Abstract
Schizophrenia (SZ) is a deleterious brain disorder affecting cognition, emotion and reality perception. The most widely accepted neurochemical-hypothesis is the imbalance of neurotransmitter-systems. Depleted GABAergic-inhibitory function might produce a regionally-located dopaminergic and glutamatergic-storm in the brain. The dopaminergic-release may underlie the positive psychotic-symptoms while the glutamatergic-release could prompt the primary negative symptoms/cognitive deficits. This may occur due to excessive synaptic-pruning during the neurodevelopmental stages of adolescence/early adulthood. Thus, although SZ is not a neurodegenerative disease, it has been suggested that exaggerated dendritic-apoptosis could explain the limited neuroprogression around its onset. This apoptotic nature of SZ highlights the potential therapeutic action of anti-apoptotic drugs, especially at prodromal stages. If dysregulation of apoptotic mechanisms underlies the molecular basis of SZ, then anti-apoptotic molecules could be a prodromal therapeutic option to halt or prevent SZ. In fact, risk alleles related in apoptotic genes have been recently associated to SZ and shared molecular apoptotic changes are common in the main neurodegenerative disorders and SZ. PRISMA-guidelines were considered. Anti-apoptotic drugs are commonly applied in classic neurodegenerative disorders with promising results. Despite both the apoptotic-hallmarks of SZ and the widespread use of anti-apoptotic targets in neurodegeneration, there is a strikingly scarce number of studies investigating anti-apoptotic approaches in SZ. We analyzed the anti-apoptotic approaches conducted in neurodegeneration and the potential applications of such anti-apoptotic therapies as a promising novel therapeutic strategy, especially during early stages.
1. Introduction
Schizophrenia (SZ) is a heterogeneous psychiatric disorder with unclear etiology affecting ~1% of the population worldwide [1]. The few pharmacological treatments available mainly target the positive psychotic symptoms, but not the negative and cognitive symptoms. To gain a complete understanding of SZ, the integration of multidisciplinary approaches through molecular biology, genetics, epigenetics, environmental factors, neuroimaging, cell and animal models, translational clinical and epidemiological research is needed. According to the neurodevelopmental hypothesis of SZ [2], early processes such as abnormal neurogenesis, neuronal migration, dendritic arborization or axonal outgrowth would affect the formation of the neural circuits. During adolescence and young adulthood, postnatal brain maturation abnormalities, including excessive dendritic spine pruning [3], could eventually account for the onset of SZ symptoms [4]. In order to study dendritic pruning during brain maturation, several key neurodevelopmental animal models of SZ have been developed. Some neurodevelopmental models of SZ include the maternal immune activation model [5] or the use of antagonists of the n-methyl-D-aspartate (NMDA) receptors, such as MK-801 [6] ketamine [7] and phencyclidine [8,9] or prenatal methylazoxymethanol acetate exposure [10,11,12,13], with the latter being one of the most validated neurodevelopmental model of SZ, in terms of face, construct and predictive validity [14,15,16,17,18].
1.1. Synaptic Pruning in SZ
Cortical pyramidal neurons from subjects with SZ exhibit different anatomical features [19] (Figure 1).
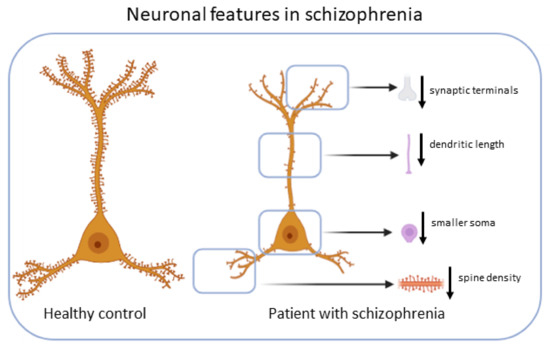
Figure 1.
Neuronal features in SZ. Cortical pyramidal neurons from subjects with schizophrenia exhibit smaller soma volume, decreased spine density, decreased dendritic length and decreased terminals.
The hypotheses of the underlying etiopathogenesis of SZ have been repeatedly reformulated and yet continue to be revisited. We recently reviewed a reassessment of the synaptic over-pruning hypothesis of SZ [20]. The glutamate hypothesis, accounting for the mostly prodromal negative symptoms, arose more recently as compatible with the initial hypothesis of a dopamine storm explaining the positive symptoms. Glutamate acts as the main neurotransmission modulator via NMDA/α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA) receptors under physiological conditions, but may trigger loss of integrity of dendritic spines upon dysregulation. During a critical neurodevelopmental period, there is dysregulation between excitatory glutamatergic pyramidal neurons and inhibitory gamma-aminobutyric acid (GABAergic) interneurons, leading to excessive local glutamate release [20]. Glutamate dysregulations, via its NMDA/AMPA receptors, negatively impact the integrity of the dendritic spines, with subsequent excessive dendritic pruning with the activation of local apoptosis machinery. Over-pruning of dendritic spines, together with aberrant synaptic plasticity may lead to neural misconnections and suboptimal synaptic function, promoting clinical symptoms [20]. Indeed, postmortem studies have reported reduced number of dendritic spines, especially on pyramidal neurons located in layer-III of the prefrontal cortex, in the superior temporal gyrus, and in hippocampal subfields (CA3) [3,21]. Taken together, the driving hypothesis of glutamate storms originating from uncontrolled extrapyramidal neuron firing due to inefficient inhibitory regulation of GABAergic-interneurons highlights the importance of pharmacological interventions during the prodromal phase of disease and apoptotic targeting. Accordingly, the use of anti-apoptotic drugs has been reported to prevent the loss of GABAergic interneurons [22].
Dendritic pruning, however, is not restricted to pathologic contexts. Physiologically, dendritic pruning corresponds to a highly regulated homeostatic neurodevelopmental process to either favor or delete certain brain connections underpinning synaptic plasticity during normal growth. Significant cell death occurs during early development of the nervous system with over half of all developing neurons dying via apoptosis [23]. Interestingly, aberrant dendritic apoptosis preceding neuronal death has also been found in the hippocampus and prefrontal cortex in the early stages of neurodegenerative diseases such as Alzheimer’s disease (AD) [24]. Even though the underlying molecular mechanisms are not fully understood [25], synaptic pruning exhibits molecular features in common with apoptosis [26]. It is worth mentioning that microglia and immune responses also contribute to the loss of dendritic spine density in SZ [27], although this is beyond the scope of this review. In any case, during dendritic apoptosis, apoptotic molecules generate the signals that attract microglia for removal of synaptic debris, suggesting that microglial phagocytosis of synapses occurs downstream in this apoptotic process [28]. Such apoptotic processes are often regulated by a complex molecular cascade of cysteine proteases, the so-called caspases (caspase-1–15, of which -2,-8,-9,-10 are initiators and -3-6-7 are effectors) [29]. The two main apoptotic pathways [24], the molecular interactions [30,31] and involvement of inhibitor-of-apoptosis (IAP) molecules [32] are herein represented (Figure 2).
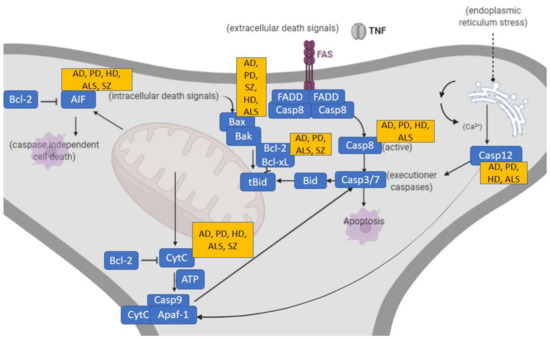
Figure 2.
Two prototypical apoptotic pathways (extrinsic and intrinsic) initiated by separate events converge at a common place to execute apoptosis. Main associations of these apoptotic pathways to the neurodegenerative disorders AD, PD, HD as well as SZ are indicated. Death receptor pathways triggered by binding of death receptors by their ligands (tumor necrosis factor (TNF, FasL) that results in receptor clustering and recruitment of adaptor proteins, leading to the activation of initiator caspase-8. The intrinsic pathway responds to a variety of cellular stress signals that act on mitochondria to cause leakage of pro-apoptotic effectors like cytochrome c and apoptosis-inducing factor (AIF). Cytochrome c binds to Apaf-1 in the presence of ATP to form the “apoptosome”, which subsequently recruits and activates initiator pro-caspase-9. AIF translocates to the nucleus to cause high molecular weight DNA fragmentation in a caspase-independent manner. Both pathways activate effector caspases-3 and -7. Endoplasmic reticulum stress causes the activation and release of caspase-12, leading to apoptotic cell death. AIF, apoptosis-inducing factor; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; Apaf, apoptotic protease activating factor; Bak, Bcl-2 homologous antagonist killer; Bax, Bcl-2 associated X protein; Bcl, B-cell lymphoma; Bid, BH3 interacting domain death agonist; Casp, caspase; CytC, cytochrome c; FADD, Fas-associated-death-domain protein; FAS, cysteine-rich transmembrane protein CD95; HD, Huntington’s disease; PD, Parkinson’s disease; SZ, schizophrenia; tBid, truncated Bid; TNF, tumor necrosis factor.
1.2. Apoptotic Molecular Studies in Neurodegenerative Disorders and Overlap with SZ
Apoptotic molecular changes and the involvement of apoptotic proteins have been observed in neurodegenerative disorders, including AD [33], Parkinson’s disease (PD) [34], Huntington’s disease (HD) [35] and amyotrophic lateral sclerosis (ALS) [36]. Some or the specific molecular apoptotic changes occurring in these neurodegenerative disorders, also observed in SZ, will be extensively discussed in Section 3 and briefly summarized in Figure 2. Importantly, at a genetic level, different known risk alleles related to apoptosis have been found to be associated with SZ in the recent literature, including apoptotic mediator BCL11B, BCL2L12, BNIP3L and ENOX1 genes [37].
The clear association between neuronal death and neurodegenerative mechanisms has led to the widespread use of anti-apoptotic therapeutic strategies in models of classic neurodegenerative disorders, including AD [38], PD [39], HD [40] and ALS [41]. However, neuronal death is a complex process that is not restricted to a unique type of mechanism, quite the opposite, it may occur via a complex and strikingly high number of molecular events [24]. While multiple molecular pathways lead to the activation of the caspases, the mitochondrial intrinsic pathway is mostly associated with neuronal apoptosis [19,31]. Regulation of mitochondrial outer membrane permeabilization and intrinsic apoptosis in neurons appears to center on control of Bax activation (Table S1). Evidence of a main role of the mitochondrial apoptotic machinery in SZ etiopathogenesis has been reported in the literature [42,43], placing mitochondrial anti-apoptotic molecules as a potential surrogate therapeutic option. Apoptotic activity can be both preceded and triggered by a broad panoply of stimuli including pro-inflammatory cytokines, excitotoxicity (including glutamate excitotoxicity earlier related to SZ), neurotrophin withdrawal, abnormal calcium concentrations and mitochondrial dysfunction with subsequent oxidative stress [44]. In fact, the inhibitory GABAergic-interneurons expressing parvalbumin are highly sensitive to oxidative stress and require a highly regulated antioxidant system to neutralize the overproduction of reactive oxygen species (ROS) generated by mitochondria [45]. Interestingly, all the aforementioned apoptotic triggers are present in SZ, suggesting apoptosis as a key factor of SZ onset.
1.3. Apoptotic Molecular Studies in SZ
Despite the underlying mechanisms leading to synaptic dysfunction in SZ being uncertain, evidence suggests that dysregulation of neuronal apoptosis may contribute to its pathophysiology [19,46]. It has been hypothesized that apoptotic activity contributes to the evidence for reduced grey matter volume and synaptic deficits in SZ [19,47]. Additionally, synaptic/dendritic neuronal loss could be explained by increased susceptibility to apoptosis in SZ. The basis for the apoptotic features in SZ is widely supported. First, there is a 50% increase in the Bax/Bcl-2 ratio as an apoptotic indicator (via cytochrome-c release) in the temporal cortex (area 21, middle temporal gyrus) of SZ patients [48]. Second, there is a 30% decrease in Bcl-2 levels in the temporal cortex in SZ [49]. This suggests that cortical neurons and synapses in patients with SZ may have less neuroprotection given that Bcl-2 can exert both neuroprotective and neurotrophic effects [19].
Although caspase activation is often considered a precursor to rapid cell death, the emerging concept of synaptic apoptosis suggests that apoptotic activation can be localized to synapses of distal neurites without inducing immediate neuronal death or involving the neuronal cell body [19,50]. Interestingly, caspase-3 activity has been associated with normal physiological activity, including synaptic plasticity [19]. It is of note that local apoptosis in SZ occurs in the absence of neuronal cell loss and without changes in the number of pyramidal neurons [51]. This is in line with animal models that have shown local caspase activity in neurons in which caspases were confined to the dendritic compartment of pruning instead of the soma or axonal areas [26]. NMDA receptor activation can trigger local dendritic apoptosis [52], and following focal application of glutamate to distal dendrites in vitro, a localized increase in caspase-3 activity was seen without propagation to the neuronal soma [50]. This local activation of apoptosis mediated by caspase-3 within distal dendrites is enough to prune dendritic spines and branches locally [53]. If apoptotic activity contributes to synaptic and neuritic elimination, then there must also be a mechanism to limit the proliferation of the caspase cascade to the rest of the neuron. The induction of endogenous caspase-inhibitors may be present in neuronal cytoplasm [54]. Importantly, the different molecular findings constituting the molecular basis of SZ, including glutamate excitotoxicity, oxidative stress or lack of neurotrophic factors may converge at the apoptotic endpoint [45]. Results from our research group add further evidence supporting the apoptotic hallmarks in the disease. We described increased apoptotic susceptibility in primary fibroblasts from a skin biopsy of naïve patients with a first-psychotic episode [55] and found a correlation between altered apoptotic markers with both the volume of certain brain regions and glutamate/glutamine-neurometabolites [56]. Additionally, we observed alterations in the expression of genes involved in apoptotic pathways [57]. The consistent findings in the literature [58], supported by our group, suggesting that dysregulation of apoptotic mechanisms underlies the molecular basis of SZ, led us to propose anti-apoptotic molecules as a challenging prodromal therapeutic option to halt onset and/or SZ progression.
This systematic review is therefore justified based on: i) the neurodevelopmental changes derived from excessive dendritic pruning via local activation of the apoptosis machinery occurring around the onset of SZ, and ii) the efficacy of apoptotic-inhibitors in classic neurodegenerative disorders, including AD, PD, HD and ALS. We have gathered the scientific data reported in the literature that used anti-apoptotic molecules to halt neurodegeneration and have analyzed those treatments with anti-apoptotic properties against SZ to further elucidate their potential as novel effective therapeutic approaches targeting SZ.
2. Materials and Methods
This systematic review was conducted in accordance with PRISMA guidelines [59]. The 24-step guide for systematic review and meta-analysis in medical research was followed [60], including the definition of research question, team and search strategy, selection criteria, data collection form, study protocol and registration (PROSPERO ID CRD42021238668). The collection of all references and abstracts, searched in multiple databases, was gathered in a single file, and elimination of duplicates was performed. Two reviewers screening title and abstract were used. Collection, comparison and selection for retrieval was developed by the two independent reviewers. The full texts of the references selected based on titles and abstracts were retrieved and the quality of the studies was considered. Articles were identified by searching for titles in Web of Science (WoS), PUBMED and SCOPUS databases by using the following research terms: “schizophrenia” OR “psychosis” OR “neurodegeneration” OR “neurodegenerative disorders” OR “neurodevelopmental disorders” AND “caspase inhibitor” OR “pan-caspase inhibitor” OR “anti-apoptotic molecules” OR “cytochrome c inhibitor” OR “apoptotic inhibitor” OR “mitochondrial apoptotic channel inhibitors”. The search was restricted to English, Spanish or German language journal articles with in vitro, human and/or animal subjects, published between 1990 and 2021. Blinding during data extraction between the review authors was achieved making use of Rayyan software for systematic reviews (Qatar Computing Research Institute, HBKU, Doha, Qatar) [61] which also helps expedite the initial screening of abstracts and titles using a process of semi-automation while incorporating a high level of usability.
The searches returned 917 records after duplicates were removed. Screening summary showed 227 included articles, 647 excluded articles and 43 potential articles for reviewer 1, and 243 included articles and 674 excluded articles, for reviewer 2. After blind was turned off, discrepancies were further solved through additional rigorous assessment criteria. The number of studies included throughout the identification, screening, eligibility and inclusion processes (n = 124) is provided as a flow diagram (Figure S1).
2.1. Inclusion and exclusion criteria
Those studies presenting neurological disorders, with a component of neurodegeneration, using interventions with anti-apoptotic compounds were included, with special focus on molecular findings at the neuronal level. Scoping, systematic and literature reviews were considered for descriptive purposes. Similarly, those studies without quantifiable data to be extracted (n = 54) and in silico studies (n = 2) were considered for descriptive purposes. Ischemic or stroke models were excluded due to a distal etiopathogenic origin with the disease of interest. Studies not controlling for major confounding factors such as drug treatment, drug abuse, and different co-morbidities, or that lacked age or gender-matched controls were not considered. Also, studies that did not allow access to the full text were excluded.
2.2. Primary and Secondary Outcomes
The main primary outcome parameters of interest for this evaluation were of a biological nature, as measured by quantification of occurrence of local neuronal viability, death, loss of dendritic arborization, spine shrinkage, synaptic pruning and function with inclusion of both continuous and dichotomous data. Since some interventions of this nature have a close relationship to mitochondria, mitochondrial parameters, such as mitochondrial respiratory chain function or oxidative stress parameters and intrinsic apoptosis with involvement of Bax, Bcl-2 and cytochrome c release were considered. Thus, the literature was selected in line with these primary outcomes: Molecular data related to mitochondrial, oxidative and apoptotic pathways. All additional outcome measures not depicted here but reported in the included studies were also examined. In addition, next to the parameters of interest, information on adverse effects following the intervention were sought to provide a full picture of the current state of knowledge and subsequent risks. The units of measurement varied depending on the data extracted; e.g., if molecular data were extracted from enzymatic activities, units would be nmol of produced or reduced substrate per minute and mg of protein.
The risk of bias was assessed by two review authors guided by the criteria recommended by the International Cochrane Collaboration: At least two external researchers were aware of the process, methodological approaches and follow-up of the study. To ensure the completeness of the outcome data, only treatment interventions using anti-apoptotic molecules in processes of neurodegeneration and neuroprogression were considered, mainly conducted in research animal models. Blinding intervention was conducted by two blinded researchers during the selection criteria and data extraction (Ryyan qcri). Different data storage (cloud and hardware) was used and access restricted to the researchers involved throughout the study. During the selective outcome reporting, primary and secondary outcomes were listed and reported, for further comparisons. Molecular and clinical findings were analyzed in order to elucidate the adequateness of the anti-apoptotic interventions in each specific context of neurodegeneration and/or neuroprogression. Any potential conflicts of interests were also explored. Any potential source of bias (risk of bias) was considered, such as bias towards favorable outcomes that may have occurred in the study. Finally, the assessment of publication bias was defined by identification of unpublished outcomes and studies, when available. Unpublished findings were searched via meeting abstracts tracked through Google Scholar, PhD theses available at the university repository, informal sources and, if needed, by contacting the authors of the studies included.
During the strategy for data synthesis, the approach plan was defined as follows: data were synthesized in a narrative manner and included studies were described according to: (i) the type of intervention, including in vivo or in vitro research of neurodegeneration and neuroprogression using anti-apoptotic molecules, and clinical trials; (ii) the type of pharmacological agent, including pan-caspase inhibitors, as well as specific anti-caspase-3 and anti-cytochrome c release; (iii) the characteristics of the study population and (iv) type of outcomes, including molecular parameters, such as neuroconnectivity, mitochondrial function and oxidative stress apoptotic markers and clinical findings of a global nature with special attention to secondary off-target reported events.
3. Results
A large number of anti-apoptotic molecules targeting apoptosis have been tested in different experimental neurodegenerative models. Most of them directly or indirectly target apoptosis through different mechanisms (Figure 3).
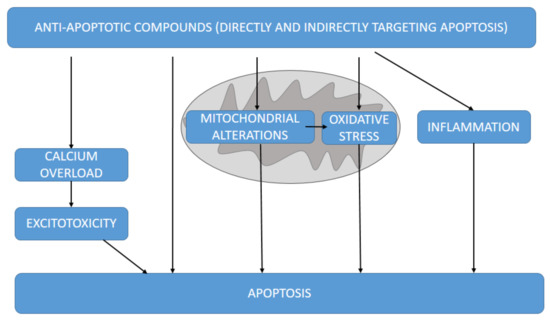
Figure 3.
Different mechanisms by which the anti-apoptotic molecules herein discussed could target apoptosis, directly or indirectly through excitotoxicity, mitochondrial alterations and derived oxidative stress and inflammation.
Some groups have suggested a neurodegenerative component of SZ [62]. In such cases, whether cell death is involved in the neuronal loss in neurodegenerative processes has important implications for the rational development of therapeutic strategies. The number of studies related to the use of specific anti-apoptotic molecules at the level of caspases in the classic neurodegenerative diseases (AD, PD, HD and ALS) vs. SZ is shown (Table S2). The outcomes indicate the lack of studies using apoptotic inhibitors in SZ with respect to those in classic neurodegenerative disorders.
3.1. Apoptotic Alterations in Classic Neurodegenerative Disorders
Accumulating data suggest essential roles for apoptotic pathways in the pathophysiology of a spectrum of neuropathological disorders [63]. Increased neuronal apoptosis has been widely demonstrated in classic neurodegenerative disorders, often via grafted or β-amyloid-induced-neurodegeneration in AD models [44,64] through 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-(MPTP)-induced-neurodegeneration in PD models [65], through 3-nitropropionic acid (NP3)- or malonate-induced neurodegeneration in HD models [46,66] and through superoxide dismutase-1 (SOD1)-mutations in ALS models [47,67].
Abnormal levels of apoptotic markers such as anti-apoptotic (Bcl-2/Bcl-xL) and pro-apoptotic proteins (Bax/Bak/Bad), Bcl-2 protein family members, initiator caspases-8 and -9 and the effector caspases-3 and -6 have been observed in experimental models of AD [68] and PD [69,70,71]. The key role of Bax in MPTP-induced-neurotoxicity is illustrated by the demonstration that mutant mice deficient in Bax are resistant to the induced toxicity of PD models [39]. Caspase-1 has also been implicated in other neurodegenerative contexts, such as HD [72]. Precisely in the context of HD, distinct studies investigated the harmful influence of human mutant huntingtin in the apoptotic cascade, specifically by triggering various Bcl-2 Homology 3 (BH3)-only proteins [73]. The functional role of caspase-1 and -3 has also been described in ALS [74]. Taking all these molecular events together, the etiopathogenesis of the four most representative classic neurodegenerative disorders is widely associated with the dysregulation of apoptotic pathways, similar to the case of SZ, for which ongoing apoptotic molecular alterations have been described in the previous section.
3.2. Intervention of Apoptotic Molecular Pathways in Neurodegenerative Disorders
The implication of caspases has been proposed as a common therapeutic target for multineurodegenerative disorders [75] and interfering at this molecular level has led to promising results. The broad-spectrum cysteine protease inhibitors (cathepsin, calpain and caspase) benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK) [76], and peptide-inhibitors of caspases-2, -3 and -9 attenuated the loss of dopaminergic ventral midbrain cell bodies (but not neurites) [77]. Similarly, Z-VAD-FMK attenuated mutant SOD1-mediated cell death in transfected PC-12 cells and in transgenic SOD1 mice [74,78], and also resulted in delayed disease onset and mortality in these ALS animals [74]. Inhibiting caspase cleavage of huntingtin reduced toxicity and aggregate formation in neuronal and non-neuronal events [79]. Expression of a dominant-negative mutant form of caspase-1 in R6/2 mice extended survival and delayed the appearance of neuronal inclusions, receptor alterations and the onset of symptoms [72], and was associated with increased resistance to the neurotoxins used to model HD [80]. Pharmacological inhibition of particular members of the caspase family, such as caspases-2, -3, -8 and -12, protects against β-amyloid-induced apoptotic cell death in vitro [38,81]. Beyond caspases, but still sticking to anti-apoptotic interventions, overexpression of Bcl-2 protected dopaminergic cells against MPTP-induced neurodegeneration [82]. The tumor suppressor protein p53, which is activated after MPTP-intoxication [83], is one of the rare molecules known to regulate Bax expression [84]. p53-inhibitors attenuated MPTP- induced Bax upregulation and the degeneration of dopaminergic neurons [85], and p53-null mice were resistant to MPTP-induced death of dopaminergic neurons (Table S3).
3.3. Mitochondrial Anti-Apoptotic Targets against Neurodegeneration
The fact that apoptosis is mostly mediated by mitochondria explains why most previously mentioned intervened apoptotic molecules are mitochondrially-related. Bcl-2 is located in the mitochondrial outer membrane, whereas cytoplasmic bax is translocated to the mitochondria upon induction of cell death, and cytochrome-c and caspase-9 are released by mitochondria [86]. Accordingly, most anti-apoptotic targets halting neuronal damage found in the literature are aimed at mitochondria or are mitochondrially-driven (Tables S4 and S5). Inhibition of cytochrome-c release was associated with therapeutic benefits in HD mice [40]. Overexpression of Bcl-2 mitigated neurodegeneration in both in vitro and in vivo models of ALS [78] and have been shown to prolong survival in the classic model of the disease consisting of transgenic SOD1 mice [41]. The carbonic anhydrase-inhibitor methazolamide, previously related to therapeutic events in HD models, prevented β-amyloid-induced mitochondrial dysfunction and caspase activation, protecting neuronal and glial cells in vitro and in vivo [87]. Microarray analysis revealed that the cyclin-dependent kinase olomoucine promoted downregulation of the protein E1B 19-kDa-interacting protein 3 (BNIP3), a pro-apoptotic Bcl-2 family protein involved in mitochondrial disruption in lipopolysaccharide and nitric oxide (NO)-cell death induced microglial cells [88]. On the other hand, mitochondrial dysfunction promotes oxidative stress which, in turn, may precede apoptosis, thus oxidative damage is an interesting target in neurodegeneration. To date, mitochondrially targeted molecules against oxidative stress is still one of the most effective therapeutic strategies in neurodegenerative disorders [40,89,90], with the presentation of a wide array of molecules (Tables S4 and S5). Anti-apoptotic-related natural compounds and antioxidants have also shown therapeutic effectiveness in HD [91]. Withanolides exerted beneficial effects on cognitive functions and ultimately neuroprotective effects in HD models [90]. Interestingly, withanolide A promoted neuritic regeneration and synaptic reconstruction in other neurodegeneration models [92]. BN-82451, a newer antioxidant, improved motor ability and attenuated neurodegeneration in a mouse model of HD [90,93]. Vitamin C and α-lipoic acid also had beneficial effects on motor symptoms and extended survival rates in rodents [93]. Coenzyme Q10, a component of mitochondrial membranes and a free radical scavenger, which has also been associated with anti-apoptotic effects [94], presents therapeutic effects in HD models. Antioxidants have also been tested in the psychiatric field. Astaxanthin has been related to antidepressant-like effects in different experimental models, including AD and PD [95]. Importantly, nicotinamide, a precursor of the coenzymes NAD and NADP involved in energy metabolism via redox reactions, protects against ketamine-induced apoptotic neurodegeneration in the infant rat brain [96], which could mimic a prodromal stage of SZ [96].
Herein we provide quantitative data of mitochondrial outcomes related to the administration of anti-apoptotic compounds, indicating less oxidative stress biomarkers in AD [97], PD [98] and HD [99], increased mitochondrial biogenesis and ATP levels in PD [100] and increased mitochondrial respiration in AD [101], among others (Table S6).
3.4. Other Anti-Apoptotic-Related Molecular Pathways Associated with Neuroprotection
Often, also related to oxidative stress, hormone metabolism plays an important role in neurodegenerative processes. This is in line with the classical use of hormone withdrawal as a model of neurodegeneration [102]. Melatonin, the circadian hormone with antioxidant features, reversed H-89 induced spatial memory deficit with involvement of oxidative stress and mitochondrial function [103]. Recent clinical trials with melatonin in PD have not led to conclusive results as yet [104], although previous studies showed a significant improvement in clinical global impression (CGI: 6.1 versus 4.6; p= 0.024) [105]. Neuroprotection by estrogen takes place in the brain, and the mitochondrial compartment is considered the presumed therapeutic target [106]. 17β-Estradiol protected retinal nerve cells against H2O2-induced apoptosis by significantly inhibiting Bax-involved mitochondrial apoptosis via the activation of the protein kinase (AKT) signal pathway [107]. Hormones involved in glucose metabolism, known to be disrupted in comorbidity with several neurodegenerative processes, have also been associated with anti-apoptotic properties and improvement of several neurodegenerative features [108], and there is even a clinical trial in PD patients that showed promising initial results [109]. The neuroprotective effects of hormonal interventions go far beyond the molecular level, leading to the improvement of neurodegenerative clinical manifestations associated with the intervention of apoptosis [110].
In summary, the wide array of aforementioned caspase-inhibitors, mitochondrial, antioxidant and hormonal molecules included in this study interact with distinct biological pathways and have been associated with anti-apoptotic properties in neurodegeneration. The multitarget effects of such compounds underlie the pharmacological mechanisms of action to tackle neurodegeneration by converging in cell death. This common anti-apoptotic link is the requirement for their inclusion in this study, and the specific mechanisms of action against cell death have been tested in several models of neurodegeneration (Table S3). This is in line with the wide array of compounds that have been studied to treat the quadriad neurodegenerative disorders by interfering with apoptosis [111] at both the mitochondrial level (Table S4) and through the interaction with a variety of targets at distinct cell death signaling pathways (Table S5).
Both representative, qualitative (Table S5) and quantitative (Table S6) outcomes using apoptotic-inhibitors in neurodegenerative models, including AD, PD, HD and ALS have been summarized. First, quantitative data of molecular outcomes showed significant improvement of synaptic function in mice [112] and decreased neuronal death in neuroblastoma in AD [113] increased neuronal survival in PD neuroblastoma [114], HD [46] and ALS rodents [115], increase of neurotransmitters [116] and decreased proinflammatory cytokines in PD rodents [117], among others. Importantly, significant quantitative findings were not restricted to molecular data but also to symptom improvement, since quantitative clinical outcomes showed significant amelioration of behavioral parameters assessed in different neurodegenerative models including AD mice [72], HD rats [109,118], as well as time of survival, time before disease onset or disease progression in ALS mice [115], upon applying anti-apoptotic approaches (Table S6).
3.5. Targeting Apoptosis in SZ
Despite the involvement of apoptotic pathways in SZ and the evidence of anti-apoptotic therapeutic effectiveness in the classic neurodegenerative disorders, the number of studies investigating anti-apoptotic molecules is lacking in SZ (Table S2). Currently, the positive symptoms of SZ are clinically attenuated through a known battery of antipsychotics. Interestingly, certain second-generation antipsychotics could have a neuroprotective role in vitro via an anti-apoptotic action, as demonstrated in a previous study from our group using a neuroblastoma cell model. The results indicated that haloperidol induces apoptosis, while risperidone and paliperidone may afford protection against it [119]. Both olanzapine and clozapine present antioxidative and anti-apoptotic activities [120]. Accordingly, antipsychotics have been related to functional changes in mitochondria as the main apoptotic orchestrator. Clozapine has been specifically reported to improve mitochondrial function by altering mitochondrial membrane potential [121]. Conversely, haloperidol, a first-generation antipsychotic, is linked to destructive changes in mitochondria [90,121]. A recent study also found that clozapine may have a neuroprotective effect on adult neural stem cells from ketamine-induced cell death in correlation with decreased apoptosis [122].
Other widespread pharmacological options, beyond antipsychotics and related to anti-apoptotic mechanisms, have been associated with neuroprotection in the context of SZ at the mitochondrial level (Table S4), mainly as adjunctive therapies in patients. Adjuntive N-acetylcysteine has been reported to reduce negative and general symptoms in SZ patients in several clinical trials [123]. N-acetylcysteine was associated with neuroprotective effects and prevented apoptosis mediated signals in various rodent models underlying mitochondrial malfunctioning in SZ pathology [121]. It protects against cadmium-induced ROS toxicity marked by reduced mitochondrial membrane potential, reduced Bcl-2 expression and p53 expression and a reduction in the caspase pathways [121].
Despite the lack of studies using caspase and apoptotic-inhibitors in SZ (Table S2), a limited proportion of previously expounded substances associated with anti-apoptotic mechanisms have been tested in this psychiatric disease (Table S7). Melatonin has been reported both as a surrogate marker and therapeutic agent in SZ [124]. SZ-like behavior was reported to be unchanged by melatonin supplementation in rodents [125], whereas other studies report attenuation of SZ-like symptoms and a protective effect on the prefrontal cortex region of brain by mitigating the alteration of neurotoxicity markers [126]. Another double-hit compound in SZ, considered both a surrogate marker and a therapeutic approach, is retinoic acid [127]. Retinoic acid, described to protect against proteasome inhibition-associated cell death in neuroblastoma cells via the survival protein kinase B (AKT) pathway [128], has been used as an add-on with antipsychotic treatment and showed a significant reduction of positive symptoms in SZ patients [129]. Finally, other adjunctive therapies with antipsychotics have also been tested with other anti-apoptotic-like substances, including estradiol, quercetin and erythropoietin, among others, all leading to promising outcomes [130,131,132] (Table S7). However, the experience using anti-apoptotic molecules exclusively rather than as an adjunctive treatment in SZ patients is null.
4. Discussion
Despite the high number of studies and growing evidence of the effectiveness of anti-apoptotic therapeutic strategies in classic neurodegenerative disorders, there is very little research on anti-apoptotic targets in SZ. This would likely be explained by the limited investment in the psychiatric field, even in developed countries [133], rather than by the lack of scientific evidence of the potential positive effects of these surrogate anti-apoptotic therapies in SZ. Considering both the neuroprogressive and dendritic apoptotic nature of SZ [20,62], through excessive synaptic pruning derived from glutamatergic storm and associated excitotoxicity, the use of apoptotic-inhibitors in SZ is likely to be a promising approach. This study arose as a proposal for the use of anti-apoptotic drugs and their potential therapeutic effect in SZ. In summary, experimental SZ models via ketamine and MK801 induction, interacting with GABA/NMDA receptors, promote subsequent apoptotic molecular pathways underlying the symptomatology of SZ. Such molecular events, related to neuronal death, may be inhibited by anti-apoptotic compounds that eventually protect against excessive synaptic pruning in the SZ brain, preventing the clinical symptoms of the disease. Specifically, anti-apoptotic molecules inhibit ROS production and promote mitochondrial membrane potential maintenance, mainly by blocking several key elements of the apoptotic cascade, including cytochrome-c release or caspase activation, ultimately resulting in protection against the accelerated dendritic apoptosis seen in SZ. The scientific evidence for the therapeutic effectiveness of anti-apoptotic strategies in neurological disorders is striking and goes far beyond the data presented herein. Targeting programmed cell death in ischemia/stroke [134] has been a common strategy which has not been considered in this study, as such mechanical models are most likely far from the molecular neurodevelopmental triggers in SZ and, subsequently, fall outside the scope of this review.
Targeting cell death is not only useful as a novel therapeutic option but also to elucidate the etiopathogenic basis of the disease. Therefore, by the use of X-linked apoptotic inhibitor (XIAP), a new relevant role of caspase-9 was elucidated in ALS [135]; or, by using the pan caspase inhibitor Z-VAD-FMK, a new role for caspase activation as a potential route rather than an obligatory initiation step of tubulin associated unit (TAU) aggregation was defined in AD [136]. The fact that the molecular basis of SZ is not fully established gives rise to another argument encouraging the use of anti-apoptotic molecules to further shed light on the unknown molecular nature of this disease.
The available anti-apoptotic approaches with great potential to restore biological pathways do not only include anti-caspase molecules, but also other apoptotic targets that could actively intervene, such as calpains [137] or cathepsins [138], among others. Upstream targets at the mitochondrial level deserve special attention [139], as well as antioxidative strategies, since they target oxidative damage preceding subsequent apoptosis [140]. Such is the case of N-acetyl-cysteine that has been tested in SZ leading to promising results [141,142]. The phosphodiesterase-type-5-inhibitor sildenafil modulated the expression of pro- and anti-apoptotic proteins of the extrinsic and intrinsic pathways and promoted remyelination in the spinal cord [143], indicating neuroprotective effects in a placebo-controlled study on cognition in SZ [144]. Thus, this review has considered multitarget molecules ultimately targeting cell death while hypothesizing that an anti-apoptotic prodromal and upstream intervention via bax inhibition and cytochrome-c release would prevent SZ-like symptoms.
There is evidence of the interaction of antipsychotic drugs with the apoptotic pathways [120]. The dopamine-stabilizer pridopidine protected cells from apoptosis, and resulted in highly improved motor performance in the HD model using R6/2 mice [145]. Chlorpromazine, an antipsychotic agent, induces G2/M phase arrest and apoptosis via regulation of the PI3K/AKT/mTOR-mediated autophagy pathways in human oral cancer [146]. Prototypical antipsychotic drugs protect hippocampal neuronal cultures against cell death induced by growth medium deprivation [147]. Additionally, the literature also reports a close interaction between some drugs altering neurotransmitter levels and mitochondria [148]. In contrast to these neuroprotective effects of some antipsychotics, haloperidol has been recently found to present pro-apoptotic features [149], which, in a paradoxical manner, would eventually aggravate the disease. Interestingly, erythropoietin prevented haloperidol-induced neuronal apoptosis through the regulation of brain-derived neurotrophic factor (BDNF) [150], and protective effects against haloperidol-toxicity have also been reported for cystamine [151]. Regardless of such controversial anti- or pro-apoptotic effects associated with different antipsychotic drugs depicted in the literature, the antipsychotic-apoptotic relationship suggests that the apoptotic pathways are indeed involved in the course of SZ and, therefore, in line with the rationale of the present study, anti-apoptotic strategies could serve as the optimal therapeutic option for SZ. However, adequate therapeutic drugs targeting apoptosis that allow us to more effectively halt, prevent or revert the development of SZ are yet to be discovered. In addition, as reviewed, the current anti-apoptotic approaches in SZ have been mainly tested as adjunctive treatments included within antipsychotic schedules rather than as exclusive interventions in SZ patients [129], probably due to the lack of preliminary in vitro and in vivo research studies.
From the main co-treatment of anti-apoptotic approaches combined with antipsychotics reported in SZ, minocycline is probably one of the most representative candidates. This antibiotic with anti-apoptotic effects has been widely studied in different neurological disorders including AD [152], PD [153], HD [154] and ALS models [155], among others, leading to promising findings in most cases [156]. The anti-apoptotic action of minocycline consists in the inhibition of cytochrome-c release from mitochondria by attenuating the mitochondrial permeability transition pore and inhibiting caspase-1 and -3 [153]. Minocycline inhibited Aβ-fibril formation [152], attenuated amyloid-induced microglial activation [157] and reduced the inflammatory events associated with the prevention of cognitive deficits. Minocycline has also been used in several clinical trials in the main neurodegenerative disorders, including AD, although, in the most recent clinical trial, it did not delay the progress of cognitive or functional impairment in people with mild AD during a two-year period [158]. Minocycline administration has also been associated with beneficial effects in HD [93] and related to delayed mortality in a transgenic mouse model of HD [159] and of ALS [155]. Accordingly, it is one of the few anti-apoptotic approaches conducted in SZ [160]. While some studies do not report any improvement associated with minocycline administration and failure of inhibition of cytochrome-c release [101], others report promising findings preventing neurodegeneration [153,156]. Controversial data have been found for the use of minocycline in SZ as well. Whereas some studies do not support a therapeutic role of minocycline in SZ [161], others cautiously report that minocycline may be helpful in treating negative and cognitive symptoms in SZ [162]. Minocycline administration has been associated with an amelioration of cognitive deficits and correlated with the remission of negative symptoms and reduction of inflammatory parameters [163]. Robust clinical improvements with minocycline treatment have also been described via the Positive and Negative-Syndrome-Scale (PANSS) for SZ [164]. Controversial data regarding minocycline administration extends to other molecules with anti-apoptotic properties in SZ. SZ-like behavior was not altered by melatonin supplementation in rodents [125]. However this compound has been reported to improve sleep disturbance, antipsychotic side effects and benzodiazepine discontinuation in SZ [124]. Together, these data demonstrate the scarcity of conclusive data and the lack of research targeting apoptotic molecules in SZ, underpinning the urgent need to further investigate this unexplored field.
When proposing anti-apoptotic therapeutic targets in SZ, special attention should be addressed to potential side effects related to such treatments, especially those concerning teratogenesis, carcinogenesis and cytotoxicity, considering their ability to intercede in cell death. Most of the studies conducted in in vitro and in vivo experimental interventions, herein reviewed, mainly investigate molecular events related to the drug-derived mechanism of action, without reporting any off-target effect [165,166]. On one hand, the FMK group of general cysteine protease inhibitors (cathepsin, calpain and caspase) cause irreversible enzyme inhibition making them unsuitable for clinical use. However, interestingly, most of the anti-apoptotic interventions addressed in other neuropathological contexts are commercially available and, therefore, their adverse secondary events have already been tested. Minocycline clinical trials demonstrated adequate tolerability and did not report adverse events after two years of administration [154]. Memantine, used in AD [167,168], has been mainly related to headache, constipation, sleepiness, and dizziness and severe side effects may include blood clots, psychosis, and heart failure [167]. High doses of methylene blue (>10µM) have been associated with cytotoxic effects in vitro [169]. It seems that all proposed uses of methylene blue entail levels that block monoamine oxidase, so cessation of serotonin reuptake inhibitors should be very carefully considered before using methylene blue [170]. Cytoprotective effects and cytochrome-P450 toxicity has been studied in a library of anti-apoptotic compounds leading to some safe molecules such as selective, reversible, small-molecule caspase-3-inhibitor (RBC1023) [171].
It is important to emphasize the challenging nature of anti-apoptotic interventions in neurological contexts. Numerous pharmacological compounds have been investigated due to their potential ability to reduce neuronal injury. Despite promising data from laboratory work, most of these agents presented disappointing clinical results. This is mainly due to the complex mechanisms involved in neuronal injury and the difficulty in controlling physiological factors, probably hampering the availability of a “perfect model of the disease” (current SZ models may be far from the real biological processes underlying the disease). One of these complexities relies on the time framework in which both the investigations and the interventions are conducted. It is most likely that prodromal interventions represent the most effective therapeutic option for SZ, before the apoptotic cascade leading to irreversible excessive synaptic pruning occurs. Moreover, heterogeneity of SZ could impact on the therapeutic efficacy of anti-apoptotic drugs when considering the loss of dendritic spines in the prefrontal cortex, while other brain regions present opposite results (such as dorsal and ventral striatum, where there are more synapses and spines). The combination of multiple strategies, including prodromal intervention, especially in patients with prominent deficit symptoms, and the use of compounds targeting different apoptosis-related pathways and the control of physiological variables, may afford the most meaningful results focused on neuroprotection in SZ.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antiox11112275/s1, Figure S1: Flow chart of the studies included in this review; Table S1: In vitro and in vivo models using Bax-/Bax- genetic manipulation, underpinning the relevance of mitochondrial apoptotic targets in the therapeutic approaches for neurodegeneration; Table S2: Number of studies using apoptotic inhibitors in the classic neurodegenerative disorders including AD, HD and PD vs. SZ; Table S3: In vivo and in vitro approaches against apoptosis in several neurodegenerative experimental models.; Table S4: Mitochondrially-targeted drugs in neurodegenerative and other neurological disorders; Table S5: Neuroprotective substances targeting cell death at distinct molecular levels in different neuropathological models; Table S6: Quantitative data of molecular and clinical outcomes by using anti-apoptotic compounds in models of neurodegeneration and neuroprogression; Table S7: Therapeutic approaches against SZ with compounds indirectly targeting apoptosis. References [39,40,72,78,85,90,96,97,98,101,103,108,111,112,113,115,117,118,128,130,132,135,137,139,140,143,144,145,151,155,163,166,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249] are cited in the supplementary materials.
Author Contributions
Conceptualization, E.P.; methodology, N.T. and C.M.; software, N.T., C.M., S.M. (Sergi Mas), M.G.; validation, N.A., S.M. (Santiago Madero), N.R., P.G.; formal analysis, E.P.; investigation, P.G., C.M., A.M.-P.; resources, A.M.-P., E.P.; data curation, A.M.-P., P.G., N.R.; writing—original draft preparation, N.T., C.M.; writing—review and editing, C.M., N.T.; visualization, P.G., S.M. (Sergi Mas), N.A., N.R., M.G., E.P.; supervision, E.P.; project administration, P.G.; funding acquisition, E.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by the Catalan Pons Balmes Grant (FCRB_PB_2018) and by the Spanish Ministry of Economy, Industry and Competitiveness, Carlos III Health Institute, Fondo de Investigación Sanitaria (FIS)—The European Regional Development Fund (FEDER – una manera de hacer Europa) (PI18/01005 and PI21/00552) and the Government of Catalonia, Secretaria d’Universitats i Recerca del Departament d’Economia I Coneixement (2017SGR1355; 2017SGR1562).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marder, S.R.; Cannon, T.D. Schizophrenia. N. Engl. J. Med. 2019, 381, 1753–1761. [Google Scholar] [CrossRef]
- Owen, M.J.; O’Donovan, M.; Thapar, A.; Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 2011, 198, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.E.; Shelton, M.A.; Sweet, R.A. Dendritic spine alterations in schizophrenia. Neurosci. Lett. 2015, 601, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Cannon, T.D. How Schizophrenia Develops: Cognitive and Brain Mechanisms Underlying Onset of Psychosis. Trends Cogn. Sci. 2015, 19, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, P.M.; Abdul, F.; Maes, M.; Binu, V.; Venkatasubramanian, G.; Kutty, B.M.; Debnath, M. Maternal Immune Activation Causes Schizophrenia-like Behaviors in the Offspring through Activation of Immune-Inflammatory, Oxidative and Apoptotic Pathways, and Lowered Antioxidant Defenses and Neuroprotection. Mol. Neurobiol. 2020, 57, 4345–4361. [Google Scholar] [CrossRef]
- Vasilescu, A.-N.; Mallien, A.; Pfeiffer, N.; Lang, U.E.; Gass, P.; Inta, D. Rapastinel alleviates the neurotoxic effect induced by NMDA receptor blockade in the early postnatal mouse brain. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 1587–1591. [Google Scholar] [CrossRef]
- Martínez-Pinteño, A.; García-Cerro, S.; Mas, S.; Torres, T.; Boloc, D.; Rodríguez, N.; Lafuente, A.; Gassó, P.; Arnaiz, J.A.; Parellada, E. The positive allosteric modulator of the mGlu2 receptor JNJ-46356479 partially improves neuropathological deficits and schizophrenia-like behaviors in a postnatal ketamine mice model. J. Psychiatr. Res. 2020, 126, 8–18. [Google Scholar] [CrossRef]
- Yuede, C.M.; Wozniak, D.F.; Creeley, C.E.; Taylor, G.T.; Olney, J.W.; Farber, N.B. Behavioral Consequences of NMDA Antagonist-Induced Neuroapoptosis in the Infant Mouse Brain. PLoS ONE 2010, 5, e11374. [Google Scholar] [CrossRef]
- Wesseling, H.; Want, E.J.; Guest, P.C.; Rahmoune, H.; Holmes, E.; Bahn, S. Hippocampal Proteomic and Metabonomic Abnormalities in Neurotransmission, Oxidative Stress, and Apoptotic Pathways in a Chronic Phencyclidine Rat Model. J. Proteome Res. 2015, 14, 3174–3187. [Google Scholar] [CrossRef]
- D’Addario, C.; Micale, V.; Di Bartolomeo, M.; Stark, T.; Pucci, M.; Sulcova, A.; Palazzo, M.; Babinska, Z.; Cremaschi, L.; Drago, F.; et al. A preliminary study of endocannabinoid system regulation in psychosis: Distinct alterations of CNR1 promoter DNA methylation in patients with schizophrenia. Schizophr. Res. 2017, 188, 132–140. [Google Scholar] [CrossRef]
- Ruda-Kucerova, J.; Babinska, Z.; Amchova, P.; Stark, T.; Drago, F.; Sulcova, A.; Micale, V. Reactivity to addictive drugs in the methylazoxymethanol (MAM) model of schizophrenia in male and female rats. World J. Biol. Psychiatry 2017, 18, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Ruda-Kucerova, J.; Babinska, Z.; Stark, T.; Micale, V. Suppression of Methamphetamine Self-Administration by Ketamine Pre-treatment Is Absent in the Methylazoxymethanol (MAM) Rat Model of Schizophrenia. Neurotox. Res. 2017, 32, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Drazanova, E.; Ruda-Kucerova, J.; Krátká, L.; Stark, T.; Kuchar, M.; Maryska, M.; Drago, F.; Starcuk, Z.; Micale, V. Different effects of prenatal MAM vs. perinatal THC exposure on regional cerebral blood perfusion detected by Arterial Spin Labelling MRI in rats. Sci. Rep. 2019, 9, 6062. [Google Scholar] [CrossRef]
- Horska, K.; Kotolova, H.; Karpisek, M.; Babinska, Z.; Hammer, T.; Prochazka, J.; Stark, T.; Micale, V.; Ruda-Kucerova, J. Metabolic profile of methylazoxymethanol model of schizophrenia in rats and effects of three antipsychotics in long-acting formulation. Toxicol. Appl. Pharmacol. 2020, 406, 115214. [Google Scholar] [CrossRef]
- Kucera, J.; Horska, K.; Hruska, P.; Kuruczova, D.; Micale, V.; Ruda-Kucerova, J.; Bienertova-Vasku, J. Interacting effects of the MAM model of schizophrenia and antipsychotic treatment: Untargeted proteomics approach in adipose tissue. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 108, 110165. [Google Scholar] [CrossRef] [PubMed]
- Stark, T.; Iannotti, F.A.; Di Martino, S.; Di Bartolomeo, M.; Ruda-Kucerova, J.; Piscitelli, F.; Wotjak, C.T.; D’Addario, C.; Drago, F.; Di Marzo, V.; et al. Early Blockade of CB1 Receptors Ameliorates Schizophrenia-like Alterations in the Neurodevelopmental MAM Model of Schizophrenia. Biomolecules 2022, 12, 108. [Google Scholar] [CrossRef]
- Micale, V.; Di Bartolomeo, M.; Di Martino, S.; Stark, T.; Dell’Osso, B.; Drago, F.; D’Addario, C. Are the epigenetic changes predictive of therapeutic efficacy for psychiatric disorders? A translational approach towards novel drug targets. Pharmacol. Ther. 2022, 108279. [Google Scholar] [CrossRef]
- Modinos, G.; Allen, P.; Grace, A.A.; McGuire, P. Translating the MAM model of psychosis to humans. Trends Neurosci. 2015, 38, 129–138. [Google Scholar] [CrossRef]
- Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A.; Jarskog, L.F. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res. 2006, 81, 47–63. [Google Scholar] [CrossRef]
- Parellada, E.; Gassó, P. Glutamate and microglia activation as a driver of dendritic apoptosis: A core pathophysiological mechanism to understand schizophrenia. Transl. Psychiatry 2021, 11, 271. [Google Scholar] [CrossRef]
- Li, W.; Ghose, S.; Gleason, K.; Begovic’, A.; Perez, J.; Bartko, J.; Russo, S.; Wagner, A.D.; Selemon, L.; Tamminga, C.A. Synaptic Proteins in the Hippocampus Indicative of Increased Neuronal Activity in CA3 in Schizophrenia. Am. J. Psychiatry 2015, 172, 373–382. [Google Scholar] [CrossRef]
- Scholz, J.; Broom, D.C.; Youn, D.-H.; Mills, C.D.; Kohno, T.; Suter, M.R.; Moore, K.A.; Decosterd, I.; Coggeshall, R.E.; Woolf, C.J. Blocking Caspase Activity Prevents Transsynaptic Neuronal Apoptosis and the Loss of Inhibition in Lamina II of the Dorsal Horn after Peripheral Nerve Injury. J. Neurosci. 2005, 25, 7317–7323. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, R.W. Cell Death During Development of the Nervous System. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Koleske, A.J. Mechanisms of Synapse and Dendrite Maintenance and Their Disruption in Psychiatric and Neurodegenerative Disorders. Annu. Rev. Neurosci. 2010, 33, 349–378. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Kondo, S.; Krzyzanowska, A.; Hiromi, Y.; Truman, J.W. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 2006, 9, 1234–1236. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Poon, I.; Lucas, C.; Rossi, A.G.; Ravichandran, K. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Wyllie, A.; Kerr, J.; Currie, A. Cell Death: The Significance of Apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Oltval, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, G.; Keane, R.W. Inhibitors of Apoptosis Proteins in Injury and Disease. IUBMB Life 2002, 54, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Apoptosis and Alzheimer’s disease. J. Neural Transm. 2000, 107, 1325–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Yang, H.; Liu, W. Balancing Apoptosis and Autophagy for Parkinson’s Disease Therapy: Targeting BCL-2. ACS Chem. Neurosci. 2019, 10, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.A.; Chesselet, M.-F. Apoptosis in Huntington’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 255–265. [Google Scholar] [CrossRef]
- Sathasivam, S.; Ince, P.; Shaw, P. Apoptosis in amyotrophic lateral sclerosis: A review of the evidence. Neuropathol. Appl. Neurobiol. 2001, 27, 257–274. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Sinha, M.; Ahmad, K.; Khalid, M.; Choi, I. Study of Caspase 8 Inhibition for the Management of Alzheimer’s Disease: A Molecular Docking and Dynamics Simulation. Molecules 2020, 25, 2071. [Google Scholar] [CrossRef]
- Vila, M.; Jackson-Lewis, V.; Vukosavic, S.; Djaldetti, R.; Liberatore, G.; Offen, D.; Korsmeyer, S.J.; Przedborski, S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 2837–2842. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, S.; Pei, Z.; Drozda, M.; Stavrovskaya, I.G.; Del Signore, S.J.; Cormier, K.; Shimony, E.M.; Wang, H.; Ferrante, R.J.; et al. Inhibitors of Cytochrome c Release with Therapeutic Potential for Huntington’s Disease. J. Neurosci. 2008, 28, 9473–9485. [Google Scholar] [CrossRef] [PubMed]
- Kostic, V.; Jackson-Lewis, V.; de Bilbao, F.; Dubois-Dauphin, M.; Przedborski, S. Bcl-2: Prolonging Life in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. Science 1997, 277, 559–563. [Google Scholar] [CrossRef]
- Faizi, M.; Salimi, A.; Rasoulzadeh, M.; Naserzadeh, P.; Pourahmad, J. Schizophrenia induces oxidative stress and cytochrome C release in isolated rat brain mitochondria: A possible pathway for induction of apoptosis and neurodegeneration. Iran. J. Pharm. Res. 2014, 13, 93–100. [Google Scholar]
- Roberts, R.C. Mitochondrial dysfunction in schizophrenia: With a focus on postmortem studies. Mitochondrion 2021, 56, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Sastry, P.S.; Rao, K.S. Apoptosis and the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar] [CrossRef]
- Perkins, D.O.; Jeffries, C.D.; Do, K.Q. Potential Roles of Redox Dysregulation in the Development of Schizophrenia. Biol. Psychiatry 2020, 88, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef]
- Berger, G.E.; Wood, S.; McGorry, P.D. Incipient neurovulnerability and neuroprotection in early psychosis. Psychopharmacol. Bull. 2003, 37, 79–101. [Google Scholar] [PubMed]
- Jarskog, L.F.; Selinger, E.S.; Lieberman, J.A.; Gilmore, J.H. Apoptotic Proteins in the Temporal Cortex in Schizophrenia: High Bax/Bcl-2 Ratio Without Caspase-3 Activation. Am. J. Psychiatry 2004, 161, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.; Gilmore, J.H.; Selinger, E.S.; Lieberman, J.A. Cortical Bcl-2 protein expression and apoptotic regulation in schizophrenia. Biol. Psychiatry 2000, 48, 641–650. [Google Scholar] [CrossRef]
- Mattson, M.P.; Keller, J.N.; Begley, J.G. Evidence for Synaptic Apoptosis. Exp. Neurol. 1998, 153, 35–48. [Google Scholar] [CrossRef]
- Bennett, M. Schizophrenia: Susceptibility genes, dendritic-spine pathology and gray matter loss. Prog. Neurobiol. 2011, 95, 275–300. [Google Scholar] [CrossRef]
- Ertürk, A.; Wang, Y.; Sheng, M. Local Pruning of Dendrites and Spines by Caspase-3-Dependent and Proteasome-Limited Mechanisms. J. Neurosci. 2014, 34, 1672–1688. [Google Scholar] [CrossRef]
- Gilman, C.P.; Mattson, M.P. Do Apoptotic Mechanisms Regulate Synaptic Plasticity and Growth-Cone Motility? Neuromol. Med. 2002, 2, 197–214. [Google Scholar] [CrossRef]
- Mattson, M.P.; Duan, W. “Apoptotic” biochemical cascades in synaptic compartments: Roles in adaptive plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 152–166. [Google Scholar] [CrossRef]
- Gassó, P.; Mas, S.; Molina, O.; Lafuente, A.; Bernardo, M.; Parellada, E. Increased susceptibility to apoptosis in cultured fibroblasts from antipsychotic-naïve first-episode schizophrenia patients. J. Psychiatr. Res. 2014, 48, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Batalla, A.; Bargallo, N.; Gassó, P.; Molina, O.; Pareto, D.; Mas, S.; Roca, J.M.; Bernardo, M.; Lafuente, A.; Parellada, E. Apoptotic markers in cultured fibroblasts correlate with brain metabolites and regional brain volume in antipsychotic-naive first-episode schizophrenia and healthy controls. Transl. Psychiatry 2015, 5, e626. [Google Scholar] [CrossRef]
- Gassó, P.; Mas, S.; Rodríguez, N.; Boloc, D.; García-Cerro, S.; Bernardo, M.; Lafuente, A.; Parellada, E. Microarray gene-expression study in fibroblast and lymphoblastoid cell lines from antipsychotic-naïve first-episode schizophrenia patients. J. Psychiatr. Res. 2017, 95, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Szymona, K.; Dudzińska, E.; Karakuła-Juchnowicz, H.; Gil-Kulik, P.; Chomik, P.; Świstowska, M.; Gałaszkiewicz, J.; Kocki, J. Analysis of the expression of BAX, BCL2, BIRC6, CASP3, CASP9 apoptosis genes during the first episode of schizophrenia. Psychiatr. Polska 2019, 53, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Muka, T.; Glisic, M.; Milic, J.; Verhoog, S.; Bohlius, J.; Bramer, W.; Chowdhury, R.; Franco, O.H. A 24-step guide on how to design, conduct, and successfully publish a systematic review and meta-analysis in medical research. Eur. J. Epidemiol. 2020, 35, 49–60. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef] [PubMed]
- Csernansky, J.G. Neurodegeneration in Schizophrenia: Evidence from In Vivo Neuroimaging Studies. Sci. World J. 2007, 7, 135–143. [Google Scholar] [CrossRef]
- Benn, S.C.; Woolf, C.J. Adult neuron survival strategies—slamming on the brakes. Nat. Rev. Neurosci. 2004, 5, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Giovanni, A.; Keramaris, E.; Morris, E.J.; Hou, S.T.; O’Hare, M.; Dyson, N.; Robertson, G.S.; Slack, R.S.; Park, D.S. E2F1 Mediates Death of B-amyloid-treated Cortical Neurons in a Manner Independent of p53 and Dependent on Bax and Caspase 3. J. Biol. Chem. 2000, 275, 11553–11560. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; de Brito, O.M.; Lombardi, V.; Louros, S.; Ribeiro, M.; Almeida, S.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. FK506 ameliorates cell death features in Huntington’s disease striatal cell models. Neurochem. Int. 2011, 59, 600–609. [Google Scholar] [CrossRef]
- Ishigaki, S.; Liang, Y.; Yamamoto, M.; Niwa, J.-I.; Ando, Y.; Yoshihara, T.; Takeuchi, H.; Doyu, M.; Sobue, G. X-Linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J. Neurochem. 2002, 82, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Correia, S.; dos Santos, R.X.C.; Cardoso, S.; Moreira, P.; Clark, T.A.; Zhu, X.; Smith, M.A.; Perry, G. Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J. Bioenerg. Biomembr. 2009, 41, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Hunot, S.; Michel, P.P.; Muriel, M.-P.; Vyas, S.; Faucheux, B.A.; Mouatt-Prigent, A.; Turmel, H.; Srinivasan, A.; Ruberg, M.; et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2875–2880. [Google Scholar] [CrossRef]
- Hartmann, A.; Michel, P.P.; Troadec, J.-D.; Mouatt-Prigent, A.; Faucheux, B.A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J. Neurochem. 2001, 76, 1785–1793. [Google Scholar] [CrossRef]
- Tatton, N.A. Increased Caspase 3 and Bax Immunoreactivity Accompany Nuclear GAPDH Translocation and Neuronal Apoptosis in Parkinson’s Disease. Exp. Neurol. 2000, 166, 29–43. [Google Scholar] [CrossRef]
- Ona, V.O.; Li, M.; Vonsattel, J.P.G.; Andrews, L.J.; Khan, S.Q.; Chung, W.M.; Frey, A.S.; Menon, A.S.; Li, X.-J.; Stieg, P.E.; et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature 1999, 399, 263–267. [Google Scholar] [CrossRef]
- Farshbaf, M.J.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Li, M.; Ona, V.O.; Guégan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.-P.; Przedborski, S.; et al. Functional Role of Caspase-1 and Caspase-3 in an ALS Transgenic Mouse Model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef]
- Khan, S.; Ahmad, K.; Alshammari, E.M.A.; Adnan, M.; Baig, M.H.; Lohani, M.; Somvanshi, P.; Haque, S. Implication of Caspase-3 as a Common Therapeutic Target for Multineurodegenerative Disorders and Its Inhibition Using Nonpeptidyl Natural Compounds. BioMed Res. Int. 2015, 2015, 379817. [Google Scholar] [CrossRef] [PubMed]
- Schotte, P.; Declercq, W.; Van Huffel, S.; Vandenabeele, P.; Beyaert, R. Non-specific effects of methyl ketone peptide inhibitors of caspases. FEBS Lett. 1999, 442, 117–121. [Google Scholar] [CrossRef]
- Bilsland, J.; Roy, S.; Xanthoudakis, S.; Nicholson, D.W.; Han, Y.; Grimm, E.; Hefti, F.; Harper, S.J. Caspase Inhibitors Attenuate 1-Methyl-4-Phenylpyridinium Toxicity in Primary Cultures of Mesencephalic Dopaminergic Neurons. J. Neurosci. 2002, 22, 2637–2649. [Google Scholar] [CrossRef]
- Vila, M.; Przedborski, S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Singaraja, R.; Ellerby, L.; Savill, J.; Roy, S.; Leavitt, B.; Cattaneo, E.; Hackam, A.; Sharp, A.; Thornberry, N.; et al. Inhibiting Caspase Cleavage of Huntingtin Reduces Toxicity and Aggregate Formation in Neuronal and Nonneuronal Cells. J. Biol. Chem. 2000, 275, 19831–19838. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Ferrante, R.J.; Hughes, D.B.; Klivenyi, P.; Dedeoglu, A.; Ona, V.O.; Friedlander, R.M.; Beal, M.F. Malonate and 3-Nitropropionic Acid Neurotoxicity Are Reduced in Transgenic Mice Expressing a Caspase-1 Dominant-Negative Mutant. J. Neurochem. 2000, 75, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Gao, Z.; Zheng, L.; Zhang, C.; Liu, Z.; Yang, Y.; Teng, H.; Hou, L.; Yin, Y.; Zou, X. Protective Effects of Fucoidan on Aβ25–35 and d-Gal-Induced Neurotoxicity in PC12 Cells and d-Gal-Induced Cognitive Dysfunction in Mice. Mar. Drugs 2017, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Matthews, R.T.; Schulz, J.B.; Klockgether, T.; Liao, A.W.; Martinou, J.C.; Penney, J.B.; Hyman, B.T.; Beal, M.F. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J. Neurosci. 1998, 18, 8145–8152. [Google Scholar] [CrossRef] [PubMed]
- Mandir, A.S.; Simbulan-Rosenthal, C.M.; Poitras, M.F.; Lumpkin, J.R.; Dawson, V.L.; Smulson, M.E.; Dawson, T.M. A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J. Neurochem. 2002, 83, 186–192. [Google Scholar] [CrossRef]
- Thornborrow, E.C.; Patel, S.; Mastropietro, A.E.; Schwartzfarb, E.M.; Manfredi, J.J. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene 2002, 21, 990–999. [Google Scholar] [CrossRef]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.-S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxidative Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef]
- Tsou, Y.-C.; Wang, H.-H.; Hsieh, C.-C.; Sun, K.-H.; Sun, G.-H.; Jhou, R.-S.; Lin, T.-I.; Lu, S.-Y.; Liu, H.-Y.; Tang, S.-J. Down-regulation of BNIP3 by olomoucine, a CDK inhibitor, reduces LPS- and NO-induced cell death in BV2 microglial cells. Neurosci. Lett. 2016, 628, 186–193. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, V.; Singh, K.; Kumar, S.; Kim, Y.-S.; Lee, Y.-M.; Kim, J.-J. Therapeutic Advances for Huntington’s Disease. Brain Sci. 2020, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Kuboyama, T.; Tohda, C.; Komatsu, K. Neuritic regeneration and synaptic reconstruction induced by withanolide A. Br. J. Pharmacol. 2005, 144, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J. Novel therapeutic targets for Huntington’s disease. Expert Opin. Ther. Targets 2005, 9, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Cialdai, F.; Bolognini, D.; Vignali, L.; Iannotti, N.; Cacchione, S.; Magi, A.; Balsamo, M.; Vukich, M.; Neri, G.; Donati, A.; et al. Effect of space flight on the behavior of human retinal pigment epithelial ARPE-19 cells and evaluation of coenzyme Q10 treatment. Cell. Mol. Life Sci. 2021, 78, 7795–7812. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Aneva, I.Y.; Farzaei, M.H.; Sobarzo-Sánchez, E. The Neuroprotective Effects of Astaxanthin: Therapeutic Targets and Clinical Perspective. Molecules 2019, 24, 2640. [Google Scholar] [CrossRef]
- Ullah, N.; Ullah, I.; Lee, H.Y.; Naseer, M.I.; Seok, P.M.; Ahmed, J.; Kim, M.O. Protective Function of Nicotinamide Against Ketamine-induced Apoptotic Neurodegeneration in the Infant Rat Brain. J. Mol. Neurosci. 2012, 47, 67–75. [Google Scholar] [CrossRef]
- Wong-Guerra, M.; Jiménez-Martin, J.; Fonseca-Fonseca, L.A.; Ramírez-Sánchez, J.; Montano-Peguero, Y.; Rocha, J.B.; D’avila, F.; De Assis, A.M.; Souza, D.; Pardo-Andreu, G.L.; et al. JM-20 protects memory acquisition and consolidation on scopolamine model of cognitive impairment. Neurol. Res. 2019, 41, 385–398. [Google Scholar] [CrossRef]
- Wang, D.; Wong, H.K.; Feng, Y.-B.; Zhang, Z.-J. Paeoniflorin, a Natural Neuroprotective Agent, Modulates Multiple Anti-Apoptotic and Pro-apoptotic Pathways in Differentiated PC12 Cells. Cell. Mol. Neurobiol. 2013, 33, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Prakash, D.; Sudhandiran, G. Neuroprotective effect of naringin, a dietary flavonoid against 3-Nitropropionic acid-induced neuronal apoptosis. Neurochem. Int. 2011, 59, 1066–1073. [Google Scholar] [CrossRef]
- Rosa, A.I.; Fonseca, I.; Nunes, M.J.; Moreira, S.; Rodrigues, E.; Carvalho, A.N.; Rodrigues, C.M.; Gama, M.J.; Castro-Caldas, M. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Biochim. Biophys. Acta—Mol. Basis Dis. 2017, 1863, 2171–2181. [Google Scholar] [CrossRef]
- Månsson, R.; Hansson, M.J.; Morota, S.; Uchino, H.; Ekdahl, C.T.; Elmér, E. Re-evaluation of mitochondrial permeability transition as a primary neuroprotective target of minocycline. Neurobiol. Dis. 2007, 25, 198–205. [Google Scholar] [CrossRef][Green Version]
- Thompson, C.K.; Brenowitz, E.A. Caspase Inhibitor Infusion Protects an Avian Song Control Circuit from Seasonal-Like Neurodegeneration. J. Neurosci. 2008, 28, 7130–7136. [Google Scholar] [CrossRef]
- Sharif, R.; Aghsami, M.; Gharghabi, M.; Sanati, M.; Khorshidahmad, T.; Vakilzadeh, G.; Mehdizadeh, H.; Gholizadeh, S.; Taghizadeh, G.; Sharifzadeh, M. Melatonin reverses H-89 induced spatial memory deficit: Involvement of oxidative stress and mitochondrial function. Behav. Brain Res. 2017, 316, 115–124. [Google Scholar] [CrossRef]
- Gilat, M.; Jackson, A.C.; Marshall, N.S.; Rn, D.H.; Mullins, A.E.; Hall, J.M.; Fang, B.A.M.; Yee, B.J.; Wong, K.K.H.; Grunstein, R.R.; et al. Melatonin for Rapid Eye Movement Sleep Behavior Disorder in Parkinson’s disease: A Randomised Controlled Trial. Mov. Disord. 2020, 35, 344–349. [Google Scholar] [CrossRef]
- Kunz, D.; Mahlberg, R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J. Sleep Res. 2010, 19, 591–596. [Google Scholar] [CrossRef]
- Arnold, S.; Beyer, C. Neuroprotection by estrogen in the brain: The mitochondrial compartment as presumed therapeutic target. J. Neurochem. 2009, 110, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, B.; Zhu, C.; Feng, Y.; Wang, S.; Shahzad, M.; Hu, C.; Mo, M.; Du, F.; Yu, X. 17β-Estradiol Impedes Bax-Involved Mitochondrial Apoptosis of Retinal Nerve Cells Induced by Oxidative Damage via the Phosphatidylinositol 3-Kinase/Akt Signal Pathway. J. Mol. Neurosci. 2013, 50, 482–493. [Google Scholar] [CrossRef]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Hölscher, C. Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jalewa, J.; Sharma, M.; Li, G.; Li, L.; Hölscher, C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2015, 303, 42–50. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Jalewa, J.; Saunders-Wood, T.; Li, L.; Hölscher, C. Neuroprotective effects of an oxyntomodulin analogue in the MPTP mouse model of Parkinson’s disease. Eur. J. Pharmacol. 2015, 765, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Stankiewicz, A.; Mocan, A.; Sah, A.; Tzvetkov, N.T.; Huminiecki, L.; Horbańczuk, J.O.; Atanasov, A.G. Ethnopharmacological Approaches for Dementia Therapy and Significance of Natural Products and Herbal Drugs. Front. Aging Neurosci. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Flores, J.; Noël, A.; Beauchet, O.; Sjöström, P.J.; Leblanc, A.C. Methylene blue inhibits Caspase-6 activity, and reverses Caspase-6-induced cognitive impairment and neuroinflammation in aged mice. Acta Neuropathol. Commun. 2019, 7, 210. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Zhang, L.-J.; Chen, Z.; Liu, L.-B. The PTEN inhibitor bpV(pic) promotes neuroprotection against amyloid β-peptide (25-35)-induced oxidative stress and neurotoxicity. Neurol. Res. 2017, 39, 758–765. [Google Scholar] [CrossRef]
- Nataraj, J.; Manivasagam, T.; Thenmozhi, A.J.; Essa, M.M. Neuroprotective effect of asiatic acid on rotenone-induced mitochondrial dysfunction and oxidative stress-mediated apoptosis in differentiated SH-SYS5Y cells. Nutr. Neurosci. 2017, 20, 351–359. [Google Scholar] [CrossRef]
- Sagot, Y.; Toni, N.; Perrelet, D.; Lurot, S.; King, B.; Rixner, H.; Mattenberger, L.; Waldmeier, P.C.; Kato, A.C. An orally active anti-apoptotic molecule (CGP 3466B) preserves mitochondria and enhances survival in an animal model of motoneuron disease. Br. J. Pharmacol. 2000, 131, 721–728. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.; Bao, J.; Yu, Z.; Cai, M.; Li, X.; Wu, T.; Xiang, J.; Cai, D. Jia-Jian-Di-Huang-Yin-Zi decoction reduces apoptosis induced by both mitochondrial and endoplasmic reticulum caspase12 pathways in the mouse model of Parkinson’s disease. J. Ethnopharmacol. 2017, 203, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, R.; Fakhfouri, G.; Mehr, S.E.; Ghia, J.-E.; Genazzani, A.A.; Payandemehr, B.; Dehpour, A.R.; Mousavizadeh, K.; Lim, D. Tropisetron attenuates amyloid-beta-induced inflammatory and apoptotic responses in rats. Eur. J. Clin. Investig. 2013, 43, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- El-Sahar, A.E.; Rastanawi, A.A.; El-Yamany, M.F.; Saad, M.A. Dapagliflozin improves behavioral dysfunction of Huntington’s disease in rats via inhibiting apoptosis-related glycolysis. Life Sci. 2020, 257, 118076. [Google Scholar] [CrossRef]
- Gassó, P.; Mas, S.; Molina, O.; Bernardo, M.; Lafuente, A.; Parellada, E. Neurotoxic/neuroprotective activity of haloperidol, risperidone and paliperidone in neuroblastoma cells. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.-L.; Ito, H.; Kondo, T.; Uehara, T.; Ikeda, M.; Abe, H.; Saitoh, J.-I.; Noguchi, K.; Suzuki, M.; Kurachi, M. Antipsychotic drugs scavenge radiation-induced hydroxyl radicals and intracellular ROS formation, and protect apoptosis in human lymphoma U937 cells. Free Radic. Res. 2019, 53, 304–312. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Lundberg, M.; Curbo, S.; Bohman, H.; Agartz, I.; Ögren, S.-O.; Patrone, C.; Mansouri, S. Clozapine protects adult neural stem cells from ketamine-induced cell death in correlation with decreased apoptosis and autophagy. Biosci. Rep. 2020, 40, BSR20193156. [Google Scholar] [CrossRef]
- Chen, A.T.; Chibnall, J.T.; Nasrallah, H.A. Placebo-controlled augmentation trials of the antioxidant NAC in schizophrenia: A review. Ann. Clin. Psychiatry 2016, 28, 190–196. [Google Scholar]
- Morera-Fumero, A.L.; Abreu-Gonzalez, P. Role of Melatonin in Schizophrenia. Int. J. Mol. Sci. 2013, 14, 9037–9050. [Google Scholar] [CrossRef]
- Afonso, A.C.; Pacheco, F.D.; Canever, L.; Wessler, P.G.; Mastella, G.A.; Godoi, A.K.; Hubbe, I.; Bischoff, L.M.; Bialecki, A.V.S.; Zugno, A.I. Schizophrenia-like behavior is not altered by melatonin supplementation in rodents. An. Acad. Bras. Cienc. 2020, 92, e20190981. [Google Scholar] [CrossRef]
- Andrabi, S.S.; Vishnoi, S.; Kaushik, M.; Parveen, K.; Tabassum, H.; Akram, M.; Parvez, S. Reversal of Schizophrenia-like Symptoms and Cholinergic Alterations by Melatonin. Arch. Med. Res. 2019, 50, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Regen, F.; Cosma, N.-C.; Otto, L.R.; Clemens, V.; Saksone, L.; Gellrich, J.; Uesekes, B.; Ta, T.M.T.; Hahn, E.; Dettling, M.; et al. Clozapine modulates retinoid homeostasis in human brain and normalizes serum retinoic acid deficit in patients with schizophrenia. Mol. Psychiatry 2021, 26, 5417–5428. [Google Scholar] [CrossRef]
- Cheng, B.; Martinez, A.A.; Morado, J.; Scofield, V.; Roberts, J.L.; Maffi, S.K. Retinoic acid protects against proteasome inhibition associated cell death in SH-SY5Y cells via the AKT pathway. Neurochem. Int. 2013, 62, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Lerner, V.; Miodownik, C.; Gibel, A.; Kovalyonok, E.; Shleifer, T.; Goodman, A.B.; Ritsner, M.S. Bexarotene as Add-On to Antipsychotic Treatment in Schizophrenia Patients. Clin. Neuropharmacol. 2008, 31, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-B.; Zheng, W.; Ning, Y.-P.; Cai, D.-B.; Yang, X.-H.; Ungvari, G.S.; Ng, C.H.; Wang, C.-Y.; Xiang, Y.-T. Erythropoietin for Cognitive Deficits Associated with Schizophrenia, Bipolar Disorder, and Major Depression: A Systematic Review. Pharmacopsychiatry 2018, 51, 100–104. [Google Scholar] [CrossRef]
- Schwartz, D.L. Quercetin as an Augmentation Agent in Schizophrenia. J. Clin. Psychopharmacol. 2016, 36, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.; Levi, L.; Zamora, D.; Biegon, A.; SanGiovanni, J.P.; Davidson, M.; Burshtein, S.; Gonen, I.; Radu, P.; Pavalache, K.S.; et al. Effect of Adjunctive Estradiol on Schizophrenia Among Women of Childbearing Age: A Randomized Clinical Trial. JAMA Psychiatry 2019, 76, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef]
- Graham, S.H.; Chen, J. Programmed Cell Death in Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tsukita, K.; Iwasato, T.; Suzuki, Y.; Tomioka, M.; Tateno, M.; Nagao, M.; Kawata, A.; Saido, T.C.; Miura, M.; et al. The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 2003, 22, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Ashton, A.Q. Proteins—Advances in Research and Application: 2012 Edition; ScholarlyEditions: Atlanta, GA, USA, 2012. [Google Scholar]
- Bizat, N.; Galas, M.-C.; Jacquard, C.; Boyer, F.; Hermel, J.-M.; Schiffmann, S.N.; Hantraye, P.; Blum, D.; Brouillet, E. Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology 2005, 49, 695–702. [Google Scholar] [CrossRef]
- Gray, J.; Haran, M.M.; Schneider, K.; Vesce, S.; Ray, A.M.; Owen, D.; White, I.R.; Cutler, P.; Davis, J.B. Evidence That Inhibition of Cathepsin-B Contributes to the Neuroprotective Properties of Caspase Inhibitor Tyr-Val-Ala-Asp-Chloromethyl Ketone. J. Biol. Chem. 2001, 276, 32750–32755. [Google Scholar] [CrossRef]
- Gallego-Sandín, S.; Novalbos, J.; Rosado, A.; Cano-Abad, M.F.; Arias, E.; Abad-Santos, F.; García, A.G. Albumin prevents mitochondrial depolarization and apoptosis elicited by endoplasmic reticulum calcium depletion of neuroblastoma cells. Eur. J. Pharmacol. 2005, 520, 1–11. [Google Scholar] [CrossRef]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagl, S.; Hener, U.; Müller, W.E.; Kögel, D. Liposome-incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim. Biophys. Acta —Biomembr. 2011, 1808, 236–243. [Google Scholar] [CrossRef]
- Naguy, A.; Naguy, C. N-acetyl-cysteine in schizophrenia—There is more than meets the eyes! CNS Spectr. 2021, 26, 446–447. [Google Scholar] [CrossRef]
- Martínez-Banaclocha, M. N-acetyl-cysteine in Schizophrenia: Potential Role on the Sensitive Cysteine Proteome. Curr. Med. Chem. 2020, 27, 6424–6439. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Silva, E.; Araújo, S.M.D.R.; Oliveira, W.H.; Lós, D.; de França, M.E.R.; Bonfanti, A.; Peron, G.; Thomaz, L.D.L.; Verinaud, L.; Nunes, A.K.D.S.; et al. Sildenafil ameliorates EAE by decreasing apoptosis in the spinal cord of C57BL/6 mice. J. Neuroimmunol. 2018, 321, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C.; Cather, C.; Freudenreich, O.; Henderson, D.C.; Evins, A.E.; Culhane, M.A.; Walsh, J.P. A placebo-controlled study of sildenafil effects on cognition in schizophrenia. Psychopharmacology 2009, 202, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- Jhou, A.-J.; Chang, H.-C.; Hung, C.-C.; Lin, H.-C.; Lee, Y.-C.; Liu, W.-T.; Han, K.-F.; Lai, Y.-W.; Lin, M.-Y.; Lee, C.-H. Chlorpromazine, an antipsychotic agent, induces G2/M phase arrest and apoptosis via regulation of the PI3K/AKT/mTOR-mediated autophagy pathways in human oral cancer. Biochem. Pharmacol. 2021, 184, 114403. [Google Scholar] [CrossRef]
- Bastianetto, S.; Danik, M.; Mennicken, F.; Williams, S.; Quirion, R. Prototypical antipsychotic drugs protect hippocampal neuronal cultures against cell death induced by growth medium deprivation. BMC Neurosci. 2006, 7, 28. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Wang, H.; Wang, X.; Narayanan, M.V.; Stavrovskaya, I.G.; Kristal, B.S.; Friedlander, R.M. Nortriptyline Protects Mitochondria and Reduces Cerebral Ischemia/Hypoxia Injury. Stroke 2008, 39, 455–462. [Google Scholar] [CrossRef]
- Papadopoulos, F.; Isihou, R.; Alexiou, G.A.; Tsalios, T.; Vartholomatos, E.; Markopoulos, G.S.; Sioka, C.; Tsekeris, P.; Kyritsis, A.P.; Galani, V. Haloperidol Induced Cell Cycle Arrest and Apoptosis in Glioblastoma Cells. Biomedicines 2020, 8, 595. [Google Scholar] [CrossRef]
- Pillai, A.; Dhandapani, K.; Pillai, B.A.; Terry, A.V.; Mahadik, S.P. Erythropoietin Prevents Haloperidol Treatment-Induced Neuronal Apoptosis through Regulation of BDNF. Neuropsychopharmacology 2008, 33, 1942–1951. [Google Scholar] [CrossRef]
- Pillai, A.; Veeranan-Karmegam, R.; Dhandapani, K.; Mahadik, S.P. Cystamine prevents haloperidol-induced decrease of BDNF/TrkB signaling in mouse frontal cortex. J. Neurochem. 2008, 107, 941–951. [Google Scholar] [CrossRef]
- Familian, A.; Boshuizen, R.S.; Eikelenboom, P.; Veerhuis, R. Inhibitory effect of minocycline on amyloid β fibril formation and human microglial activation. Glia 2006, 53, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Hödl, A.K.; Hofmann, P.; Kapfhammer, H.-P. Neuroprotection in Huntington’s disease: A 2-year study on minocycline. Int. Clin. Psychopharmacol. 2004, 19, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Stavrovskaya, I.G.; Drozda, M.; Kim, B.Y.S.; Ona, V.; Li, M.; Sarang, S.; Liu, A.S.; Hartley, D.M.; Wu, D.C.; et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417, 74–78. [Google Scholar] [CrossRef]
- Du, Y.; Ma, Z.; Lin, S.; Dodel, R.C.; Gao, F.; Bales, K.R.; Triarhou, L.C.; Chernet, E.; Perry, K.W.; Nelson, D.L.G.; et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 14669–14674. [Google Scholar] [CrossRef]
- Fan, R.; Xu, F.; Previti, M.L.; Davis, J.; Grande, A.M.; Robinson, J.K.; Van Nostrand, W.E. Minocycline Reduces Microglial Activation and Improves Behavioral Deficits in a Transgenic Model of Cerebral Microvascular Amyloid. J. Neurosci. 2007, 27, 3057–3063. [Google Scholar] [CrossRef]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients With Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef]
- Solmi, M.; Veronese, N.; Thapa, N.; Facchini, S.; Stubbs, B.; Fornaro, M.; Carvalho, A.F.; Correll, C.U. Systematic review and meta-analysis of the efficacy and safety of minocycline in schizophrenia. CNS Spectr. 2017, 22, 415–426. [Google Scholar] [CrossRef]
- Deakin, B.; Suckling, J.; Dazzan, P.; Joyce, E.; Lawrie, S.M.; Upthegrove, R.; Husain, N.; Chaudhry, I.B.; Dunn, G.; Jones, P.B.; et al. Minocycline for Negative Symptoms of Schizophrenia and Possible Mechanistic Actions: The BeneMin RCT. Southampton (UK): NIHR Journals Library. 2019. Available online: http://www.ncbi.nlm.nih.gov/books/NBK545707/ (accessed on 17 June 2022).
- Hovens, J.E.; Onderwater, T.A.M. Minocycline for schizophrenia: A brief overview. Tijdschr. Voor Psychiatr. 2014, 56, 402–406. [Google Scholar]
- Zhang, L.; Zheng, H.; Wu, R.; Kosten, T.R.; Zhang, X.-Y.; Zhao, J. The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophr. Res. 2019, 212, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, T.; Yasukawa, R.; Yasuda, H.; Hayashida, M.; Inagaki, T.; Horiguchi, J. Minocycline as Adjunctive Therapy for Schizophrenia. Clin. Neuropharmacol. 2008, 31, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Mouri, A.; Lee, H.; Mamiya, T.; Aoyama, Y.; Matsumoto, Y.; Kubota, H.; Huang, W.; Chiou, L.; Nabeshima, T. Hispidulin attenuates the social withdrawal in isolated disrupted-in-schizophrenia-1 mutant and chronic phencyclidine-treated mice. Br. J. Pharmacol. 2020, 177, 3210–3224. [Google Scholar] [CrossRef] [PubMed]
- Timpe, J.M.; Wang, C.Z.; Kim, J.; Johnson, K.M. α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptor activation protects against phencyclidine-induced caspase-3 activity by activating voltage-gated calcium channels. J. Neurosci. Res. 2014, 92, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine Monotherapy for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Li, Y.; Lin, L.; Cao, Y. Anti-autophagic and anti-apoptotic effects of memantine in a SH-SY5Y cell model of Alzheimer’s disease via mammalian target of rapamycin-dependent and -independent pathways. Mol. Med. Rep. 2015, 12, 7615–7622. [Google Scholar] [CrossRef]
- Zhang, X.; Rojas, J.C.; Gonzalez-Lima, F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox. Res. 2006, 9, 47–57. [Google Scholar] [CrossRef]
- Gillman, P.K. CNS toxicity involving methylene blue: The exemplar for understanding and predicting drug interactions that precipitate serotonin toxicity. J. Psychopharmacol. 2011, 25, 429–436. [Google Scholar] [CrossRef]
- Ma, H.; Wu, J.; Wang, Y.; Liang, S. Cytoprotective effect of selective small-molecule caspase inhibitors against staurosporine-induced apoptosis. Drug Des. Dev. Ther. 2014, 8, 583–600. [Google Scholar] [CrossRef][Green Version]
- Zhu, L.-F.; Wu, X.-Y.; Liu, X.; Ma, L.; Zhang, Q.; Yu, Z.; He, Z.-J.; Ying, Y.; Zhang, Z.-T.; Pan, X.-Y.; et al. Absence of BAX differentially affects astrocyte density in the mouse cortex and hippocampus. Sheng Li Xue Bao Acta Physiol. Sin. 2021, 73, 1–9. [Google Scholar]
- D’Orsi, B.; Kilbride, S.M.; Chen, G.; Alvarez, S.P.; Bonner, H.P.; Pfeiffer, S.; Plesnila, N.; Engel, T.; Henshall, D.C.; Düssmann, H.; et al. Bax Regulates Neuronal Ca2+ Homeostasis. J. Neurosci. 2015, 35, 1706–1722. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.I.; Deshmukh, M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007, 14, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Tolkovsky, A.M. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J. Neurochem. 2006, 96, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Xiang, H.; London, S.; Kinoshita, Y.; Knudson, M.; Mayberg, M.; Korsmeyer, S.J.; Morrison, R.S. Evidence for involvement of Bax and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J. Neurosci. Res. 1998, 54, 721–733. [Google Scholar] [CrossRef]
- Miller, T.M.; Moulder, K.L.; Knudson, C.M.; Creedon, D.J.; Deshmukh, M.; Korsmeyer, S.J.; Johnson, E.M. Bax Deletion Further Orders the Cell Death Pathway in Cerebellar Granule Cells and Suggests a Caspase-independent Pathway to Cell Death. J. Cell Biol. 1997, 139, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Shin, J.; Nam, K.; An, J.S.; Yang, S.; Hong, S.; Bae, M.; Moon, K.; Cho, Y.; Woo, J.; et al. Rhizolutin, a Novel 7/10/6-Tricyclic Dilactone, Dissociates Misfolded Protein Aggregates and Reduces Apoptosis/Inflammation Associated with Alzheimer’s Disease. Angew. Chem. Int. Ed. 2020, 59, 22994–22998. [Google Scholar] [CrossRef]
- Rohn, T.T.; Kokoulina, P.; Eaton, C.R.; Poon, W.W. Caspase activation in transgenic mice with Alzheimer-like pathology: Results from a pilot study utilizing the caspase inhibitor, Q-VD-OPh. Int. J. Clin. Exp. Med. 2009, 2, 300–308. [Google Scholar]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Kanthasamy, A.G.; Anantharam, V.; Zhang, D.; Latchoumycandane, C.; Jin, H.; Kaul, S.; Kanthasamy, A. A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCδ) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic. Biol. Med. 2006, 41, 1578–1589. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.; Höllerhage, M.; Höglinger, G.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef] [PubMed]
- Aharony, I.; Ehrnhoefer, D.E.; Shruster, A.; Qiu, X.; Franciosi, S.; Hayden, M.; Offen, D. A Huntingtin-based peptide inhibitor of caspase-6 provides protection from mutant Huntingtin-induced motor and behavioral deficits. Hum. Mol. Genet. 2015, 24, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-W.; Jeon, G.S.; Kim, M.-J.; Shon, J.-H.; Kim, J.-E.; Shin, J.-Y.; Kim, S.-M.; Kim, S.H.; Ye, I.-H.; Lee, K.-W.; et al. Neuroprotective effects of JGK-263 in transgenic SOD1-G93A mice of amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Hawking, Z.L. Alzheimer’s disease: The role of mitochondrial dysfunction and potential new therapies: The International Journal of Student Research. Biosci. Horizons 2016, 9. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Sun, X.-Q.; Zhang, R.; Zhang, H.-D.; Yuan, P.; Wang, X.-J.; Zhao, Q.-H.; Wang, L.; Jiang, R.; Bogaard, H.J.; Jing, Z.-C. Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria-dependent apoptosis. Hypertens. Res. 2016, 39, 302–311. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J. The Contribution of Mitochondria to Sensory Processing and Pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell. Mol. Biol. 2017, 328, 49–103. [Google Scholar] [CrossRef]
- Marchionini, D.M.; Collier, T.J.; Pitzer, M.R.; Sortwell, C.E. Reassessment of caspase inhibition to augment grafted dopamine neuron survival. Cell Transplant. 2004, 13, 273–282. [Google Scholar] [CrossRef]
- Sivasangari, K.; Rajan, K.E. Standardized Bacopa monnieri Extract Ameliorates Learning and Memory Impairments through Synaptic Protein, Neurogranin, Pro-and Mature BDNF Signaling, and HPA Axis in Prenatally Stressed Rat Offspring. Antioxidants 2020, 9, 1229. [Google Scholar] [CrossRef]
- Lee, J.-E.; Sim, H.; Yoo, H.M.; Lee, M.; Baek, A.; Jeon, Y.-J.; Seo, K.-S.; Son, M.-Y.; Yoon, J.S.; Kim, J. Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease. Molecules 2020, 25, 3602. [Google Scholar] [CrossRef]
- Ravishankar, D.; Corona, G.; Hogan, S.M.; Spencer, J.P.; Greco, F.; Osborn, H.M. Thioflavones as novel neuroprotective agents. Bioorganic Med. Chem. 2016, 24, 5513–5520. [Google Scholar] [CrossRef]
- Nuñez-Figueredo, Y.; Pardo-Andreu, G.L.; Ramírez-Sánchez, J.; Delgado-Hernández, R.; Ochoa-Rodríguez, E.; Verdecia-Reyes, Y.; Naal, Z.; Muller, A.P.; Portela, L.V.; Souza, D.O. Antioxidant effects of JM-20 on rat brain mitochondria and synaptosomes: Mitoprotection against Ca2+-induced mitochondrial impairment. Brain Res. Bull. 2014, 109, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Momeni, H.R.; Kanje, M. Calpain inhibitors delay injury-induced apoptosis in adult mouse spinal cord motor neurons. Neuroreport 2006, 17, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Manigandan, V.; Nataraj, J.; Karthik, R.; Manivasagam, T.; Saravanan, R.; Thenmozhi, A.J.; Essa, M.M.; Guillemin, G.J. Low Molecular Weight Sulfated Chitosan: Neuroprotective Effect on Rotenone-Induced In Vitro Parkinson’s Disease. Neurotox. Res. 2019, 35, 505–515. [Google Scholar] [CrossRef]
- Casarejos, M.J.; Menéndez, J.; Solano, R.M.; Rodríguez-Navarro, J.A.; De Yébenes, J.G.; Mena, M.A. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J. Neurochem. 2006, 97, 934–946. [Google Scholar] [CrossRef]
- Sirianni, A.C.; Jiang, J.; Zeng, J.; Mao, L.L.; Zhou, S.; Sugarbaker, P.; Zhang, X.; Li, W.; Friedlander, R.M.; Wang, X. N-acetyl-l -tryptophan, but not N-acetyl-d -tryptophan, rescues neuronal cell death in models of amyotrophic lateral sclerosis. J. Neurochem. 2015, 134, 956–968. [Google Scholar] [CrossRef]
- Mookherjee, P.; Johnson, G.V. Tau phosphorylation during apoptosis of human SH-SY5Y neuroblastoma cells. Brain Res. 2001, 921, 31–43. [Google Scholar] [CrossRef]
- Gu, C.; Zhang, Y.; Hu, Q.; Wu, J.; Ren, H.; Liu, C.-F.; Wang, G. P7C3 inhibits GSK3β activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell Death Dis. 2017, 8, e2858. [Google Scholar] [CrossRef]
- Hu, X.; Weng, Z.; Chu, C.; Zhang, L.; Cao, G.; Gao, Y.; Signore, A.; Zhu, J.; Hastings, T.; Greenamyre, J.T.; et al. Peroxiredoxin-2 Protects against 6-Hydroxydopamine-Induced Dopaminergic Neurodegeneration via Attenuation of the Apoptosis Signal-Regulating Kinase (ASK1) Signaling Cascade. J. Neurosci. 2011, 31, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, K.; Bavithra, S.; Suganthi, M.; Benson, C.S.; Elumalai, P.; Arunkumar, R.; Krishnamoorthy, G.; Venkataraman, P.; Arunakaran, J. Protective Role of Quercetin on PCBs-Induced Oxidative Stress and Apoptosis in Hippocampus of Adult Rats. Neurochem. Res. 2012, 37, 708–721. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Deleglise, B.; Lassus, B.; Soubeyre, V.; Alleaume-Butaux, A.; Hjorth, J.J.; Vignes, M.; Schneider, B.; Brugg, B.; Viovy, J.-L.; Peyrin, J.-M. Synapto-Protective Drugs Evaluation in Reconstructed Neuronal Network. PLoS ONE 2013, 8, e71103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.; Reiriz, J.; Piqué, M.; Gil, J.; Ferrer, I.; Ambrosio, S. Low concentrations of 1-methyl-4-phenylpyridinium ion induce caspase-mediated apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 2001, 63, 421–428. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Hammami, M.; Khalouf, A.; Al Shaikh, Y.; Mohammed, A.K.; Hamad, M.; Salehi, A.; Taneera, J. Dimethyloxalylglycine (DMOG) and the Caspase Inhibitor “Ac-LETD-CHO” Protect Neuronal ND7/23 Cells of Gluocotoxicity. Exp. Clin. Endocrinol. Diabetes 2021, 129, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Park, H.Y.; Kim, M.O. Anthocyanins Protect against Kainic Acid-induced Excitotoxicity and Apoptosis via ROS-activated AMPK Pathway in Hippocampal Neurons. CNS Neurosci. Ther. 2014, 20, 327–338. [Google Scholar] [CrossRef]
- Song, D.; Jiang, X.; Liu, Y.; Sun, Y.; Cao, S.; Zhang, Z. Asiaticoside Attenuates Cell Growth Inhibition and Apoptosis Induced by Aβ1-42 via Inhibiting the TLR4/NF-κB Signaling Pathway in Human Brain Microvascular Endothelial Cells. Front. Pharmacol. 2018, 9, 28. [Google Scholar] [CrossRef]
- Werth, J.L.; Deshmukh, M.; Cocabo, J.; Johnson, E.M.; Rothman, S.M. Reversible Physiological Alterations in Sympathetic Neurons Deprived of NGF but Protected from Apoptosis by Caspase Inhibition or Bax Deletion. Exp. Neurol. 2000, 161, 203–211. [Google Scholar] [CrossRef]
- Jung, E.B.; Lee, C.S. Baicalein attenuates proteasome inhibition-induced apoptosis by suppressing the activation of the mitochondrial pathway and the caspase-8- and Bid-dependent pathways. Eur. J. Pharmacol. 2014, 730, 116–124. [Google Scholar] [CrossRef]
- Gliyazova, N.S.; Ibeanu, G.C. The Chemical Molecule B355252 is Neuroprotective in an In Vitro Model of Parkinson’s Disease. Cell. Mol. Neurobiol. 2016, 36, 1109–1122. [Google Scholar] [CrossRef]
- Silva, J.; Alves, C.; Pinteus, S.; Mendes, S.; Pedrosa, R. Seaweeds’ neuroprotective potential set in vitro on a human cellular stress model. Mol. Cell. Biochem. 2020, 473, 229–238. [Google Scholar] [CrossRef]
- Tseng, Y.-T.; Tsai, Y.-H.; Fülöp, F.; Chang, F.-R.; Lo, Y.-C. 2-Iodo-4′-Methoxychalcone Attenuates Methylglyoxal-Induced Neurotoxicity by Activation of GLP-1 Receptor and Enhancement of Neurotrophic Signal, Antioxidant Defense and Glyoxalase Pathway. Molecules 2019, 24, 2249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Boulton, A.A.; Zuo, D.-M.; Yu, P.H. MK-801 induces apoptotic neuronal death in the rat retrosplenial cortex: Prevention by cycloheximide and R(?)-2-Hexyl-N- Methylpropargylamine. J. Neurosci. Res. 1996, 46, 82–89. [Google Scholar] [CrossRef]
- Busquets, O.; Ettcheto, M.; Verdaguer, E.; Castro-Torres, R.D.; Auladell, C.; Beas-Zarate, C.; Folch, J.; Camins, A. JNK1 inhibition by Licochalcone A leads to neuronal protection against excitotoxic insults derived of kainic acid. Neuropharmacology 2018, 131, 440–452. [Google Scholar] [CrossRef]
- Xian, H.; Zhao, J.; Zheng, Y.; Wang, M.; Huang, J.; Wu, B.; Sun, C.; Yang, Y. MADP, a salidroside analog, protects hippocampal neurons from glutamate induced apoptosis. Life Sci. 2014, 103, 34–40. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.-Y.; Detaille, D.; R-Villanueva, G.; Esteban, M.D.; Guigas, B.; Attia, S.; Fontaine, E.; Almeida, A.; Leverve, X. Neuroprotective Role of Antidiabetic Drug Metformin Against Apoptotic Cell Death in Primary Cortical Neurons. J. Mol. Neurosci. 2008, 34, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.C.; John, J.M.; Lee, J.; Gonzalez-Lima, F. Methylene Blue Provides Behavioral and Metabolic Neuroprotection Against Optic Neuropathy. Neurotox. Res. 2009, 15, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; McInnis, J.; West, J.B.; Bao, J.; Anastasio, N.; Guidry, J.A.; Ye, Y.; Salvemini, D.; Johnson, K.M. Blockade of Phencyclidine-Induced Cortical Apoptosis and Deficits in Prepulse Inhibition by M40403, a Superoxide Dismutase Mimetic. J. Pharmacol. Exp. Ther. 2003, 304, 266–271. [Google Scholar] [CrossRef]
- Kumar, S.N.K.; Deepthy, J.; Saraswathi, U.; Thangarajeswari, M.; Kanna, S.Y.; Ezhil, P.; Kalaiselvi, P. Morinda citrifolia mitigates rotenone-induced striatal neuronal loss in male Sprague-Dawley rats by preventing mitochondrial pathway of intrinsic apoptosis. Redox Rep. 2017, 22, 418–429. [Google Scholar] [CrossRef]
- Liu, W.-B.; Zhou, J.; Qu, Y.; Li, X.; Lu, C.-T.; Xie, K.-L.; Sun, X.-L.; Fei, Z. Neuroprotective effect of osthole on MPP+-induced cytotoxicity in PC12 cells via inhibition of mitochondrial dysfunction and ROS production. Neurochem. Int. 2010, 57, 206–215. [Google Scholar] [CrossRef]
- De Sarno, P.; Shestopal, S.A.; King, T.D.; Zmijewska, A.; Song, L.; Jope, R.S. Muscarinic Receptor Activation Protects Cells from Apoptotic Effects of DNA Damage, Oxidative Stress, and Mitochondrial Inhibition. J. Biol. Chem. 2003, 278, 11086–11093. [Google Scholar] [CrossRef]
- Dong, H.; Li, R.; Yu, C.; Xu, T.; Zhang, X.; Dong, M. Paeoniflorin inhibition of 6-hydroxydopamine-induced apoptosis in PC12 cells via suppressing reactive oxygen species-mediated PKCδ/NF-κB pathway. Neuroscience 2015, 285, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Sanz, E.; Quintana, A.; Battaglia, V.; Toninello, A.; Hidalgo, J.; Ambrosio, S.; Valoti, M.; Marco, J.L.; Tipton, K.F.; Unzeta, M. Anti-apoptotic effect of Mao-B inhibitor PF9601N [N-(2-propynyl)-2-(5-benzyloxy-indolyl) methylamine] is mediated by p53 pathway inhibition in MPP+-treated SH-SY5Y human dopaminergic cells. J. Neurochem. 2008, 105, 2404–2417. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Jeong, W.-S.; Jun, M. Protective Effects of the Key Compounds Isolated from Corni fructus against β-Amyloid-Induced Neurotoxicity in PC12 Cells. Molecules 2012, 17, 10831–10845. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Sugama, S.; Mischak, R.P.; Kiaei, M.; Bizat, N.; Brouillet, E.; Joh, T.H.; Beal, M.F. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol. Dis. 2004, 17, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Dionne, K.R.; Leser, J.S.; Lorenzen, K.A.; Beckham, J.D.; Tyler, K.L. A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp. Neurol. 2011, 228, 222–231. [Google Scholar] [CrossRef]
- Sifringer, M.; Bendix, I.; Borner, C.; Endesfelder, S.; Von Haefen, C.; Kalb, A.; Holifanjaniaina, S.; Prager, S.; Schlager, G.W.; Keller, M.; et al. Prevention of neonatal oxygen-induced brain damage by reduction of intrinsic apoptosis. Cell Death Dis. 2012, 3, e250. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Han, E.S.; Han, Y.S.; Bang, H. Differential effect of calmodulin antagonists on MG132-induced mitochondrial dysfunction and cell death in PC12 cells. Brain Res. Bull. 2005, 67, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Choi, D.-K.; Jung, H.J. Neuroprotective Effects of Vanillyl Alcohol in Gastrodia elata Blume Through Suppression of Oxidative Stress and Anti-Apoptotic Activity in Toxin-Induced Dopaminergic MN9D Cells. Molecules 2011, 16, 5349–5361. [Google Scholar] [CrossRef]
- Perche, O.; Doly, M.; Ranchon-Cole, I. Transient protective effect of caspase inhibitors in RCS rat. Exp. Eye Res. 2008, 86, 519–527. [Google Scholar] [CrossRef]
- Cutillas, B.; Espejo, M.; Gil, J.; Ferrer, I.; Ambrosio, S. Caspase inhibition protects nigral neurons against 6-OHDA-induced retrograde degeneration. Neuroreport 1999, 10, 2605–2608. [Google Scholar] [CrossRef]
- Chen, W.-F.; Chakraborty, C.; Sung, C.-S.; Feng, C.-W.; Jean, Y.-H.; Lin, Y.-Y.; Hung, H.-C.; Huang, T.-Y.; Huang, S.-Y.; Su, T.-M.; et al. Neuroprotection by marine-derived compound, 11-dehydrosinulariolide, in an in vitro Parkinson’s model: A promising candidate for the treatment of Parkinson’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Bozzatello, P.; Brignolo, E.; De Grandi, E.; Bellino, S. Supplementation with Omega-3 Fatty Acids in Psychiatric Disorders: A Review of Literature Data. J. Clin. Med. 2016, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Ben-Azu, B.; Aderibigbe, A.O.; Omogbiya, I.A.; Ajayi, A.M.; Iwalewa, E.O. Morin Pretreatment Attenuates Schizophrenia-Like Behaviors in Experimental Animal Models. Drug Res. 2018, 68, 159–167. [Google Scholar] [CrossRef]
- Morera-Fumero, A.L.; Abreu-Gonzalez, P. Diazepam Discontinuation Through Agomelatine in Schizophrenia With Insomnia and Depression. J. Clin. Psychopharmacol. 2010, 30, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.; Laudon, M.; Barak, Y.; Anis, Y.; Rotenberg, V.; Elizur, A.; Zisapel, N. Melatonin Improves Sleep Quality of Patients With Chronic Schizophrenia. J. Clin. Psychiatry 2000, 61, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.; Barak, Y.; Shalman, I.; Laudon, M.; Zisapel, N.; Tarrasch, R.; Elizur, A.; Weizman, R. Melatonin Treatment for Tardive Dyskinesia: A double-blind, placebo-controlled, crossover study. Arch. Gen. Psychiatry 2001, 58, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Hassanpour, F.; Zarghami, M.; Mouodi, S.; Moosazadeh, M.; Barzegar, F.; Bagheri, M.; Hendouei, N. Adjunctive Memantine Treatment of Schizophrenia: A Double-Blind, Randomized Placebo-Controlled Study. J. Clin. Psychopharmacol. 2019, 39, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Sepehrmanesh, Z.; Heidary, M.; Akasheh, N.; Akbari, H.; Heidary, M. Therapeutic effect of adjunctive N-acetyl cysteine (NAC) on symptoms of chronic schizophrenia: A double-blind, randomized clinical trial. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kang, S.-S.; Kim, J.Y.; Tchah, H. The Antioxidant N-Acetylcysteine Inhibits Inflammatory and Apoptotic Processes in Human Conjunctival Epithelial Cells in a High-Glucose Environment. Investig. Opthalmology Vis. Sci. 2015, 56, 5614–5621. [Google Scholar] [CrossRef]
- A Wittkopp, T.; Abuzzahab, F.S. Nicotinic acid and nicotinamide adenine dinucleotide (NAD) therapy in schizophrenia: A review. Behav. Neuropsychiatry 1972, 4, 6–12. [Google Scholar] [PubMed]
- Lerner, V.; Miodownik, C.; Gibel, A.; Sirota, P.; Bush, I.; Elliot, H.; Benatov, R.; Ritsner, M.S. The Retinoid X Receptor Agonist Bexarotene Relieves Positive Symptoms of Schizophrenia. J. Clin. Psychiatry 2013, 74, 1224–1232. [Google Scholar] [CrossRef]
- Dakhale, G.N.; Khanzode, S.D.; Saoji, A. Supplementation of vitamin C with atypical antipsychotics reduces oxidative stress and improves the outcome of schizophrenia. Psychopharmacology 2005, 182, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.J. Vitamin therapy in schizophrenia. Isr. J. Psychiatry Relat. Sci. 2008, 45, 3–10. [Google Scholar] [PubMed]
- Sandyk, R.; Kanofsky, J.D. Vitamin C in the Treatment of Schizophrenia. Int. J. Neurosci. 1993, 68, 67–71. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).