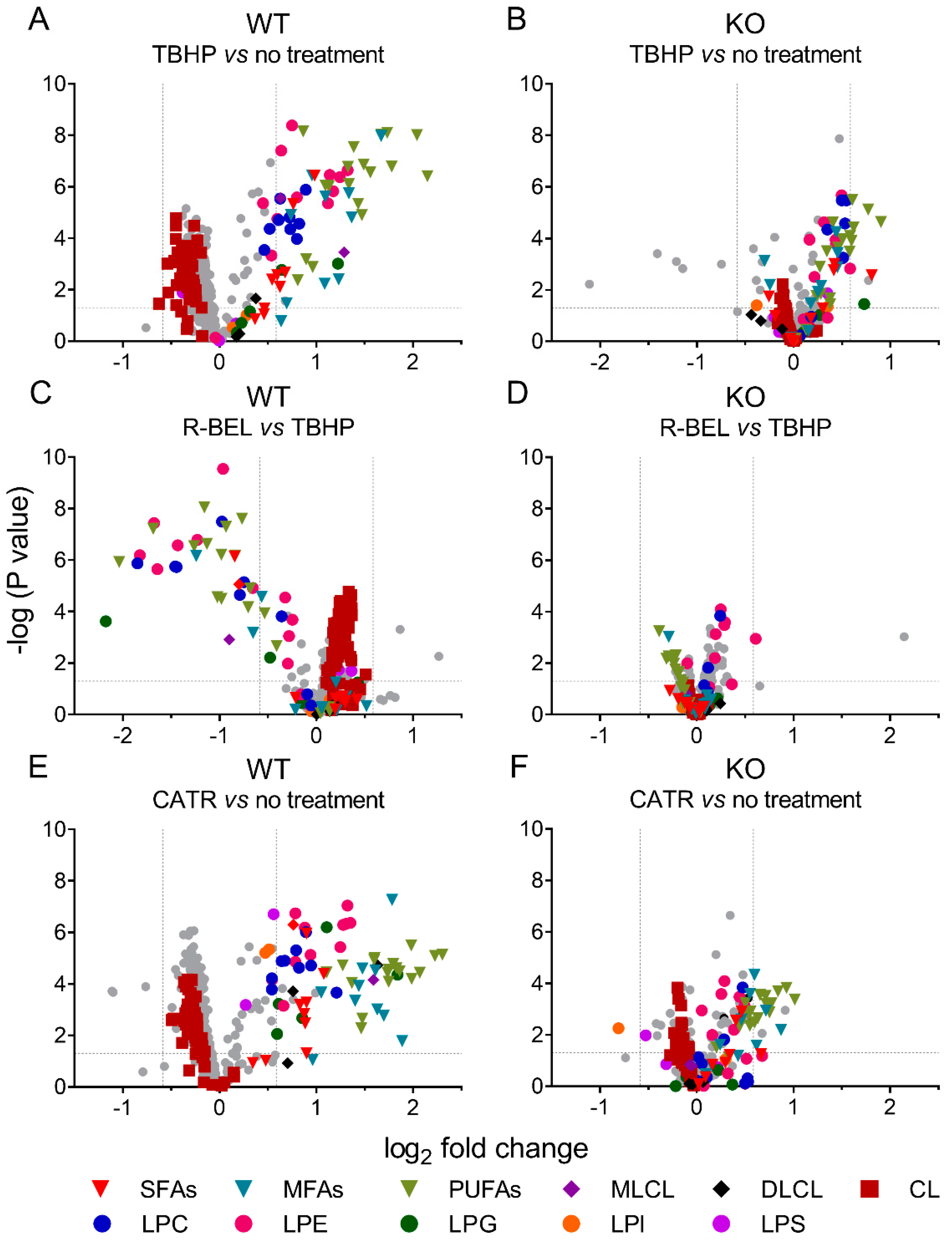
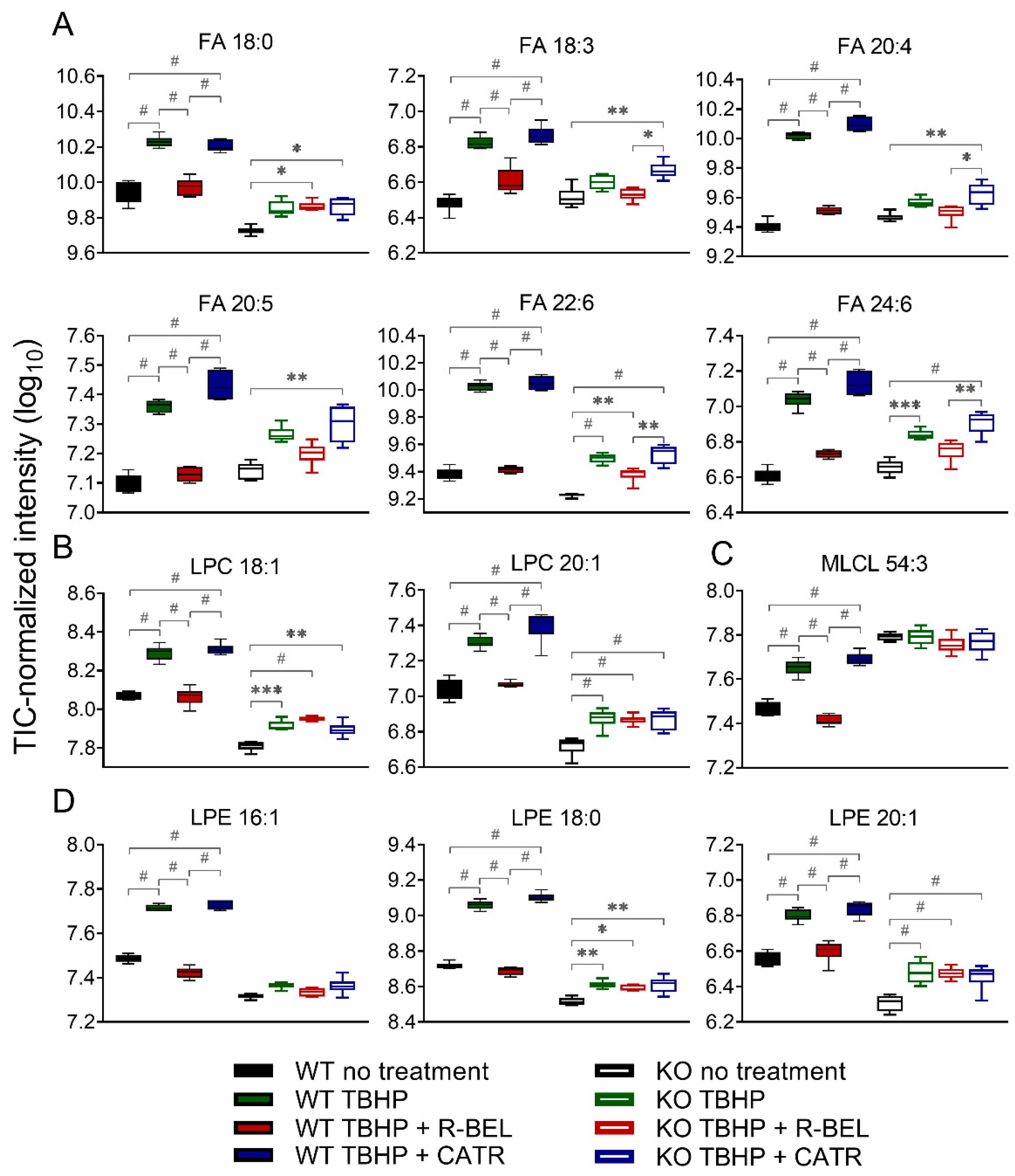
3.1.2. Detailed Lipidomic Analyses Reveal the Main Products of the TBHP-Activated iPLA2γ in Brain Mitochondria
3.1.3. Univariate Analyses of Lipidomic Data Demonstrate a More Detailed Pattern of iPLA2γ Reaction Products
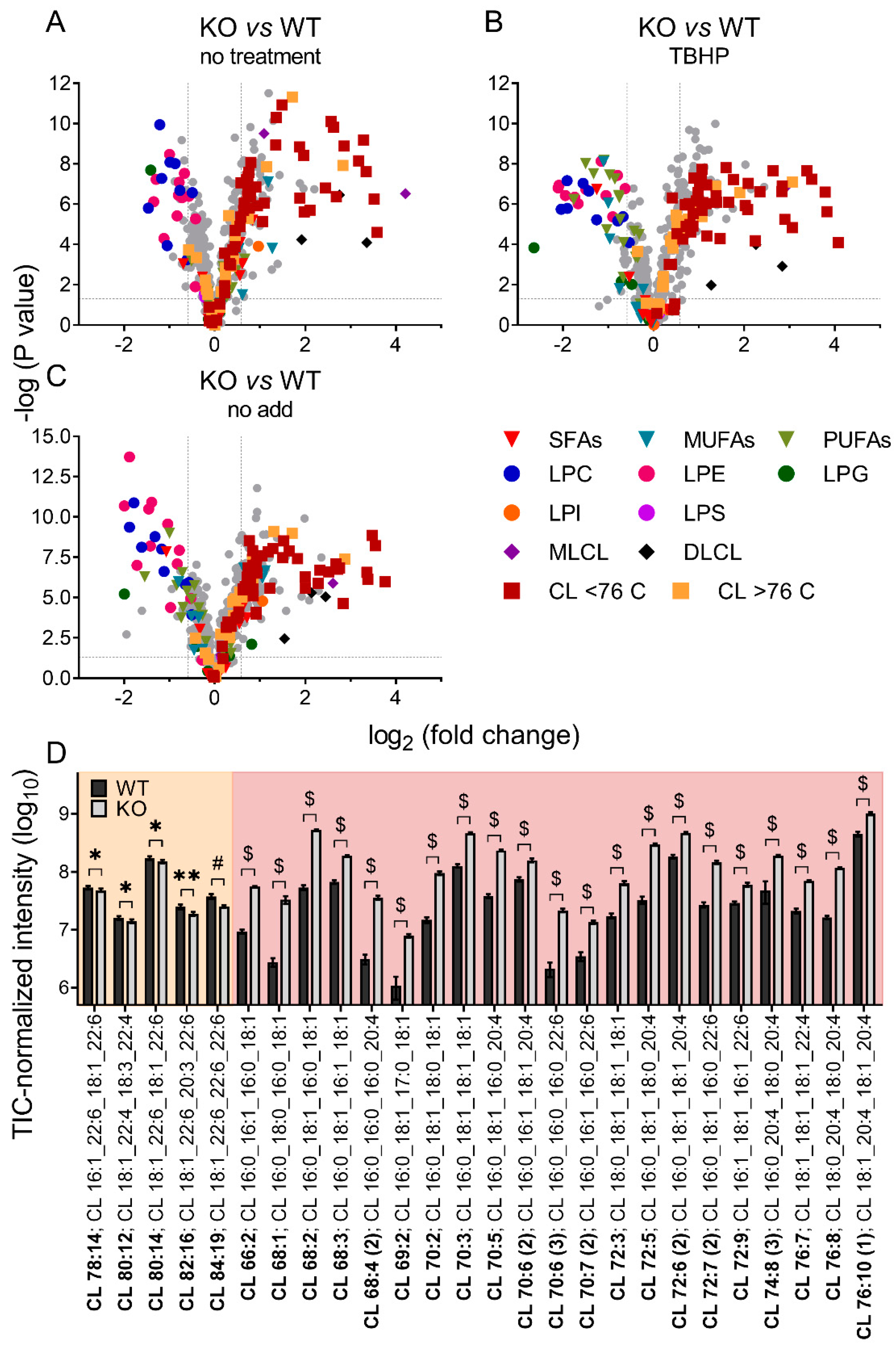
3.2. Differences in Brain Mitochondrial Lipid Composition of iPLA2γ- KO Mice
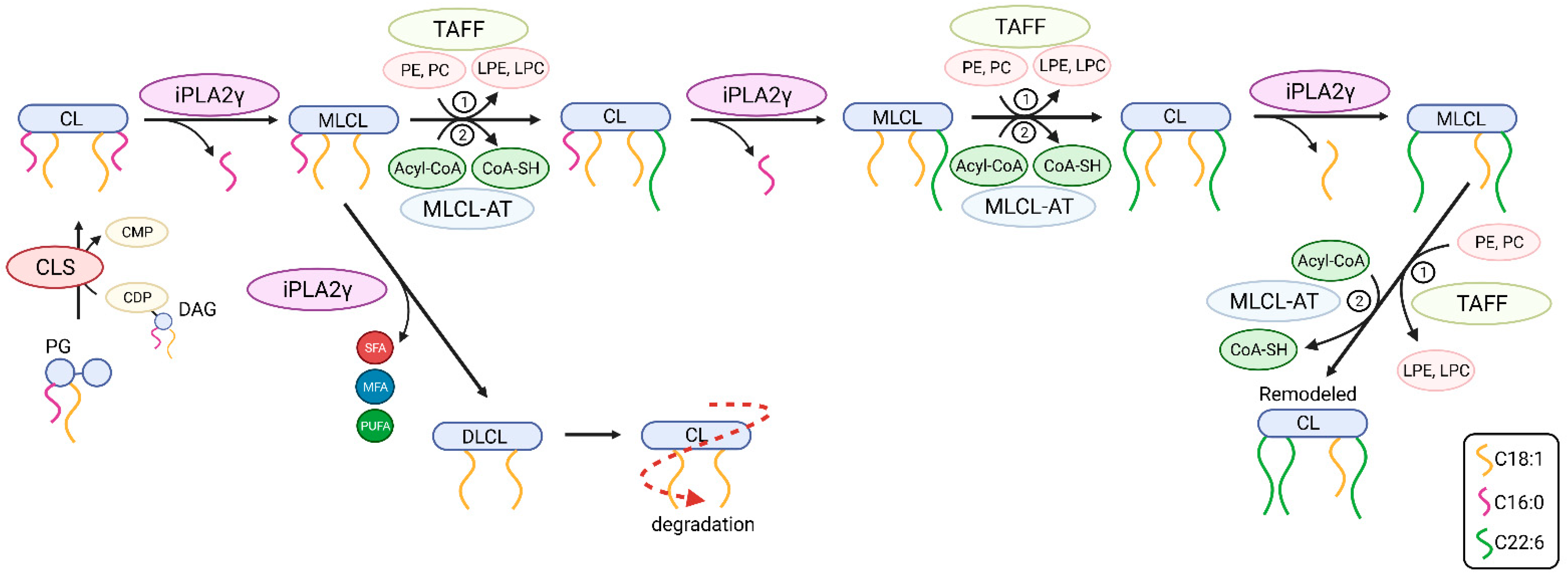
Mitochondrial iPLA2γ Participates in Cardiolipin Remodeling
3.3. Antioxidant Role of iPLA2γ-Released Fatty Acids in Isolated Brain Mitochondria
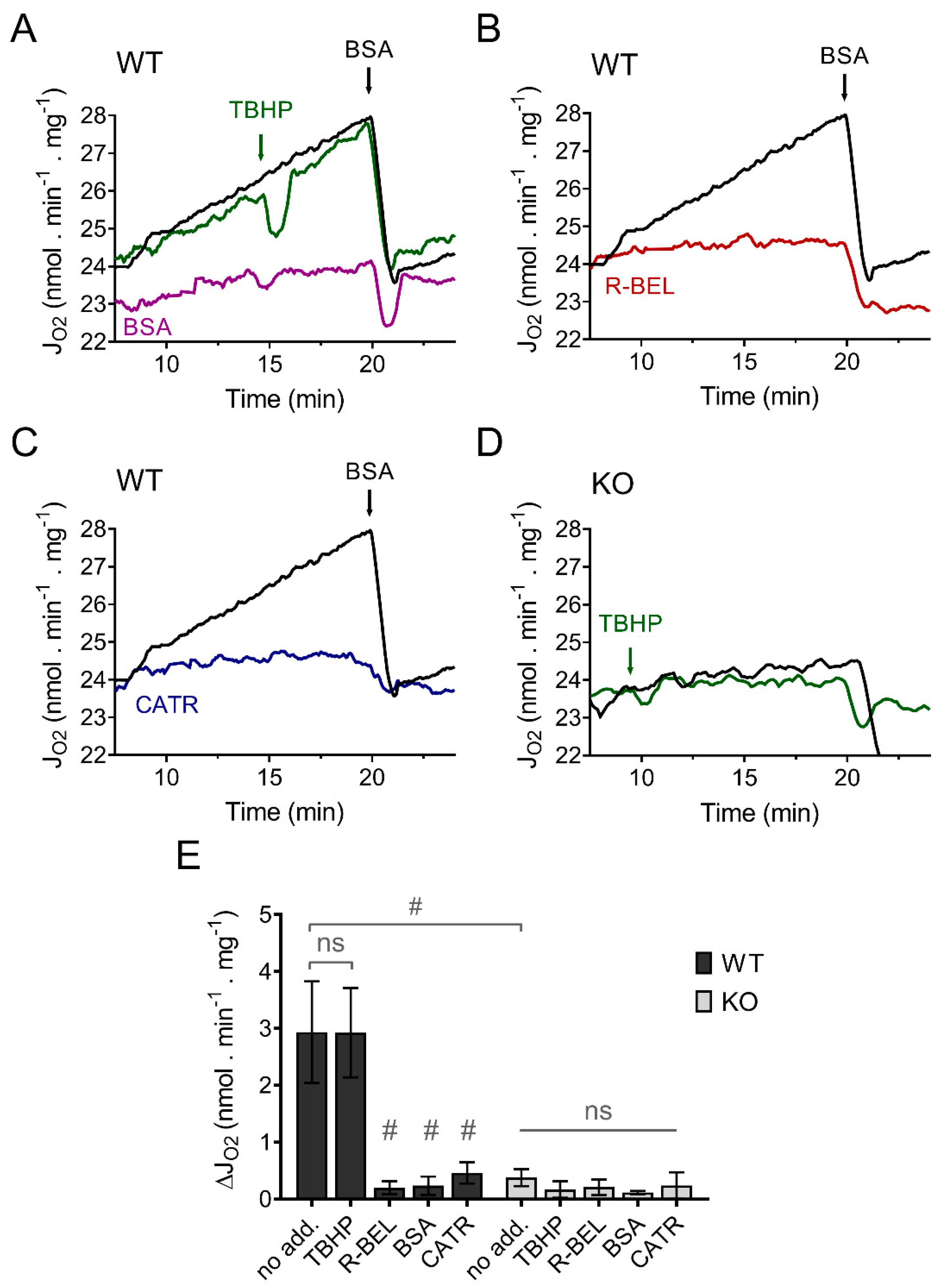
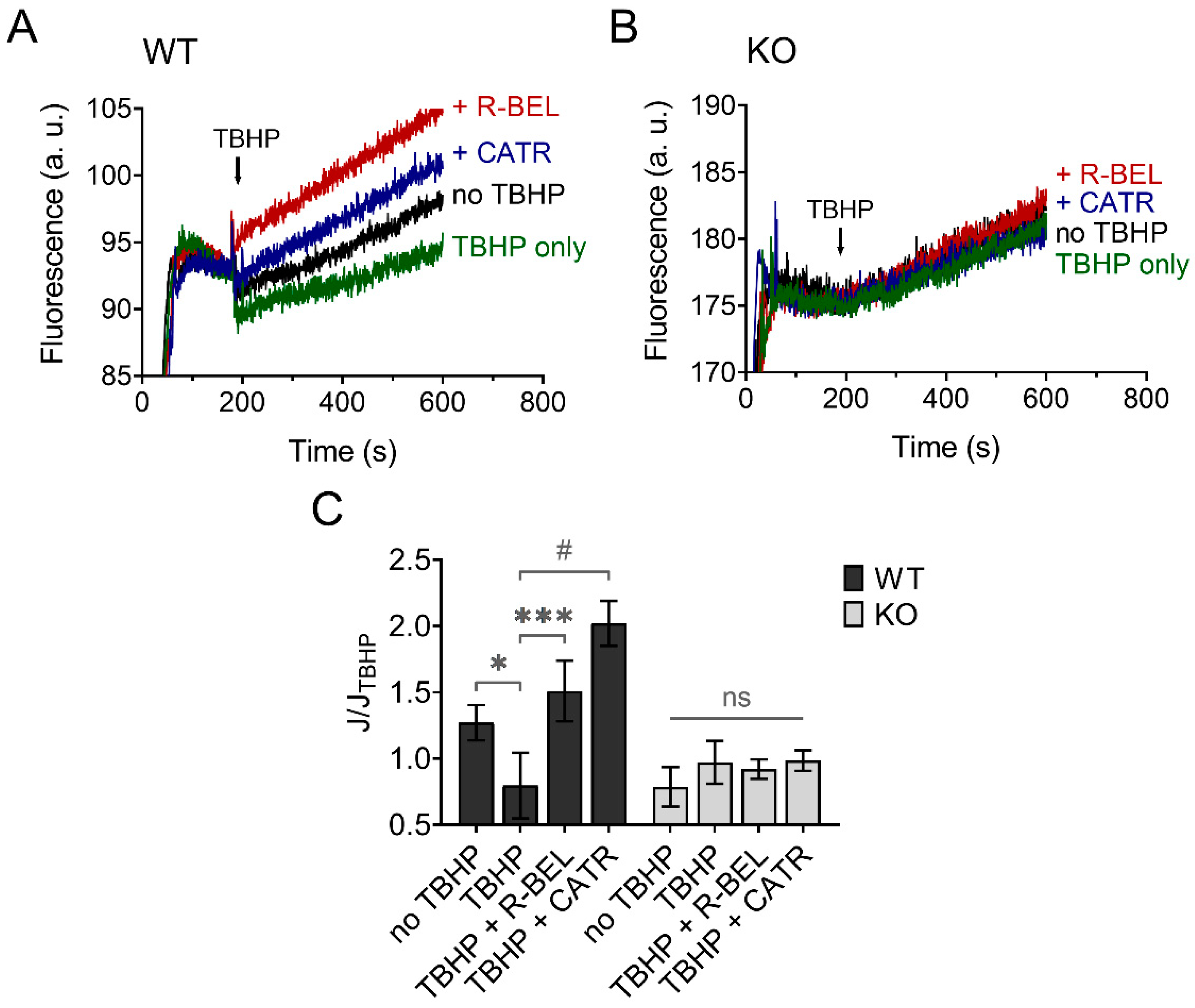
3.3.1. Mitochondrial Superoxide Release into the Matrix Decreases following Redox Activation of iPLA2γ
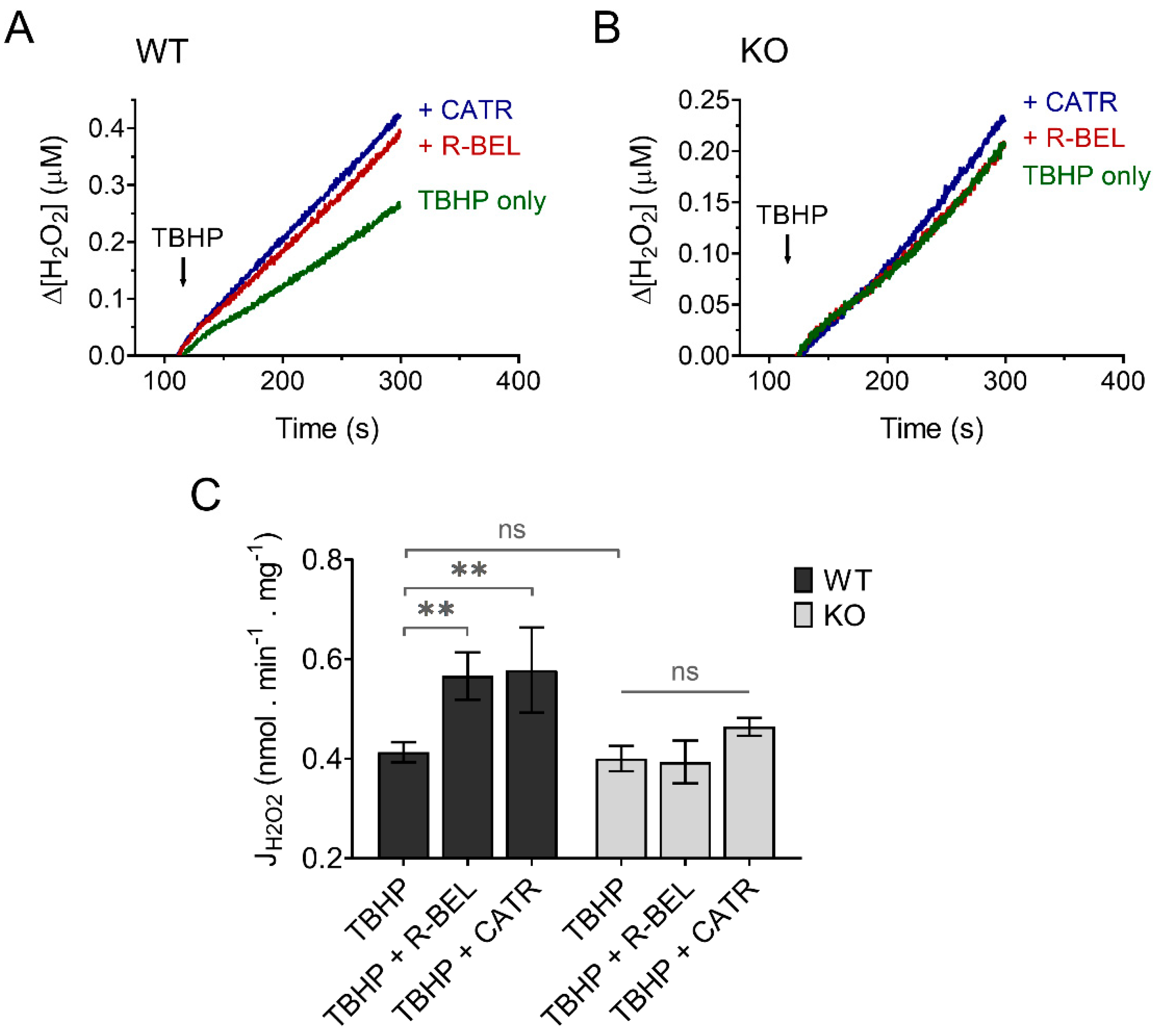
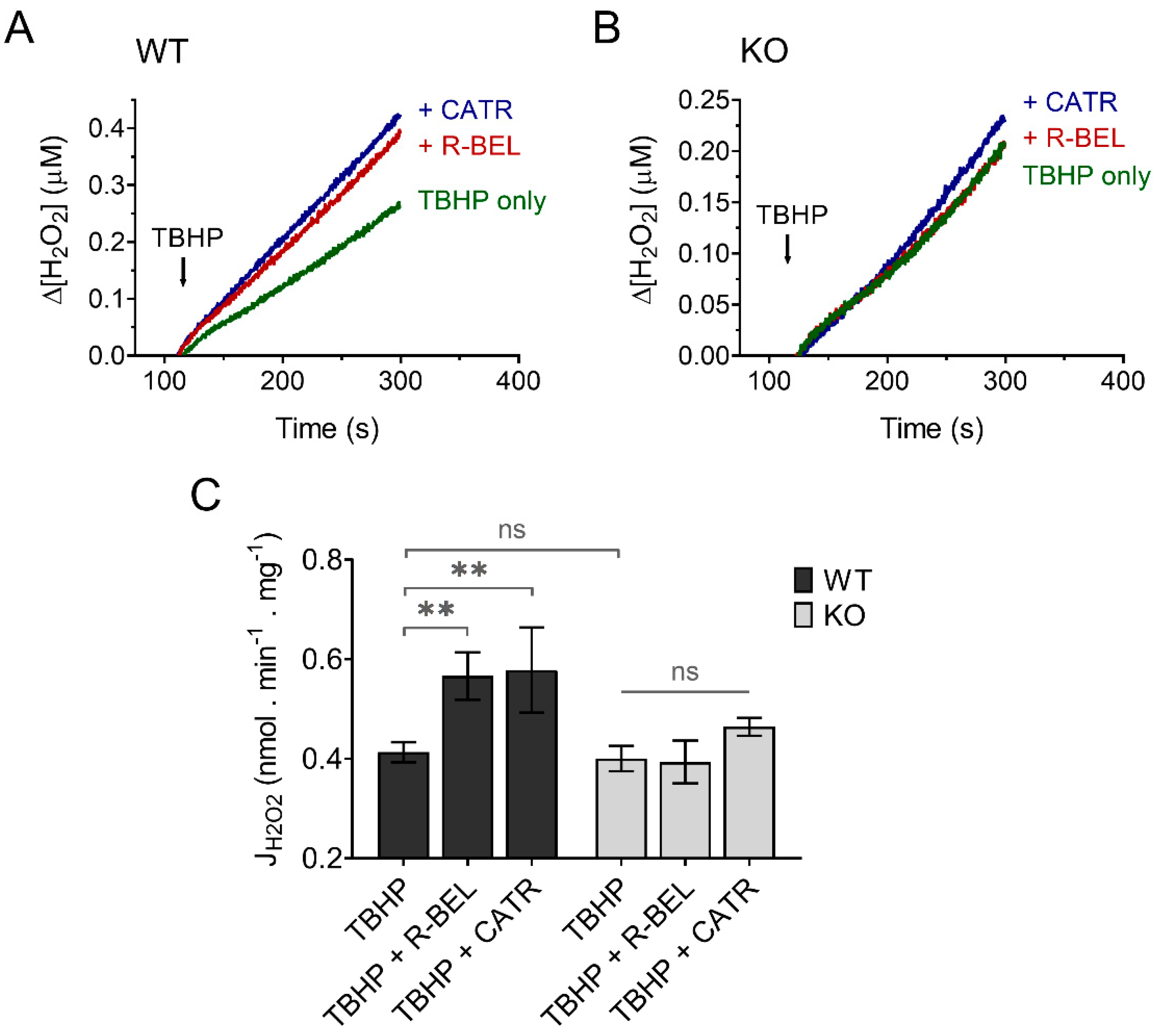
3.3.2. Extramitochondrial H2O2 Release Decreases following Redox Activation of iPLA2γ
3.4. Antioxidant Role of iPLA2γ In Vivo
3.4.1. iPLA2γ—Dependent Antioxidant Function Leads to Decreased Protein Carbonyl Content in the Brain
3.4.2. iPLA2γ–ANT Antioxidant Synergy Decreases the Levels of Inflammatory Marker IL-6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Crosstalk between Neuron and Glial Cells in Oxidative Injury and Neuroprotection. Int. J. Mol. Sci. 2021, 22, 13315. [Google Scholar] [CrossRef]
- Vinokurov, A.Y.; Stelmashuk, O.A.; Ukolova, P.A.; Zherebtsov, E.A.; Abramov, A.Y. Brain region specificity in reactive oxygen species production and maintenance of redox balance. Free Radic. Biol. Med. 2021, 174, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.J.; Yousefian-Jazi, A.; Hyeon, S.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef]
- Cantó-Santos, J.; Grau-Junyent, J.M.; Garrabou, G. The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Vargas, M.R. Redox Biology in Neurological Function, Dysfunction, and Aging. Antioxid. Redox Signal. 2018, 28, 1583–1586. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. How the brain fights fatty acids’ toxicity. Neurochem. Int. 2021, 148, 105050. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, A.J.; Tian, J.; Zimmerman, M.C. Increased mitochondrial superoxide in the brain, but not periphery, sensitizes mice to angiotensin II-mediated hypertension. Redox Biol. 2017, 11, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.S.; Sullivan, K.A.; Wilkinson, J.E.; Backus, C.; Hayes, J.M.; Sakowski, S.A.; Feldman, E.L. Neurodegeneration and early lethality in superoxide dismutase 2-deficient mice: A comprehensive analysis of the central and peripheral nervous systems. Neuroscience 2012, 212, 201–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinerfeld, D.; Traini, M.D.; Weinberger, R.P.; Cochran, B.; Doctrow, S.R.; Harry, J.; Melov, S. Endogenous mitochondrial oxidative stress: Neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J. Neurochem. 2004, 88, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Ježek, P.; Plecitá-Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Ježek, P.; Plecitá-Hlavatá, L. Mitochondrial reticulum network dynamics in relation to oxidative stress, redox regulation, and hypoxia. Int. J. Biochem. Cell Biol. 2009, 41, 1790–1804. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Plecitá-Hlavatá, L. Redox Signaling from Mitochondria: Signal Propagation and Its Targets. Biomolecules 2020, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 Removal in Brain Mitochondria via the Thioredoxin/Peroxiredoxin System. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Micioni Di Bonaventura, M.V.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 2017, 109, 68–77. [Google Scholar] [CrossRef]
- Niki, E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Izumi, Y.; Zorumski, C.F.; Gross, R.W. Long-term potentiation requires activation of calcium-independent phospholipase A2. FEBS Lett. 1995, 377, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Jenkins, C.M.; Gross, R.W. The Genomic Organization, Complete mRNA Sequence, Cloning, and Expression of a Novel Human Intracellular Membrane-associated Calcium-independent Phospholipase A2. J. Biol. Chem. 2000, 275, 9937–9945. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Jenkins, C.M.; Han, X.; Mancuso, D.J.; Sims, H.F.; Yang, K.; Gross, R.W. The Highly Selective Production of 2-Arachidonoyl Lysophosphatidylcholine Catalyzed by Purified Calcium-independent Phospholipase A2γ: Identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signali. J. Biol. Chem. 2005, 280, 26669–26679. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-Y.; Moon, S.H.; Jenkins, C.M.; Li, M.; Sims, H.F.; Guan, S.; Gross, R.W. The phospholipase iPLA2γ is a major mediator releasing oxidized aliphatic chains from cardiolipin, integrating mitochondrial bioenergetics and signaling. J. Biol. Chem. 2017, 292, 10672–10684. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, D.J.; Kotzbauer, P.; Wozniak, D.F.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Han, X.; Yang, K.; Sun, G.; Malik, I.; et al. Genetic Ablation of Calcium-independent Phospholipase A2γ Leads to Alterations in Hippocampal Cardiolipin Content and Molecular Species Distribution, Mitochondrial Degeneration, Autophagy, and Cognitive Dysfunction. J. Biol. Chem. 2009, 284, 35632–35644. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.; Liu, Y.; Fu, X.; Xu, X.; Bao, Z.; Lin, C.; Li, Z.; Liu, Y.; Wang, X.; You, Y.; et al. Lowered iPLA2γ activity causes increased mitochondrial lipid peroxidation and mitochondrial dysfunction in a rotenone-induced model of Parkinson’s disease. Exp. Neurol. 2018, 300, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Saneto, R.P.; Hebbar, M.; Mirzaa, G.; Girisha, K.M. A neurodegenerative mitochondrial disease phenotype due to biallelic loss-of-function variants in PNPLA8 encoding calcium-independent phospholipase A2γ. Am. J. Med. Genet. Part A 2018, 176, 1232–1237. [Google Scholar] [CrossRef]
- Chao, H.; Anthonymuthu, T.S.; Kenny, E.M.; Amoscato, A.A.; Cole, L.K.; Hatch, G.M.; Ji, J.; Kagan, V.E.; Bayır, H. Disentangling oxidation/hydrolysis reactions of brain mitochondrial cardiolipins in pathogenesis of traumatic injury. JCI Insight 2018, 3, e97677. [Google Scholar] [CrossRef] [Green Version]
- Ježek, J.; Jabůrek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [CrossRef]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H2O2-Activated Mitochondrial Phospholipase iPLA2γ Prevents Lipotoxic Oxidative Stress in Synergy with UCP2, Amplifies Signaling via G-Protein–Coupled Receptor GPR40, and Regulates Insulin Secretion in Pancreatic β-Cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaburek, M.; Jezek, J.; Zelenka, J.; Ježek, P. Antioxidant activity by a synergy of redox-sensitive mitochondrial phospholipase A2 and uncoupling protein-2 in lung and spleen. Int. J. Biochem. Cell Biol. 2013, 45, 816–825. [Google Scholar] [CrossRef]
- Jaburek, M.; Garlid, K.D. Reconstitution of Recombinant Uncoupling Proteins. UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [Green Version]
- Jabůrek, M.; Varecha, M.; Gimeno, R.E.; Dembski, M.; Jezek, P.; Zhang, M.; Burn, P.; Tartaglia, L.A.; Garlid, K.D. Transport Function and Regulation of Mitochondrial Uncoupling Proteins 2 and 3. J. Biol. Chem. 1999, 274, 26003–26007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaburek, M.; Miyamoto, S.; Di Mascio, P.; Garlid, K.; Ježek, P. Hydroperoxy Fatty Acid Cycling Mediated by Mitochondrial Uncoupling Protein UCP2. J. Biol. Chem. 2004, 279, 53097–53102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Kazak, L.; Chouchani, E.T.; Bogaczyńska, M.G.; Paranjpe, I.; Wainwright, G.L.; Bétourné, A.; Kajimura, S.; Spiegelman, B.M.; Kirichok, Y. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab. 2017, 25, 811–822.e4. [Google Scholar] [CrossRef] [Green Version]
- Wojtczak, L.; Wiȩckowski, M.R. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. J. Bioenerg. Biomembr. 1999, 31, 447–455. [Google Scholar] [CrossRef]
- Wojtczak, L.; Wiȩckowski, M.R.; Schönfeld, P. Protonophoric Activity of Fatty Acid Analogs and Derivatives in the Inner Mitochondrial Membrane: A Further Argument for the Fatty Acid Cycling Model. Arch. Biochem. Biophys. 1998, 357, 76–84. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef]
- Ježek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Jabůrek, M.; Průchová, P.; Holendová, B.; Galkin, A.; Ježek, P. Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters. Antioxidants 2021, 10, 678. [Google Scholar] [CrossRef]
- Kašpárek, P.; Krausova, M.; Haneckova, R.; Kriz, V.; Zbodakova, O.; Korinek, V.; Sedlacek, R. Efficient gene targeting of theRosa26locus in mouse zygotes using TALE nucleases. FEBS Lett. 2014, 588, 3982–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, A.; Kahl, A.; Konrad, C.; Ten, V.; Starkov, A.; Galkin, A. Reverse electron transfer results in a loss of flavin from mitochondrial complex I: Potential mechanism for brain ischemia reperfusion injury. J. Cereb. Blood Flow Metab. 2017, 37, 3649–3658. [Google Scholar] [CrossRef] [Green Version]
- Plecitá-Hlavatá, L.; Engstová, H.; Holendová, B.; Tauber, J.; Špaček, T.; Petrásková, L.; Křen, V.; Špačková, J.; Gotvaldová, K.; Ježek, J.; et al. Mitochondrial Superoxide Production Decreases on Glucose-Stimulated Insulin Secretion in Pancreatic β Cells Due to Decreasing Mitochondrial Matrix NADH/NAD+Ratio. Antioxid. Redox Signal. 2020, 33, 789–815. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A Stable Nonfluorescent Derivative of Resorufin for the Fluorometric Determination of Trace Hydrogen Peroxide: Applications in Detecting the Activity of Phagocyte NADPH Oxidase and Other Oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Janovska, P.; Melenovsky, V.; Svobodova, M.; Havlenova, T.; Kratochvilova, H.; Haluzik, M.; Hoskova, E.; Pelikanova, T.; Kautzner, J.; Monzo, L.; et al. Dysregulation of epicardial adipose tissue in cachexia due to heart failure: The role of natriuretic peptides and cardiolipin. J. Cachexia Sarcopenia Muscle 2020, 11, 1614–1627. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990, 186, 464–478. [Google Scholar] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. Spec. Publ. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Grace, S.C.; Hudson, D.A. Processing and Visualization of Metabolomics Data Using R. In Metabolomics—Fundamentals and Applications; IntechOpen, 2016; ISBN 978-953-51-2854-0. Available online: https://www.intechopen.com/chapters/52527 (accessed on 27 December 2021). [CrossRef] [Green Version]
- Pollard, A.K.; Ortori, C.A.; Stöger, R.; Barrett, D.; Chakrabarti, L. Mouse mitochondrial lipid composition is defined by age in brain and muscle. Aging 2017, 9, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Munro, D.; Banh, S.; Sotiri, E.; Tamanna, N.; Treberg, J.R. The thioredoxin and glutathione-dependent H2O2 consumption pathways in muscle mitochondria: Involvement in H2O2 metabolism and consequence to H2O2 efflux assays. Free Radic. Biol. Med. 2016, 96, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Jaburek, M.; Jezek, J.; Ježek, P. Cytoprotective activity of mitochondrial uncoupling protein-2 in lung and spleen. FEBS Open Bio. 2018, 8, 692–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef]
- Nordmann, C.; Strokin, M.; Schönfeld, P.; Reiser, G. Putative roles of Ca2+ -independent phospholipase A2 in respiratory chain-associated ROS production in brain mitochondria: Influence of docosahexaenoic acid and bromoenol lactone. J. Neurochem. 2014, 131, 163–176. [Google Scholar] [CrossRef]
- Semenovich, D.S.; Plotnikov, E.Y.; Titko, O.V.; Lukiyenko, E.P.; Kanunnikova, N.P. Effects of Panthenol and N-Acetylcysteine on Changes in the Redox State of Brain Mitochondria under Oxidative Stress In Vitro. Antioxidants 2021, 10, 1699. [Google Scholar] [CrossRef]
- Cheng, H.; Mancuso, D.J.; Jiang, X.; Guan, S.; Yang, J.; Yang, K.; Sun, G.; Gross, R.W.; Han, X. Shotgun Lipidomics Reveals the Temporally Dependent, Highly Diversified Cardiolipin Profile in the Mammalian Brain: Temporally Coordinated Postnatal Diversification of Cardiolipin Molecular Species with Neuronal Remodeling. Biochemistry 2008, 47, 5869–5880. [Google Scholar] [CrossRef] [Green Version]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291.e4. [Google Scholar] [CrossRef]
- Hayashi, D.; Mouchlis, V.D.; Dennis, E.A. Each phospholipase A2 type exhibits distinct selectivity toward sn-1 ester, alkyl ether, and vinyl ether phospholipids. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2022, 1867, 159067. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layé, S.; Nadjar, A.; Joffre, C.; Bazinet, R.P. Anti-Inflammatory Effects of Omega-3 Fatty Acids in the Brain: Physiological Mechanisms and Relevance to Pharmacology. Pharmacol. Rev. 2018, 70, 12–38. [Google Scholar] [CrossRef]
- Yang, B.; Fritsche, K.L.; Beversdorf, D.Q.; Gu, Z.; Lee, J.C.; Folk, W.R.; Greenlief, C.M.; Sun, G.Y. Yin-Yang Mechanisms Regulating Lipid Peroxidation of Docosahexaenoic Acid and Arachidonic Acid in the Central Nervous System. Front. Neurol. 2019, 10, 642. [Google Scholar] [CrossRef]
- Green, J.T.; Orr, S.K.; Bazinet, R.P. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J. Lipid Res. 2008, 49, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, Y.; Kim, H.-W.; Igarashi, M.; Modi, H.R.; Chang, L.; Ma, K.; Greenstein, D.; Wohltmann, M.; Turk, J.; Rapoport, S.I.; et al. Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A2-VIA (iPLA2β)-knockout mice. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, H.; Taha, A.Y.; Cheon, Y.; Kim, H.-W.; Turk, J.; Rapoport, S.I. iPLA2β Knockout Mouse, a Genetic Model for Progressive Human Motor Disorders, Develops Age-Related Neuropathology. Neurochem. Res. 2014, 39, 1522–1532. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Průchová, P.; Gotvaldová, K.; Smolková, K.; Alán, L.; Holendová, B.; Tauber, J.; Galkin, A.; Ježek, P.; Jabůrek, M. Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain. Antioxidants 2022, 11, 198. https://doi.org/10.3390/antiox11020198
Průchová P, Gotvaldová K, Smolková K, Alán L, Holendová B, Tauber J, Galkin A, Ježek P, Jabůrek M. Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain. Antioxidants. 2022; 11(2):198. https://doi.org/10.3390/antiox11020198
Chicago/Turabian StylePrůchová, Pavla, Klára Gotvaldová, Katarína Smolková, Lukáš Alán, Blanka Holendová, Jan Tauber, Alexander Galkin, Petr Ježek, and Martin Jabůrek. 2022. "Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain" Antioxidants 11, no. 2: 198. https://doi.org/10.3390/antiox11020198
APA StylePrůchová, P., Gotvaldová, K., Smolková, K., Alán, L., Holendová, B., Tauber, J., Galkin, A., Ježek, P., & Jabůrek, M. (2022). Antioxidant Role and Cardiolipin Remodeling by Redox-Activated Mitochondrial Ca2+-Independent Phospholipase A2γ in the Brain. Antioxidants, 11(2), 198. https://doi.org/10.3390/antiox11020198