

Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress

Abstract
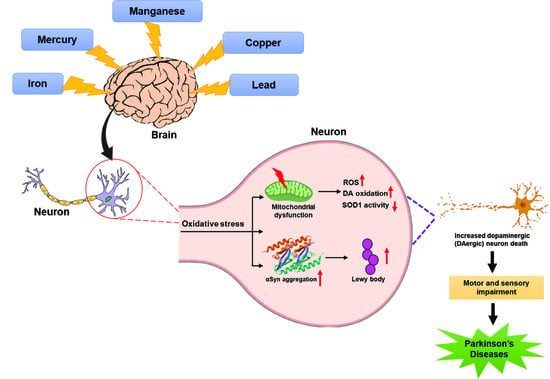
1. Introduction

{kind=link}
{kind=link}
{kind=link}
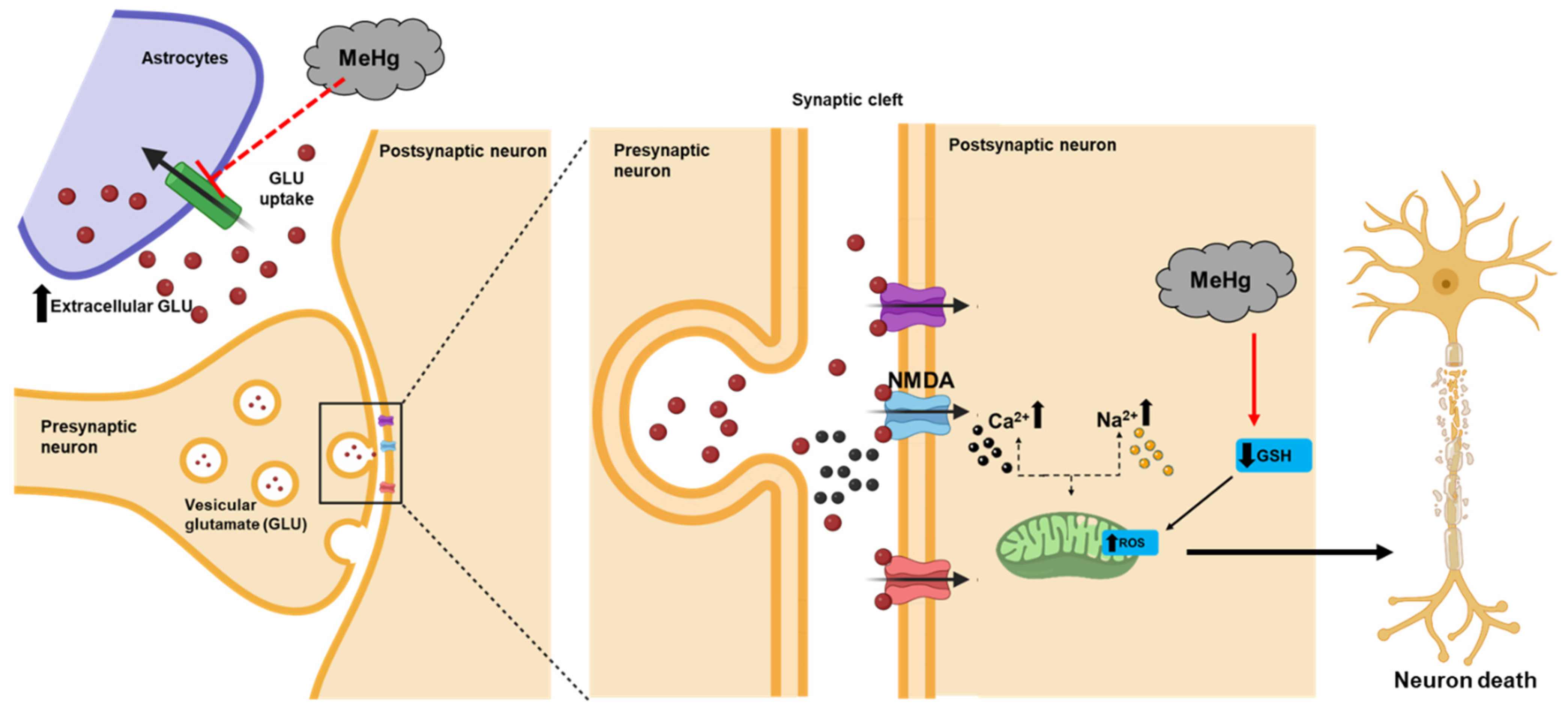{kind=link}
{kind=link}
{kind=link}
| Metal | Type of Study | Sample Size (Case/Control) | Geographical Region | Main Findings | Reference |
|---|---|---|---|---|---|
| Fe, Cu, Pb | Case-control | 150/170 | India | Level of metals in plasma was positively correlated with PD: Fe (r = 0.29, p < 0.001), Pb (r = 0.16, p = 0.007), and Cu (r = 0.11, p = 0.047) | [12] |
| Fe, Mn, Cu, Pb | Case-control | 144/464 | USA | Exposure exceeding 20 years of exposure to Mn (OR = 10.61, 95% CI = 1.06, 105.83) and Cu (OR = 2.49, 95% CI = 1.06, 5.89) was associated with PD. Combination of Pb and Cu (OR = 5.24, 95% CI = 0.59, 17.21), Pb and Fe (OR = 2.83, 95% CI = 1.07, 7.50), and Fe and Cu (OR = 5.24, 95% CI = 1.40, 9.71) was associated with PD | [13] |
| Pb | Case-control | 121/414 | USA | Pb exposure for lifetime exposure increase for PD (OR = 2.27, 95% CI = 1.13, 4.55). | [14] |
| Pb | Case-control | 330/308 | USA | Compared with the lowest quartile of tibia Pb, the OR for PD in the highest quartile was 3.21 (95% CI = 1.17, 8.83) | [15] |
| Hg | Case-control | 54/95 | Singapore | The logarithmic unit elevate in blood and urine Hg is associated with 21.0 (p < 0.05) or 18.65 times increase in risk of PD. | [16] |
| Hg | Case-control | 17/15 | Taiwan | A significantly negative correlation between urine Hg level and uptake ratio in the striatum, caudate, putamen (r = −0.501, p = 0.040; r = −0.635, p = 0.006; r = −0.559, p = 0.020, respectively) | [17] |
2. Metals and Parkinson’s Disease
2.1. Iron and Parkinson’s Disease
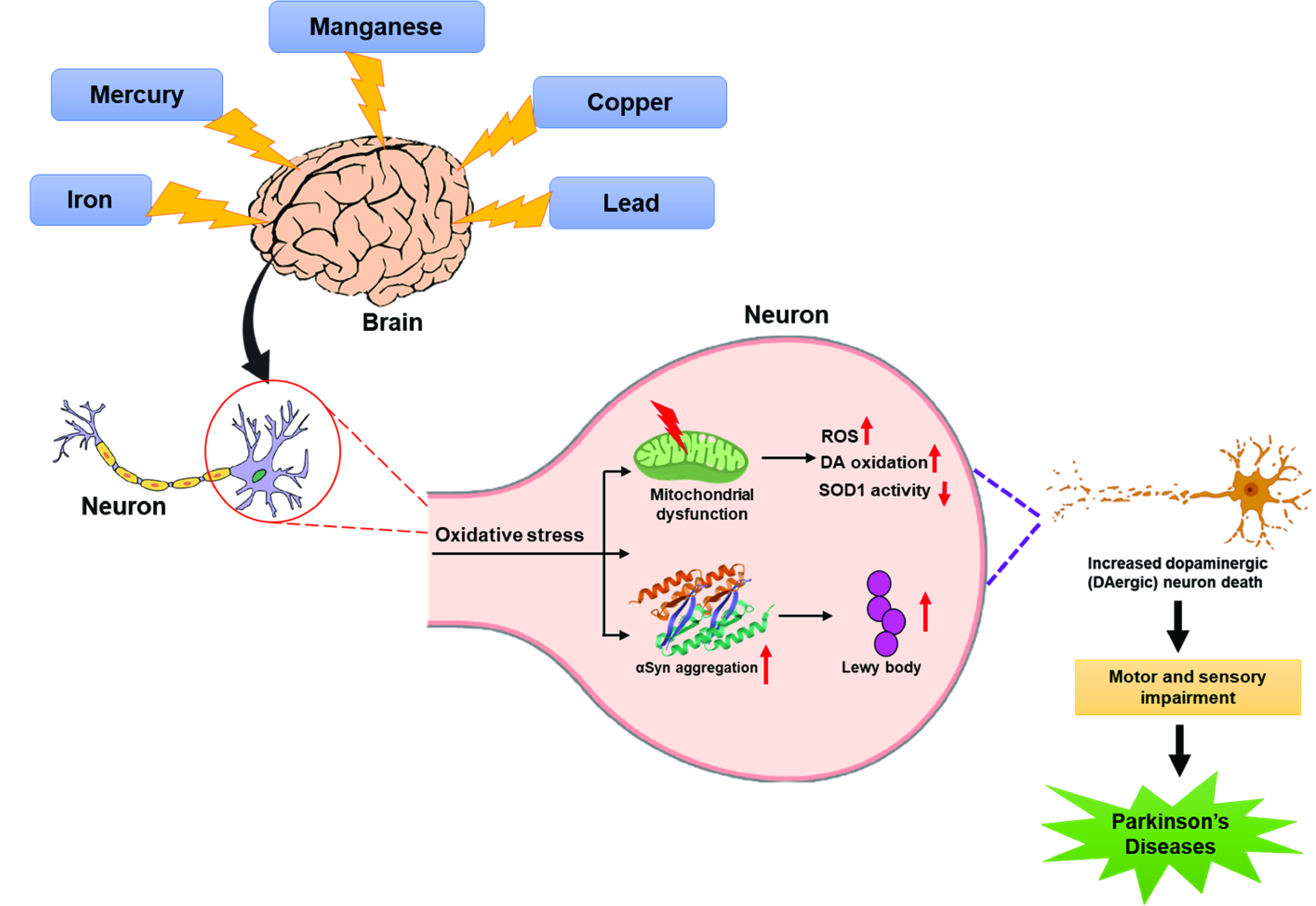
2.2. Manganese and Parkinson’s Disease
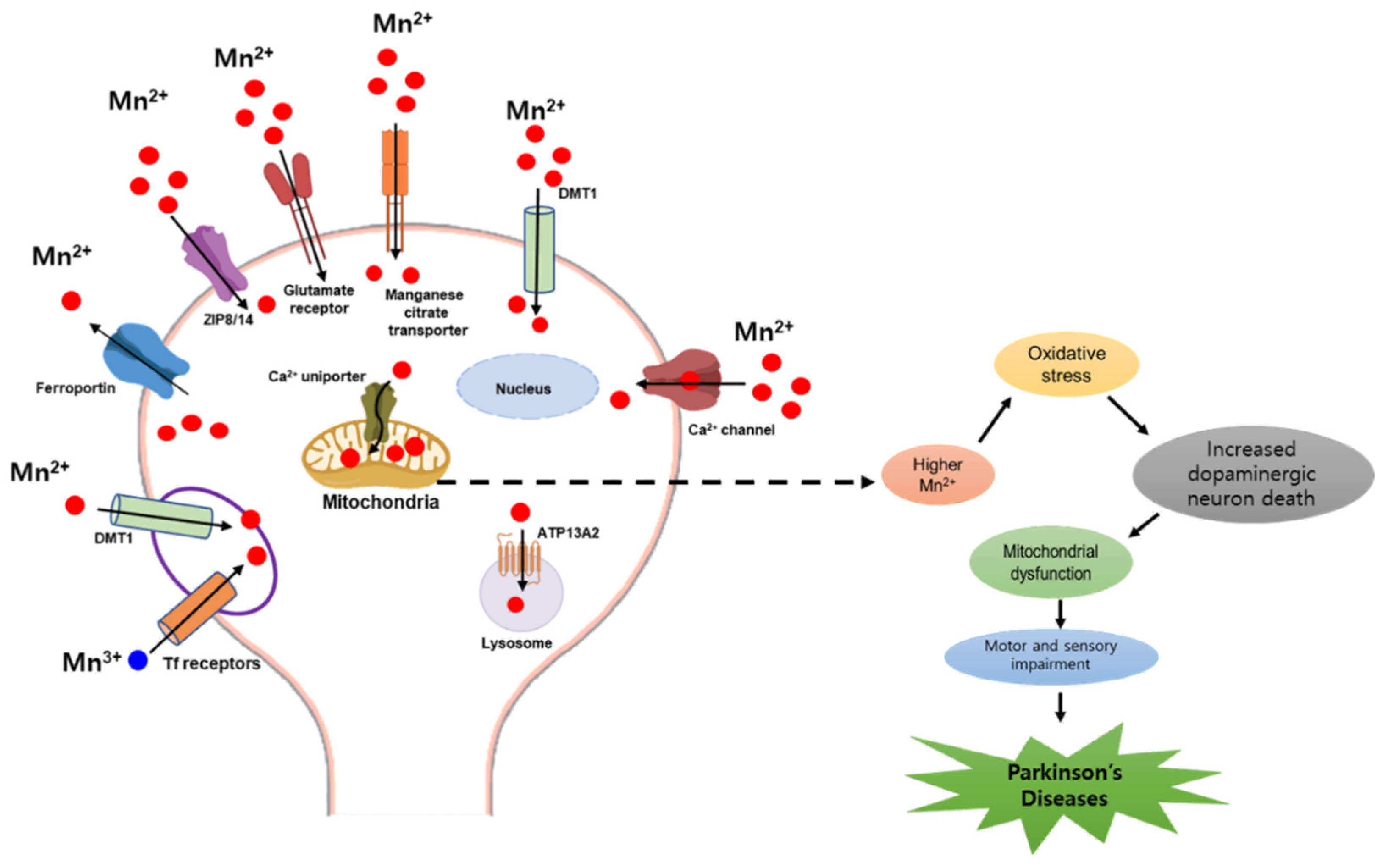
2.3. Mercury and Parkinson’s Disease
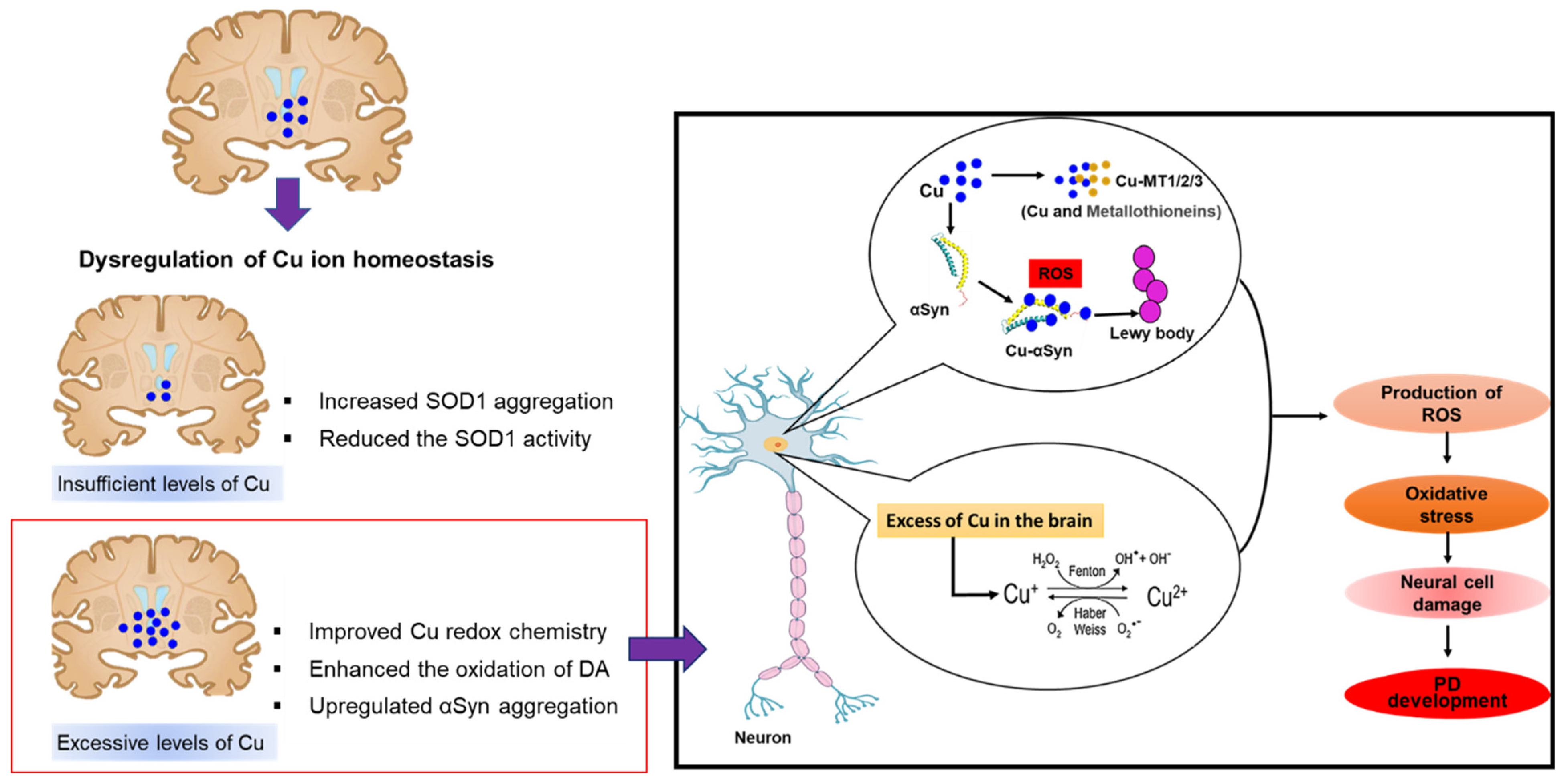
2.4. Copper and Parkinson’s Disease
2.5. Lead and Parkinson’s Disease
3. Metal Mixtures and Parkinson’s Disease
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The emerging evidence of the Parkinson pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, D.; Jeong, J.H.; Yook, S. Development of immunotherapy and nanoparticles-based strategies for the treatment of Parkinson’s disease. J. Pharm. Investig. 2021, 51, 465–481. [Google Scholar]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, A.; Hiba, O.E.; Gamrani, H. Copper poisoning induces neurobehavioral features of Parkinson’s disease in rat: Alters dopaminergic system and locomotor performance. Park. Relat. Disord. 2016, 22, e188. [Google Scholar]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s disease: Mechanisms and biochemical processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Tanner, C.M.; Ottman, R.; Goldman, S.M.; Ellenberg, J.; Chan, P.; Mayeux, R.; Langston, J.W. Parkinson disease in twins: An etiologic study. JAMA 1999, 281, 341–346. [Google Scholar] [CrossRef]
- Pezzoli, G.; Cereda, E. Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 2013, 80, 2035–2041. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Kumudini, N.; Uma, A.; Devi, Y.P.; Naushad, S.M.; Mridula, R.; Borgohain, R.; Kutala, V.K. Association of Parkinson’s disease with altered serum levels of lead and transition metals among South Indian subjects. Indian J. Biochem. Biophys. 2014, 51, 121–126. [Google Scholar] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar] [PubMed]
- Coon, S.; Stark, A.; Peterson, E.; Gloi, A.; Kortsha, G.; Pounds, J.; Chettle, D.; Gorell, J. Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ. Health Perspect. 2006, 114, 1872–1876. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; Weuve, J.; Nie, H.; Saint-Hilaire, M.H.; Sudarsky, L.; Simon, D.K.; Hersh, B.; Schwartz, J.; Wright, R.O.; Hu, H. Association of cumulative lead exposure with Parkinson’s disease. Environ. Health Perspect. 2010, 118, 1609–1613. [Google Scholar] [CrossRef]
- Ngim, C.H.; Devathasan, G. Epidemiologic study on the association between body burden mercury level and idiopathic Parkinson’s disease. Neuroepidemiology 1989, 8, 128–141. [Google Scholar] [CrossRef]
- Lin, C.Y.; Liou, S.H.; Hsiech, C.M.; Ku, M.C.; Tsai, S.Y. Dose-response relationship between cumulative mercury exposure index and specific uptake ratio in the striatum on Tc-99m TRODAT SPECT. Clin. Nucl. Med. 2011, 36, 689–693. [Google Scholar] [CrossRef]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Çubukçu, H.C.; Yurtdaş, M.; Durak, Z.E.; Aytaç, B.; Güneş, H.N.; Çokal, B.G.; Yoldaş, T.K.; Durak, İ. Oxidative and nitrosative stress in serum of patients with Parkinson’s disease. Neurol. Sci. 2016, 37, 1793–1798. [Google Scholar] [CrossRef]
- Ullah, I.; Zhao, L.; Hai, Y.; Fahim, M.; Alwayli, D.; Wang, X.; Li, H. Metal elements and pesticides as risk factors for Parkinson’s disease—A review. Toxicol. Rep. 2021, 8, 607–616. [Google Scholar] [CrossRef]
- Fukushima, T.; Tan, X.; Luo, Y.; Wang, P.; Song, J.; Kanda, H.; Hayakawa, T.; Kumagai, T.; Kakamu, T.; Tsuji, M.; et al. Heavy metals in blood and urine and its relation to depressive symptoms in Parkinson’s disease patients. Fukushima J. Med. Sci. 2013, 59, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.S.; Lian, T.H.; Hu, Y.; Zuo, L.J.; Guo, P.; Yu, S.Y.; Liu, L.; Jin, Z.; Zhao, H.; Li, L.X.; et al. Restless legs syndrome in Parkinson disease: Clinical characteristics, abnormal iron metabolism and altered neurotransmitters. Sci. Rep. 2017, 7, 10547. [Google Scholar] [CrossRef]
- Xuan, M.; Guan, X.; Gu, Q.; Shen, Z.; Yu, X.; Qiu, T.; Luo, X.; Song, R.; Jiaerken, Y.; Xu, X.; et al. Different iron deposition patterns in early- and middle-late-onset Parkinson’s disease. Park. Relat. Disord. 2017, 44, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Björkblom, B.; Adilbayeva, A.; Maple-Grødem, J.; Piston, D.; Ökvist, M.; Xu, X.M.; Brede, C.; Larsen, J.P.; Møller, S.G. Parkinson disease protein DJ-1 binds metals and protects against metal-induced cytotoxicity. J. Biol. Chem. 2013, 288, 22809–22820. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.E.; Double, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Cass, W.A.; Grondin, R.; Andersen, A.H.; Zhang, Z.; Hardy, P.A.; Hussey-Andersen, L.K.; Rayens, W.S.; Gerhardt, G.A.; Gash, D.M. Iron accumulation in the striatum predicts aging-related decline in motor function in rhesus monkeys. Neurobiol. Aging 2007, 28, 258–271. [Google Scholar] [CrossRef]
- Zhukovskaya, E.; Karelin, A.; Rumyantsev, A. Neurocognitive Dysfunctions in Iron Deficiency Patients; IntechOpen: London, UK, 2019; pp. 83–113. [Google Scholar]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Dobson, B.R.; Jones, G.R.; Clarke, D.T. Iron in the basal ganglia in Parkinson’s disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain 1999, 122, 667–673. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity linked to dysfunctional metal ion homeostasis and xenobiotic metal exposure: Redox signaling and oxidative stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Rogers, E.; Xia, Y.; Liu, W.; Ma, W.; Zeng, W.; Gong, L.; Fang, J.; et al. The association of iron and the pathologies of Parkinson’s diseases in MPTP/MPP+-induced neuronal degeneration in non-human primates and in cell culture. Front. Aging Neurosci. 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for Parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Gerlach, M.; Schünemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.; Riederer, P.; Ben-Shachar, D. Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2003, 86, 1142–1148. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Lucas, H.R. Iron redox chemistry and implications in the Parkinson’s disease brain. Oxidative Med. Cell. Longev. 2019, 2019, 4609702. [Google Scholar] [CrossRef]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef] [PubMed]
- Erikson, K.M.; Syversen, T.; Aschner, J.L.; Aschner, M. Interactions between excessive manganese exposures and dietary iron-deficiency in neurodegeneration. Environ. Toxicol. Pharmacol. 2005, 19, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Fitsanakis, V.A.; Au, C.; Erikson, K.M.; Aschner, M. The effects of manganese on glutamate, dopamine and gamma-aminobutyric acid regulation. Neurochem. Int. 2006, 48, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A. Manganese action in brain function. Brain Res. Rev. 2003, 41, 79–87. [Google Scholar] [CrossRef]
- Archibald, F.S.; Tyree, C. Manganese poisoning and the attack of trivalent manganese upon catecholamines. Arch. Biochem. Biophys. 1987, 256, 638–650. [Google Scholar] [CrossRef]
- Horning, K.J.; Caito, S.W.; Tipps, K.G.; Bowman, A.B.; Aschner, M. Manganese is essential for neuronal health. Annu. Rev. Nutr. 2015, 35, 71–108. [Google Scholar] [CrossRef]
- Pal, P.K.; Samii, A.; Calne, D.B. Manganese neurotoxicity: A review of clinical features, imaging and pathology. Neurotoxicology 1999, 20, 227–238. [Google Scholar]
- Aschner, M.; Dorman, D.C. Manganese: Pharmacokinetics and molecular mechanisms of brain uptake. Toxicol. Rev. 2006, 25, 147–154. [Google Scholar] [CrossRef]
- Martinez-Finley, E.J.; Gavin, C.E.; Aschner, M.; Gunter, T.E. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic. Biol. Med. 2013, 62, 65–75. [Google Scholar] [CrossRef]
- Au, C.; Benedetto, A.; Aschner, M. Manganese transport in eukaryotes: The role of DMT1. Neurotoxicology 2008, 29, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Erikson, K.M.; John, C.E.; Jones, S.R.; Aschner, M. Manganese accumulation in striatum of mice exposed to toxic doses is dependent upon a functional dopamine transporter. Environ. Toxicol. Pharmacol. 2005, 20, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Mattei, C.; Kelsell, R.E.; Medhurst, A.D.; Calver, A.R.; Randall, A.D.; Davis, J.B.; Benham, C.D.; Pangalos, M.N. Cloning and functional expression of human short TRP7, a candidate protein for store-operated Ca2+ influx. J. Biol. Chem. 2002, 277, 12302–12309. [Google Scholar] [CrossRef] [PubMed]
- Kannurpatti, S.S.; Joshi, P.G.; Joshi, N.B. Calcium sequestering ability of mitochondria modulates influx of calcium through glutamate receptor channel. Neurochem. Res. 2000, 25, 1527–1536. [Google Scholar] [CrossRef]
- Crossgrove, J.S.; Allen, D.D.; Bukaveckas, B.L.; Rhineheimer, S.S.; Yokel, R.A. Manganese distribution across the blood-brain barrier. I. Evidence for carrier-mediated influx of managanese citrate as well as manganese and manganese transferrin. Neurotoxicology 2003, 24, 3–13. [Google Scholar] [CrossRef]
- Fujishiro, H.; Doi, M.; Enomoto, S.; Himeno, S. High sensitivity of RBL-2H3 cells to cadmium and manganese: An implication of the role of ZIP8. Metallomics 2011, 3, 710–718. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef]
- Harischandra, D.S.; Ghaisas, S.; Zenitsky, G.; Jin, H.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Manganese-induced neurotoxicity: New insights into the triad of protein misfolding, mitochondrial impairment, and neuroinflammation. Front. Neurosci. 2019, 13, 654. [Google Scholar] [CrossRef]
- Spadoni, F.; Stefani, A.; Morello, M.; Lavaroni, F.; Giacomini, P.; Sancesario, G. Selective vulnerability of pallidal neurons in the early phases of manganese intoxication. Exp. Brain Res. 2000, 135, 544–551. [Google Scholar] [CrossRef]
- Gavin, C.E.; Gunter, K.K.; Gunter, T.E. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol. Appl. Pharmacol. 1992, 115, 1–5. [Google Scholar] [CrossRef]
- HaMai, D.; Campbell, A.; Bondy, S.C. Modulation of oxidative events by multivalent manganese complexes in brain tissue. Free Radic. Biol. Med. 2001, 31, 763–768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bouabid, S.; Delaville, C.; De Deurwaerdère, P.; Lakhdar-Ghazal, N.; Benazzouz, A. Manganese-induced atypical parkinsonism is associated with altered Basal Ganglia activity and changes in tissue levels of monoamines in the rat. PLoS ONE 2014, 9, e98952. [Google Scholar] [CrossRef] [PubMed]
- Klos, K.J.; Chandler, M.; Kumar, N.; Ahlskog, J.E.; Josephs, K.A. Neuropsychological profiles of manganese neurotoxicity. Eur. J. Neurol. 2006, 13, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Calne, D.B.; Chu, N.S.; Huang, C.C.; Lu, C.S.; Olanow, W. Manganism and idiopathic parkinsonism: Similarities and differences. Neurology 1994, 44, 1583–1586. [Google Scholar] [CrossRef]
- Lai, B.C.; Marion, S.A.; Teschke, K.; Tsui, J.K. Occupational and environmental risk factors for Parkinson’s disease. Park. Relat. Disord. 2002, 8, 297–309. [Google Scholar] [CrossRef]
- Racette, B.A.; Searles Nielsen, S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 2017, 88, 344–351. [Google Scholar] [CrossRef]
- Gonzalez, O.A.; Novak, M.J.; Kirakodu, S.; Stromberg, A.J.; Shen, S.; Orraca, L.; Gonzalez-Martinez, J.; Ebersole, J.L. Effects of aging on apoptosis gene expression in oral mucosal tissues. Apoptosis 2013, 18, 249–259. [Google Scholar] [CrossRef]
- Kanthasamy, A.G.; Anantharam, V.; Zhang, D.; Latchoumycandane, C.; Jin, H.; Kaul, S.; Kanthasamy, A. A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCdelta) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic. Biol. Med. 2006, 41, 1578–1589. [Google Scholar] [CrossRef]
- Tomás-Camardiel, M.; Herrera, A.J.; Venero, J.L.; Cruz Sánchez-Hidalgo, M.; Cano, J.; Machado, A. Differential regulation of glutamic acid decarboxylase mRNA and tyrosine hydroxylase mRNA expression in the aged manganese-treated rats. Mol. Brain Res. 2002, 103, 116–129. [Google Scholar] [CrossRef]
- Autissier, N.; Rochette, L.; Dumas, P.; Beley, A.; Loireau, A.; Bralet, J. Dopamine and norepinephrine turnover in various regions of the rat brain after chronic manganese chloride administration. Toxicology 1982, 24, 175–182. [Google Scholar] [CrossRef]
- Díaz-Véliz, G.; Mora, S.; Gómez, P.; Dossi, M.T.; Montiel, J.; Arriagada, C.; Aboitiz, F.; Segura-Aguilar, J. Behavioral effects of manganese injected in the rat substantia nigra are potentiated by dicumarol, a DT-diaphorase inhibitor. Pharmacol. Biochem. Behav. 2004, 77, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, T.R. Manganese and Parkinson’s disease: A critical review and new findings. Environ. Health Perspect. 2010, 118, 1071–1080. [Google Scholar] [CrossRef]
- Chandra, S.V.; Shukla, G.S. Concentrations of striatal catecholamines in rats given manganese chloride through drinking water. J. Neurochem. 1981, 36, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Li, Z.; Sridhar, S.; Khoshbouei, H. The effect of manganese on dopamine toxicity and dopamine transporter (DAT) in control and DAT transfected HEK cells. Neurotoxicology 2013, 35, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Leenders, K.L. Significance of non-presynaptic SPECT tracer methods in Parkinson’s disease. Mov. Disord. 2003, 18, S39–S42. [Google Scholar] [CrossRef]
- Felicio, A.C.; Shih, M.C.; Godeiro-Junior, C.; Andrade, L.A.; Bressan, R.A.; Ferraz, H.B. Molecular imaging studies in Parkinson disease: Reducing diagnostic uncertainty. Neurologist 2009, 15, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.M.; Walker, E.M., Jr.; Wu, M.; Gillette, C.; Blough, E.R. Environmental mercury and its toxic effects. J. Prev. Med. Public Health 2014, 47, 74–83. [Google Scholar] [CrossRef]
- Aschner, M.; Erikson, K.M.; Herrero Hernández, E.; Tjalkens, R. Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef]
- Kerper, L.E.; Ballatori, N.; Clarkson, T.W. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am. J. Physiol. 1992, 262, R761–R765. [Google Scholar] [CrossRef]
- Jan, A.T.; Azam, M.; Siddiqui, K.; Ali, A.; Choi, I.; Haq, Q.M. Heavy metals and human health: Mechanistic insight into toxicity and counter defense system of antioxidants. Int. J. Mol. Sci. 2015, 16, 29592–29630. [Google Scholar] [CrossRef]
- Watanabe, C.; Yoshida, K.; Kasanuma, Y.; Kun, Y.; Satoh, H. In utero methylmercury exposure differentially affects the activities of selenoenzymes in the fetal mouse brain. Environ. Res. 1999, 80, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Johnson, V.J.; Sharma, R.P. Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: Differential modulation of NF-kappaB and p38 MAPK signaling pathways. Nitric Oxide 2002, 7, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Singh, S.; Siddiqi, N.J. Biomedical implications of heavy metals induced imbalances in redox systems. Biomed. Res. Int. 2014, 2014, 640754. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chang, C.W.; Lee, H.L.; Chuang, C.C.; Chiu, H.C.; Li, W.Y.; Horng, J.T.; Fu, E. Association between history of dental amalgam fillings and risk of Parkinson’s disease: A population-based retrospective cohort study in Taiwan. PLoS ONE 2016, 11, e0166552. [Google Scholar] [CrossRef]
- Ni, M.; Li, X.; Yin, Z.; Sidoryk-Węgrzynowicz, M.; Jiang, H.; Farina, M.; Rocha, J.B.; Syversen, T.; Aschner, M. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 2011, 59, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.L.; Teixeira, A.; Meotti, F.C.; Ribas, C.M.; Stringari, J.; Garcia Pomblum, S.C.; Moro, A.M.; Bohrer, D.; Bairros, A.V.; Dafre, A.L.; et al. Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ. Res. 2006, 102, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Stringari, J.; Nunes, A.K.; Franco, J.L.; Bohrer, D.; Garcia, S.C.; Dafre, A.L.; Milatovic, D.; Souza, D.O.; Rocha, J.B.; Aschner, M.; et al. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol. Appl. Pharmacol. 2008, 227, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Aschner, M.; Rocha, J.B. Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2011, 256, 405–417. [Google Scholar] [CrossRef]
- Franco, J.L.; Posser, T.; Dunkley, P.R.; Dickson, P.W.; Mattos, J.J.; Martins, R.; Bainy, A.C.; Marques, M.R.; Dafre, A.L.; Farina, M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic. Biol. Med. 2009, 47, 449–457. [Google Scholar] [CrossRef]
- Franco, J.L.; Braga, H.C.; Stringari, J.; Missau, F.C.; Posser, T.; Mendes, B.G.; Leal, R.B.; Santos, A.R.; Dafre, A.L.; Pizzolatti, M.G.; et al. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: Protective effects of quercetin. Chem. Res. Toxicol. 2007, 20, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Ke, T.; Gonçalves, F.M.; Gonçalves, C.L.; Dos Santos, A.A.; Rocha, J.B.T.; Farina, M.; Skalny, A.; Tsatsakis, A.; Bowman, A.B.; Aschner, M. Post-translational modifications in MeHg-induced neurotoxicity. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Aschner, J.L. Mercury neurotoxicity: Mechanisms of blood-brain barrier transport. Neurosci. Biobehav. Rev. 1990, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.N.; Racz, W.J. Effects of methylmercury on the spontaneous and potassium-evoked release of endogenous amino acids from mouse cerebellar slices. Can. J. Physiol. Pharmacol. 1987, 65, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Matyja, E. Glutamate: A potential mediator of inorganic mercury neurotoxicity. Metab. Brain Dis. 1996, 11, 175–184. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Dantzig, P.I. Parkinson’s disease, macular degeneration and cutaneous signs of mercury toxicity. J. Occup. Environ. Med. 2006, 48, 656. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Ochudło, S.; Opala, G.; Smolicha, W.; Siuda, J. Parkinsonism in chronic occupational metallic mercury intoxication. Neurol. Neurochir. Pol. 2003, 37, 31–38. [Google Scholar]
- Lewińska-Preis, L.; Jabłońska, M.; Fabiańska, M.J.; Kita, A. Bioelements and mineral matter in human livers from the highly industrialized region of the Upper Silesia Coal Basin (Poland). Environ. Geochem. Health 2011, 33, 595–611. [Google Scholar] [CrossRef]
- Szerdahelyi, P.; Kása, P. Histochemical demonstration of copper in normal rat brain and spinal cord. Evidence of localization in glial cells. Histochemistry 1986, 85, 341–347. [Google Scholar] [CrossRef]
- Bulcke, F.; Dringen, R.; Scheiber, I.F. Neurotoxicity of copper. Adv. Neurobiol. 2017, 18, 313–343. [Google Scholar] [PubMed]
- Navarro, J.A.; Schneuwly, S. Copper and zinc homeostasis: Lessons from Drosophila melanogaster. Front. Genet. 2017, 8, 223. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Copper and copper nanoparticles toxicity and their impact on basic functions in the body. Bratisl. Lek. Listy 2019, 120, 397–409. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, M.; Wang, B.; Li, M.; Chen, H.; Yu, X.; Zhao, Y.; Feng, W.; Chai, Z. The distribution profile and oxidation states of biometals in APP transgenic mouse brain: Dyshomeostasis with age and as a function of the development of Alzheimer’s disease. Metallomics 2012, 4, 289–296. [Google Scholar] [CrossRef]
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241. [Google Scholar] [CrossRef]
- Boll, M.C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723. [Google Scholar] [CrossRef]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef]
- De Lazzari, F.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide radical dismutation as new therapeutic strategy in Parkinson’s disease. Aging Dis. 2018, 9, 716–728. [Google Scholar] [CrossRef]
- Letelier, M.E.; Faúndez, M.; Jara-Sandoval, J.; Molina-Berríos, A.; Cortés-Troncoso, J.; Aracena-Parks, P.; Marín-Catalán, R. Mechanisms underlying the inhibition of the cytochrome P450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702. [Google Scholar] [CrossRef]
- Scheuhammer, A.M.; Cherian, M.G. Effects of heavy metal cations, sulfhydryl reagents and other chemical agents on striatal D2 dopamine receptors. Biochem. Pharmacol. 1985, 34, 3405–3413. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, oxidative stress and protein-quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew. Chem. Int. Ed. Engl. 2019, 58, 6512–6527. [Google Scholar] [CrossRef] [PubMed]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction between alpha-synuclein and metal ions, still looking for a role in the pathogenesis of Parkinson’s disease. Neuromol. Med. 2009, 11, 239–251. [Google Scholar] [CrossRef]
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat. Disord. 2018, 55, 117–121. [Google Scholar] [CrossRef]
- Uitti, R.J.; Rajput, A.H.; Rozdilsky, B.; Bickis, M.; Wollin, T.; Yuen, W.K. Regional metal concentrations in Parkinson’s disease, other chronic neurological diseases, and control brains. Can. J. Neurol. Sci. 1989, 16, 310–314. [Google Scholar] [CrossRef]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar] [CrossRef]
- Zayed, J.; Ducic, S.; Campanella, G.; Panisset, J.C.; André, P.; Masson, H.; Roy, M. Environmental factors in the etiology of Parkinson’s disease. Can. J. Neurol. Sci. 1990, 17, 286–291. [Google Scholar] [CrossRef]
- Rybicki, B.A.; Johnson, C.C.; Uman, J.; Gorell, J.M. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov. Disord. 1993, 8, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 1997, 48, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M. Occupational metal exposure and Parkinsonism. Adv. Neurobiol. 2017, 18, 143–158. [Google Scholar] [PubMed]
- Csavina, J.; Field, J.; Taylor, M.P.; Gao, S.; Landázuri, A.; Betterton, E.A.; Sáez, A.E. A review on the importance of metals and metalloids in atmospheric dust and aerosol from mining operations. Sci. Total Environ. 2012, 433, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed. Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef]
- Loikkanen, J.J.; Naarala, J.; Savolainen, K.M. Modification of glutamate-induced oxidative stress by lead: The role of extracellular calcium. Free Radic. Biol. Med. 1998, 24, 377–384. [Google Scholar] [CrossRef]
- Gurer, H.; Ercal, N. Can antioxidants be beneficial in the treatment of lead poisoning? Free Radic. Biol. Med. 2000, 29, 927–945. [Google Scholar] [CrossRef]
- Patra, R.C.; Rautray, A.K.; Swarup, D. Oxidative stress in lead and cadmium toxicity and its amelioration. Vet. Med. Int. 2011, 2011, 457327. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Rozek, L.S.; Dolinoy, D.C.; Paulson, H.L.; Hu, H. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Curr. Alzheimer Res. 2012, 9, 563–573. [Google Scholar] [CrossRef]
- Rahman, A.; Brew, B.J.; Guillemin, G.J. Lead dysregulates serine/threonine protein phosphatases in human neurons. Neurochem. Res. 2011, 36, 195–204. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Zawia, N.H. Alzheimer’s disease biomarkers and epigenetic intermediates following exposure to Pb in vitro. Curr. Alzheimer Res. 2012, 9, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Dosunmu, R.; Alashwal, H.; Zawia, N.H. Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mech. Ageing Dev. 2012, 133, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Bihaqi, S.W.; Cui, L.; Zawia, N.H. In vitro Pb exposure disturbs the balance between Aβ production and elimination: The role of AβPP and neprilysin. Neurotoxicology 2011, 32, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Zawia, N.H. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 2013, 39, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Bahmani, A.; Subaiea, G.M.; Zawia, N.H. Infantile exposure to lead and late-age cognitive decline: Relevance to AD. Alzheimer’s Dement. 2014, 10, 187–195. [Google Scholar] [CrossRef]
- Ashafaq, M.; Tabassum, H.; Vishnoi, S.; Salman, M.; Raisuddin, S.; Parvez, S. Tannic acid alleviates lead acetate-induced neurochemical perturbations in rat brain. Neurosci. Lett. 2016, 617, 94–100. [Google Scholar] [CrossRef]
- Fairbrother, A.; Wenstel, R.; Sappington, K.; Wood, W. Framework for metals risk assessment. Ecotoxicol. Environ. Saf. 2007, 68, 145–227. [Google Scholar] [CrossRef]
- Lorscheider, F.L.; Vimy, M.J.; Summers, A.O. Mercury exposure from “silver” tooth fillings: Emerging evidence questions a traditional dental paradigm. FASEB J. 1995, 9, 504–508. [Google Scholar] [CrossRef]
- Peng, J.; Peng, L.; Stevenson, F.F.; Doctrow, S.R.; Andersen, J.K. Iron and paraquat as synergistic environmental risk factors in sporadic Parkinson’s disease accelerate age-related neurodegeneration. J. Neurosci. 2007, 27, 6914–6922. [Google Scholar] [CrossRef]
- Haley, B.E. Mercury toxicity: Genetic susceptibility and synergistic effects. Med. Veritas 2005, 2, 535–542. [Google Scholar] [CrossRef]
- Binolfi, A.; Quintanar, L.; Bertoncini, C.W.; Griesinger, C.; Fernández, C.O. Bioinorganic chemistry of copper coordination to alpha-synuclein: Relevance to Parkinson’s disease. Coord. Chem. Rev. 2012, 256, 2188–2201. [Google Scholar] [CrossRef]
- Rai, A.; Maurya, S.K.; Khare, P.; Srivastava, A.; Bandyopadhyay, S. Characterization of developmental neurotoxicity of As, Cd, and Pb mixture: Synergistic action of metal mixture in glial and neuronal functions. Toxicol. Sci. 2010, 118, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Karri, V.; Schuhmacher, M.; Kumar, V. Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environ. Toxicol. Pharmacol. 2016, 48, 203–213. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyatha, S.; Kim, H.; Lee, D.; Kim, K. Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress. Antioxidants 2022, 11, 2467. https://doi.org/10.3390/antiox11122467
Pyatha S, Kim H, Lee D, Kim K. Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress. Antioxidants. 2022; 11(12):2467. https://doi.org/10.3390/antiox11122467
Chicago/Turabian StylePyatha, Sarita, Haesoo Kim, Daeun Lee, and Kisok Kim. 2022. "Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress" Antioxidants 11, no. 12: 2467. https://doi.org/10.3390/antiox11122467
APA StylePyatha, S., Kim, H., Lee, D., & Kim, K. (2022). Association between Heavy Metal Exposure and Parkinson’s Disease: A Review of the Mechanisms Related to Oxidative Stress. Antioxidants, 11(12), 2467. https://doi.org/10.3390/antiox11122467