
Hydropersulfides (RSSH) and Nitric Oxide (NO) Signaling: Possible Effects on S-Nitrosothiols (RS-NO)

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Physiological Relationship between H2S and NO
3. S-Nitrosothiols (RS-NOs) and NO Signaling
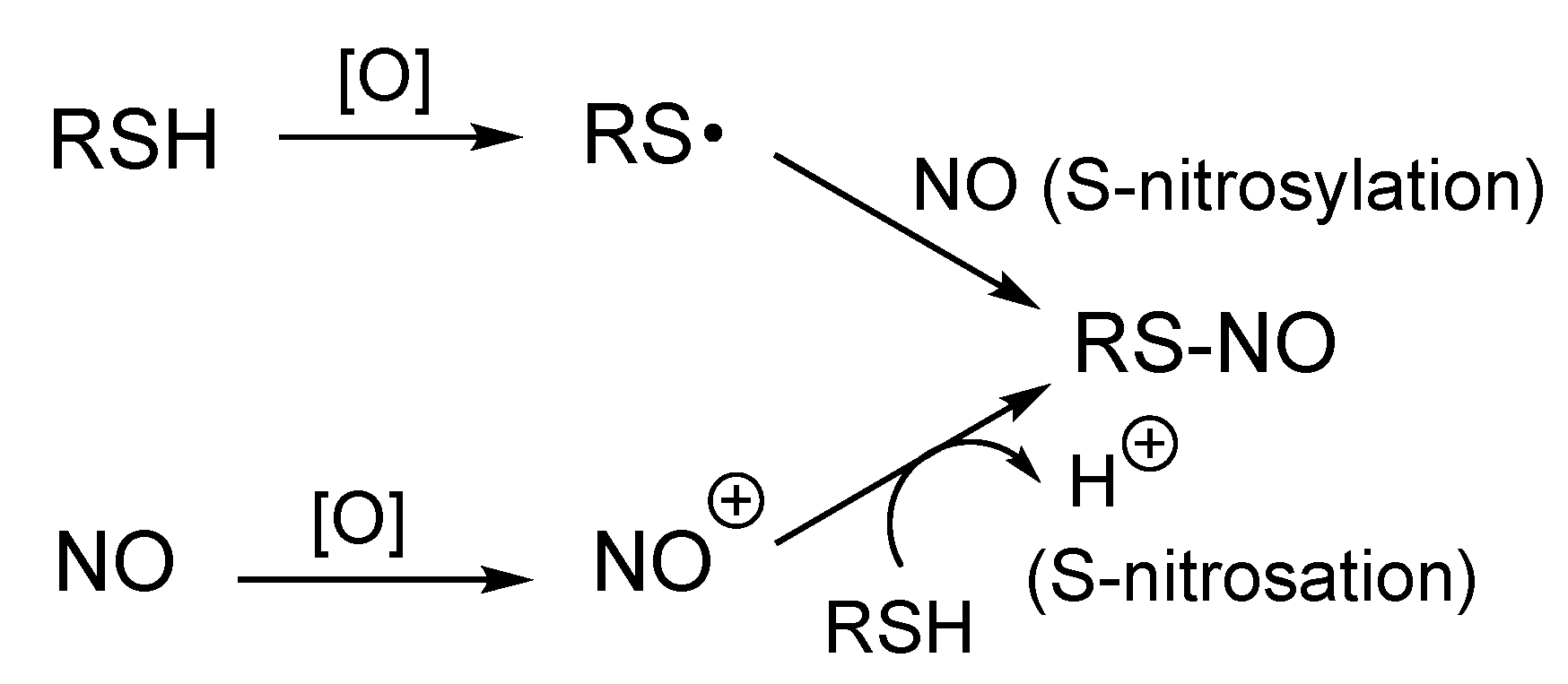
4. Possible Mechanisms of Formation of RS-NO
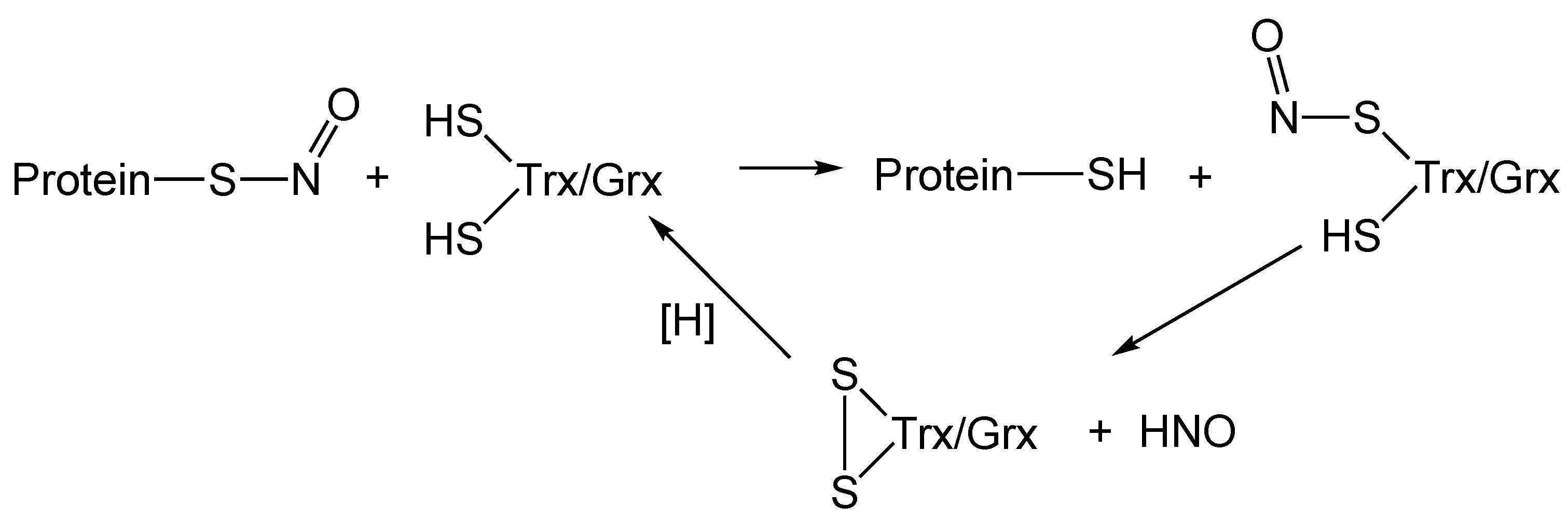
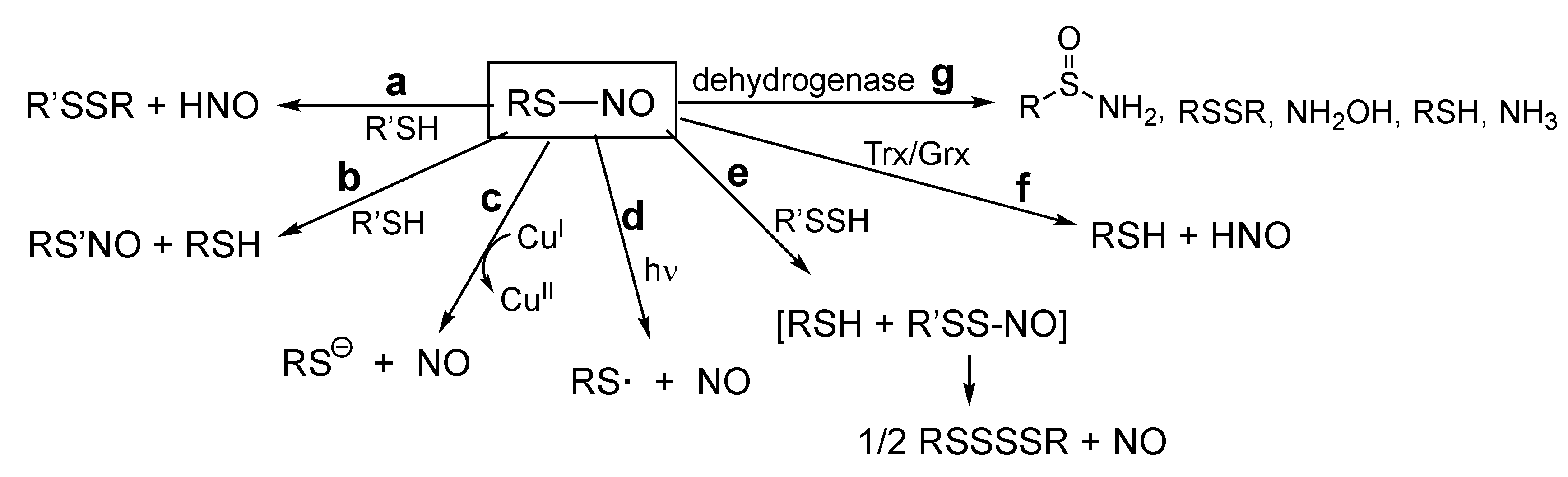
5. RS-NO Degradation Pathways
6. Steady State Levels of RS-NO and Signaling Pathways
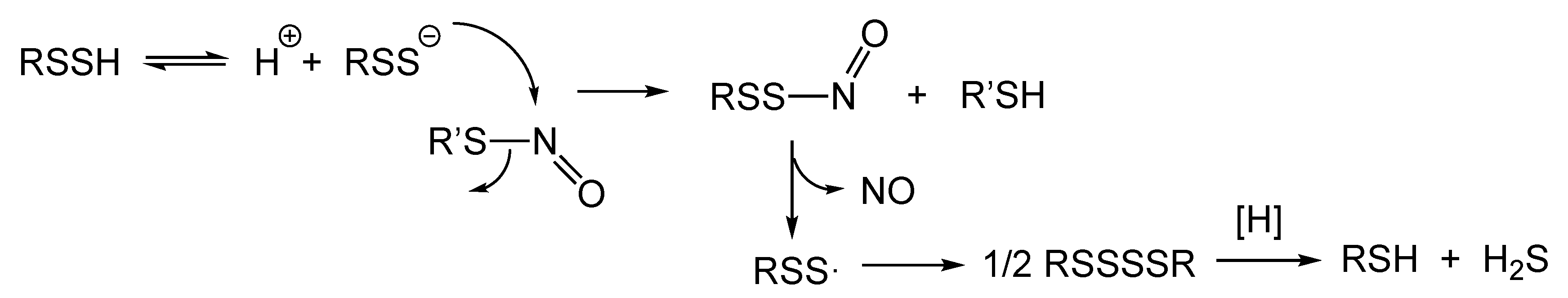
7. Hydropersulfides (RSSH) and Possible Importance to RS-NO Degradation
8. RS-NO as a Storage Form of NO and Regulated NO Release
9. H2S/RS-NO Interactions and ONSS• Formation
10. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Hobbs, A.J. Soluble guanylate cyclase: The forgotten sibling. Trends Pharmacol. Sci. 1997, 18, 484–491. [Google Scholar] [CrossRef]
- Ignarro, L.J. Nitric oxide: A unique endogenous signaling molecule in vascular biology. Biosci. Rep. 1999, 19, 51–71. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Ridnour, L.A.; Cheng, R.; Kesarwala, A.H.; Heinecke, J.; Wink, D.A. Biological signaling by small inorganic molecules. Coord. Chem. Rev. 2016, 306, 708–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Snyder, S.H. Signaling by gasotransmitters. Sci. Signal. 2009, 2, re2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal. 2010, 13, 157–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpure, B.V.; Bian, J.S. Interaction of Hydrogen Sulfide with Nitric Oxide in the Cardiovascular System. Oxid. Med. Cell. Longev. 2016, 2016, 6904327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibli, S.I.; Szabo, C.; Chatzianastasiou, A.; Luck, B.; Zukunft, S.; Fleming, I.; Papapetropoulos, A. Hydrogen Sulfide Preserves Endothelial Nitric Oxide Synthase Function by Inhibiting Proline-Rich Kinase 2: Implications for Cardiomyocyte Survival and Cardioprotection. Mol. Pharmacol. 2017, 92, 718–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, R.; Palinkas, Z.; Basell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Marozkina, N.V.; Gaston, B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim. Biophys. Acta 2012, 1820, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T.; Scott, J.D. Protein phosphorylation in signaling—50 years and counting. Trends Biochem. Sci. 2005, 30, 286–290. [Google Scholar] [CrossRef]
- Heinrich, T.A.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signalling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.J.; Atochina-Vasserman, E.N.; Abramova, E.; Foley, J.P.; Zaman, A.; Crouch, E.; Beers, M.F.; Savani, R.C.; Gow, A.J. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008, 6, e266. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.; van der Vliet, A.; Janssen-Heininger, Y.M. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Won, J.S.; Singh, A.K.; Sharma, A.K.; Singh, I. STAT3 regulation by S-nitrosylation: Implication for inflammatory disease. Antioxid. Redox Signal. 2014, 20, 2514–2527. [Google Scholar] [CrossRef]
- Nakazawa, H.; Chang, K.; Shinozaki, S.; Yasukawa, T.; Ishimaru, K.; Yasuhara, S.; Yu, Y.M.; Martyn, J.A.; Tompkins, R.G.; Shimokado, K.; et al. iNOS as a Driver of Inflammation and Apoptosis in Mouse Skeletal Muscle after Burn Injury: Possible Involvement of Sirt1 S-Nitrosylation-Mediated Acetylation of p65 NF-kappaB and p53. PLoS ONE 2017, 12, e0170391. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. ‘SNO’-Storms Compromise Protein Activity and Mitochondrial Metabolism in Neurodegenerative Disorders. Trends Endocrinol. Metab. 2017, 28, 879–892. [Google Scholar] [CrossRef]
- Wink, D.A.; Darbyshire, J.F.; Nims, R.W.; Saavedra, J.E.; Ford, P.C. Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: Determination of the kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem. Res. Toxicol. 1993, 6, 23–27. [Google Scholar] [CrossRef]
- Keshive, M.; Singh, S.; Wishnok, J.S.; Tannenbaum, S.R.; Deen, W.M. Kinetics of S-nitrosation of thiols in nitric oxide solutions. Chem. Res. Toxicol. 1996, 9, 988–993. [Google Scholar] [CrossRef]
- Goldstein, S.; Czapski, G. Mechanism of the Nitrosation of Thiols and Amines by Oxygenated NO Solutions: The Nature of the Nitrosating Intermediates. J. Am. Chem. Soc. 1996, 118, 3419–3425. [Google Scholar] [CrossRef]
- Liu, X.; Miller, M.J.; Joshi, M.S.; Thomas, D.D.; Lancaster, J.R., Jr. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 2175–2179. [Google Scholar] [CrossRef] [Green Version]
- Wayland, B.B.; Olson, L.W. Spectroscopic studies and bonding model for nitric oxide complexes of iron porphyrins. J. Am. Chem. Soc. 1974, 96, 6037–6041. [Google Scholar] [CrossRef]
- Wade, R.S.; Castro, C.E. Redox reactivity of iron(III) porphyrins and heme proteins with nitric oxide. Nitrosyl transfer to carbon, oxygen, nitrogen, and sulfur. Chem. Res. Toxicol. 1990, 3, 289–291. [Google Scholar] [CrossRef]
- Castro, C.E.; Bartnicki, E.W. The Interconversion of Nucleic Acid Bases by Iron(III) Porphyrins and Nitric Oxide. J. Org. Chem. 1994, 59, 4051–4052. [Google Scholar] [CrossRef]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Stubbe, J.; van der Donk, W.A. Ribonucleotide reductases: Radical enzymes with suicidal tendencies. Chem. Biol. 1995, 2, 793–801. [Google Scholar] [CrossRef] [Green Version]
- Schoneich, C. Mechanisms of protein damage induced by cysteine thiyl radical formation. Chem. Res. Toxicol. 2008, 21, 1175–1179. [Google Scholar] [CrossRef]
- Schöneich, C.; Dillinger, U.; von Bruchhausen, F.; Asmus, K.-D. Oxidation of polyunsaturated fatty acids and lipids through thiyl and sulfonyl radicals: Reaction kinetics, and influence of oxygen and structure of thiyl radicals. Arch. Biochem. Biophys. 1992, 292, 456–467. [Google Scholar] [CrossRef]
- Lancaster, J.R., Jr. How are nitrosothiols formed de novo in vivo? Arch. Biochem. Biophys. 2017, 617, 137–144. [Google Scholar] [CrossRef]
- Singh, R.J.; Hogg, N.; Joseph, J.; Kalyanaraman, B. Mechanism of nitric oxide release from S-nitrosothiols. J. Biol. Chem. 1996, 271, 18596–18603. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.L.H. The Chemistry of S-Nitrosothiols. Acc. Chem. Res. 1999, 32, 869–876. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O‘Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Sexton, D.J.; Muruganandam, A.; McKenney, D.J.; Mutus, B. Visible light photochemical release of nitric oxide from S-nitrosoglutathione: Potential photochemotherapeutic applications. Photochem. Photobiol. 1994, 59, 463–467. [Google Scholar] [CrossRef]
- Oplander, C.; Deck, A.; Volkmar, C.M.; Kirsch, M.; Liebmann, J.; Born, M.; van Abeelen, F.; van Faassen, E.E.; Kroncke, K.D.; Windolf, J.; et al. Mechanism and biological relevance of blue-light (420–453 nm)-induced nonenzymatic nitric oxide generation from photolabile nitric oxide derivates in human skin in vitro and in vivo. Free Radic. Biol. Med. 2013, 65, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.E.; Belka, G.K.; Du Bois, G.C. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. Biochem. J. 1998, 331 Pt 2, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [Google Scholar] [CrossRef]
- Liu, Z.; Rudd, M.A.; Freedman, J.E.; Loscalzo, J. S-Transnitrosation Reactions Are Involved in the Metabolic Fate and Biological Actions of Nitric Oxide. J. Pharmacol. Exp. Ther. 1998, 284, 526–534. [Google Scholar] [PubMed]
- Hogg, N. Biological chemistry and clinical potential of S-nitrosothiols. Free Radic. Biol. Med. 2000, 28, 1478–1486. [Google Scholar] [CrossRef]
- Hogg, N. The kinetics of S-transnitrosation—A reversible second-order reaction. Anal. Biochem. 1999, 272, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Mahapatro, S.N.; Broene, R.D.; Guy, J.K. Oxidation and reduction of hemoproteins by trioxodinitrate(II). The role of nitrosyl hydride and nitrite. J. Am. Chem. Soc. 1988, 110, 593–599. [Google Scholar] [CrossRef]
- Wong, P.S.; Hyun, J.; Fukuto, J.M.; Shirota, F.N.; DeMaster, E.G.; Shoeman, D.W.; Nagasawa, H.T. Reaction between S-nitrosothiols and thiols: Generation of nitroxyl (HNO) and subsequent chemistry. Biochemistry 1998, 37, 5362–5371. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Cisneros, C.J.; Kinkade, R.L. A comparison of the chemistry associated with the biological signaling and actions of nitroxyl (HNO) and nitric oxide (NO). J. Inorg. Biochem. 2013, 118, 201–208. [Google Scholar] [CrossRef]
- Fukuto, J.M. A recent history of nitroxyl chemistry, pharmacology and therapeutic potential. Br. J. Pharmacol. 2019, 176, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Sengupta, R.; Lu, J.; Lundberg, J.O.; Holmgren, A. Characterization of mammalian glutaredoxin isoforms as S-denitrosylases. FEBS Lett. 2019, 593, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, R.; Holmgren, A. Thioredoxin and thioredoxin reductase in relation to reversible S-nitrosylation. Antioxid. Redox Signal. 2013, 18, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, A.; Sengupta, R. Cellular S-denitrosylases: Potential role and interplay of Thioredoxin, TRP14, and Glutaredoxin systems in thiol-dependent protein denitrosylation. Int. J. Biochem. Cell Biol. 2021, 131, 105904. [Google Scholar] [CrossRef] [PubMed]
- Timerghazin, Q.K.; Talipov, M.R. Unprecedented External Electric Field Effects on S-Nitrosothiols: Possible Mechanism of Biological Regulation? J. Phys. Chem. Lett. 2013, 4, 1034–1038. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef]
- Scorza, G.; Pietraforte, D.; Minetti, M. Role of Ascorbate and Protein Thiols in the Release of Nitric oxide from S-Nitroso-Albumin and S-Nitroso-Glutathione in Human Plasma. Free Radic. Biol. Med. 1997, 22, 633–642. [Google Scholar] [CrossRef]
- Kashiba-Iwatsuki, M.; Kitoh, K.; Kasahara, E.; Yu, H.; Nisikawa, M.; Matsuo, M.; Inoue, M. Ascorbic Acid and Reducing Agents Regulate the Fates and Functions of S-Nitrosothiols. J. Biochem. 1997, 122, 1208–1214. [Google Scholar] [CrossRef]
- Kashiba-Iwatsuki, M.; Yamaguchi, M.; Inoue, M. Role of ascorbic acid in the metabolism ofS-nitroso-glutathione. FEBS Lett. 1996, 389, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.J.; Williams, D.L.H. Reaction of ascorbic acid with S-nitrosothiols: Clear evidence for two distinct reaction pathways. J. Chem. Soc. Perkin Trans. 2000, 2, 1639–1644. [Google Scholar] [CrossRef]
- Smith, J.N.; Dasgupta, T.P. Kinetics and mechanism of the decomposition of S-nitrosoglutathione by l-ascorbic acid and copper ions in aqueous solution to produce nitric oxide. Nitric Oxide 2000, 4, 57–66. [Google Scholar] [CrossRef]
- Zhang, Y.; Keszler, A.; Broniowska, K.A.; Hogg, N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic. Biol. Med. 2005, 38, 874–881. [Google Scholar] [CrossRef]
- Kirsch, M.; Buscher, A.M.; Aker, S.; Schulz, R.; de Groot, H. New insights into the S-nitrosothiol-ascorbate reaction. The formation of nitroxyl. Org. Biomol. Chem. 2009, 7, 1954–1962. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Mishra, D.; Patel, V.; Banerjee, D. Nitric Oxide and S-Nitrosylation in Cancers: Emphasis on Breast Cancer. Breast Cancer Basic Clin. Res. 2020, 14, 1178223419882688. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, L.B.; Maiwald, T.; Conzelmann, H.; Lauffenburger, D.A.; Sorger, P.K. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell 2011, 43, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelens, L.; Saurin, A.T. Exploring the Function of Dynamic Phosphorylation-Dephosphorylation Cycles. Dev. Cell 2018, 44, 659–663. [Google Scholar] [CrossRef] [Green Version]
- Fukuto, J.M.; Ignarro, L.J.; Nagy, P.; Wink, D.A.; Kevil, C.G.; Feelisch, M.; Cortese-Krott, M.M.; Bianco, C.L.; Kumagai, Y.; Hobbs, A.J.; et al. Biological hydropersulfides and related polysulfides—A new concept and perspective in redox biology. FEBS Lett. 2018, 592, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Fukuto, J.M. The Biological/Physiological Utility of Hydropersulfides (RSSH) and Related Species: What Is Old Is New Again. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J.; et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Ezerina, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef] [Green Version]
- Bianco, C.L.; Akaike, T.; Ida, T.; Nagy, P.; Bogdandi, V.; Toscano, J.P.; Kumagai, Y.; Henderson, C.F.; Goddu, R.N.; Lin, J.; et al. The reaction of hydrogen sulfide with disulfides: Formation of a stable trisulfide and implications for biological systems. Br. J. Pharmacol. 2019, 176, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.F.; Bica, I.; Long, F.T.; Irwin, D.D.; Stull, C.H.; Baker, B.W.; Suarez Vega, V.; Taugher, Z.M.; Fletes, E.D.; Bartleson, J.M.; et al. Cysteine Trisulfide Protects E. coli from Electrophile-Induced Death through the Generation of Cysteine Hydropersulfide. Chem. Res. Toxicol. 2020, 33, 678–686. [Google Scholar] [CrossRef]
- Doka, E.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenfeld, P.; Cordova, F.; Duran, W.N.; Sanchez, F.A. S-nitrosylation and its role in breast cancer angiogenesis and metastasis. Nitric Oxide 2019, 87, 52–59. [Google Scholar] [CrossRef]
- Bianco, C.L.; Chavez, T.A.; Sosa, V.; Saund, S.S.; Nguyen, Q.N.N.; Tantillo, D.J.; Ichimura, A.S.; Toscano, J.P.; Fukuto, J.M. The chemical biology of the persulfide (RSSH)/perthiyl (RSS.) redox couple and possible role in biological redox signaling. Free Radic. Biol. Med. 2016, 101, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauvin, J.R.; Griesser, M.; Pratt, D.A. Hydropersulfides: H-Atom Transfer Agents Par Excellence. J. Am. Chem. Soc. 2017, 139, 6484–6493. [Google Scholar] [CrossRef]
- Sevilla, M.D.; Becker, D.; Yan, M. The formation and structure of the sulfoxyl radicals RSO., RSOO., RSO2. and RSO2OO from the reaction of cysteine, glutathione and penicillamine thiyl radicals with molecular oxygen. Int. J. Radiat. Biol. 1990, 57, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Shimatani, K.; Fujita, K.; Abe, T.; Shimizu, M.; Fujii, T.; Hoshino, T.; Takaya, N. Glutathione reductase/glutathione is responsible for cytotoxic elemental sulfur tolerance via polysulfide shuttle in fungi. J. Biol. Chem. 2011, 286, 20283–20291. [Google Scholar] [CrossRef] [Green Version]
- Doka, E.; Pader, I.; Biro, A.; Johansson, K.; Cheng, Q.; Ballago, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.R.; Gao, Y. Effects of inhibiting antioxidant pathways on cellular hydrogen sulfide and polysulfide metabolism. Free Radic. Biol. Med. 2019, 135, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cuevasanta, E.; Lange, M.; Bonanata, J.; Coitino, E.L.; Ferrer-Sueta, G.; Filipovic, M.R.; Alvarez, B. Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J. Biol. Chem. 2015, 290, 26866–26880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, S.A.; Wardman, P. Perthiols as antioxidants: Radical-scavenging andprooxidative mechanisms. Methods Enzymol. 1995, 251, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Benchoam, D.; Semelak, J.A.; Cuevasanta, E.; Mastrogiovanni, M.; Grassano, J.S.; Ferrer-Sueta, G.; Zeida, A.; Trujillo, M.; Moller, M.N.; Estrin, D.A.; et al. Acidity and nucleophilic reactivity of glutathione persulfide. J. Biol. Chem. 2020, 295, 15466–15481. [Google Scholar] [CrossRef] [PubMed]
- Fina, N.J.; Edwards, J.O. The alpha effect. A review. Int. J. Chem. Kinet. 1973, 5, 1–26. [Google Scholar] [CrossRef]
- Thomas, D.D.; Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Kesarwala, A.H.; Switzer, C.H.; McVicar, D.W.; Roberts, D.D.; Glynn, S.; Fukuto, J.M.; et al. Signaling and stress: The redox landscape in NOS2 biology. Free Radic. Biol. Med. 2015, 87, 204–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Filomeni, G. Exploiting S-nitrosylation for cancer therapy: Facts and perspectives. Biochem. J. 2020, 477, 3649–3672. [Google Scholar] [CrossRef]
- Gorelenkova Miller, O.; Mieyal, J.J. Sulfhydryl-mediated redox signaling in inflammation: Role in neurodegenerative diseases. Arch. Toxicol. 2015, 89, 1439–1467. [Google Scholar] [CrossRef]
- Yang, L.; Calay, E.S.; Fan, J.; Arduini, A.; Kunz, R.C.; Gygi, S.P.; Yalcin, A.; Fu, S.; Hotamisligil, G.S. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015, 349, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Fukuto, J.M.; Hobbs, A.J. A comparison of the chemical biology of hydropersulfides (RSSH) with other protective biological antioxidants and nucleophiles. Nitric Oxide 2021, 107, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L. Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys. 2017, 617, 3–8. [Google Scholar] [CrossRef]
- Stamler, J.S.; Jaraki, O.; Osborne, J.; Simon, D.I.; Keaney, J.; Vita, J.; Singel, D.; Valeri, C.R.; Loscalzo, J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. USA 1992, 89, 7674–7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Maloney, R.E.; Rassaf, T.; Bryan, N.S.; Feelisch, M. Chemical nature of nitric oxide storage forms in rat vascular tissue. Proc. Natl. Acad. Sci. USA 2003, 100, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Ng, E.S.; Kubes, P. The physiology of S-nitrosothiols: Carrier molecules for nitric oxide. Can. J. Physiol. Pharmacol. 2003, 81, 759–764. [Google Scholar] [CrossRef]
- Rayner, B.S.; Wu, B.J.; Raftery, M.; Stocker, R.; Witting, P.K. Human S-nitroso oxymyoglobin is a store of vasoactive nitric oxide. J. Biol. Chem. 2005, 280, 9985–9993. [Google Scholar] [CrossRef] [Green Version]
- Singel, D.J.; Stamler, J.S. Chemical physiology of blood flow regulation by red blood cells: The role of nitric oxide and S-nitrosohemoglobin. Annu. Rev. Physiol. 2005, 67, 99–145. [Google Scholar] [CrossRef]
- Banerjee, R. Catalytic promiscuity and heme-dependent redox regulation of H2S synthesis. Curr. Opin. Chem. Biol. 2017, 37, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.K.; Martinov, M.; Vitvitsky, V.; Seravalli, J.; Wedmann, R.; Filipovic, M.R.; Banerjee, R. Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J. Am. Chem. Soc. 2016, 138, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Stipanuk, M.H. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr. 1986, 6, 179–209. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallini, D.; Federici, G.; Barboni, E. Interaction of proteins with sulfide. Eur. J. Biochem. 1970, 14, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Francoleon, N.E.; Carrington, S.J.; Fukuto, J.M. The reaction of H2S with oxidized thiols: Generation of persulfides and implications to H2S biology. Arch. Biochem. Biophys. 2011, 516, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Fernandez, B.O.; Santos, J.L.; Mergia, E.; Grman, M.; Nagy, P.; Kelm, M.; Butler, A.; Feelisch, M. Nitrosopersulfide (SSNO-) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biol. 2014, 2, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Bogdandi, V.; Ditroi, T.; Batai, I.Z.; Sandor, Z.; Minnion, M.; Vasas, A.; Galambos, K.; Buglyo, P.; Pinter, E.; Feelisch, M.; et al. Nitrosopersulfide (SSNO-) Is a Unique Cysteine Polysulfidating Agent with Reduction-Resistant Bioactivity. Antioxid. Redox Signal. 2020, 33, 1277–1294. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Kuhnle, G.G.; Dyson, A.; Fernandez, B.O.; Grman, M.; DuMond, J.F.; Barrow, M.P.; McLeod, G.; Nakagawa, H.; Ondrias, K.; et al. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. USA 2015, 112, E4651–E4660. [Google Scholar] [CrossRef] [Green Version]
- Wedmann, R.; Zahl, A.; Shubina, T.E.; Durr, M.; Heinemann, F.W.; Bugenhagen, B.E.; Burger, P.; Ivanovic-Burmazovic, I.; Filipovic, M.R. Does perthionitrite (SSNO(-)) account for sustained bioactivity of NO? A (bio)chemical characterization. Inorg. Chem. 2015, 54, 9367–9380. [Google Scholar] [CrossRef]
- Bolden, C.; King, S.B.; Kim-Shapiro, D.B. Reactions between nitrosopersulfide and heme proteins. Free Radic. Biol. Med. 2016, 99, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Marcolongo, J.P.; Morzan, U.N.; Zeida, A.; Scherlis, D.A.; Olabe, J.A. Nitrosodisulfide [S2NO](-) (perthionitrite) is a true intermediate during the “cross-talk” of nitrosyl and sulfide. Phys. Chem. Chem. Phys. 2016, 18, 30047–30052. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuto, J.M.; Perez-Ternero, C.; Zarenkiewicz, J.; Lin, J.; Hobbs, A.J.; Toscano, J.P. Hydropersulfides (RSSH) and Nitric Oxide (NO) Signaling: Possible Effects on S-Nitrosothiols (RS-NO). Antioxidants 2022, 11, 169. https://doi.org/10.3390/antiox11010169
Fukuto JM, Perez-Ternero C, Zarenkiewicz J, Lin J, Hobbs AJ, Toscano JP. Hydropersulfides (RSSH) and Nitric Oxide (NO) Signaling: Possible Effects on S-Nitrosothiols (RS-NO). Antioxidants. 2022; 11(1):169. https://doi.org/10.3390/antiox11010169
Chicago/Turabian StyleFukuto, Jon M., Cristina Perez-Ternero, Jessica Zarenkiewicz, Joseph Lin, Adrian J. Hobbs, and John P. Toscano. 2022. "Hydropersulfides (RSSH) and Nitric Oxide (NO) Signaling: Possible Effects on S-Nitrosothiols (RS-NO)" Antioxidants 11, no. 1: 169. https://doi.org/10.3390/antiox11010169
APA StyleFukuto, J. M., Perez-Ternero, C., Zarenkiewicz, J., Lin, J., Hobbs, A. J., & Toscano, J. P. (2022). Hydropersulfides (RSSH) and Nitric Oxide (NO) Signaling: Possible Effects on S-Nitrosothiols (RS-NO). Antioxidants, 11(1), 169. https://doi.org/10.3390/antiox11010169