
Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Anti-Inflammatory Actions of Rosemary and Carnosic Acid
1.2. Uses of Rosemary and Rosemary Extract
2. Anti-Inflammatory Mechanisms
2.1. Anti-Inflammatory Effects of CA on Tissue Macrophages
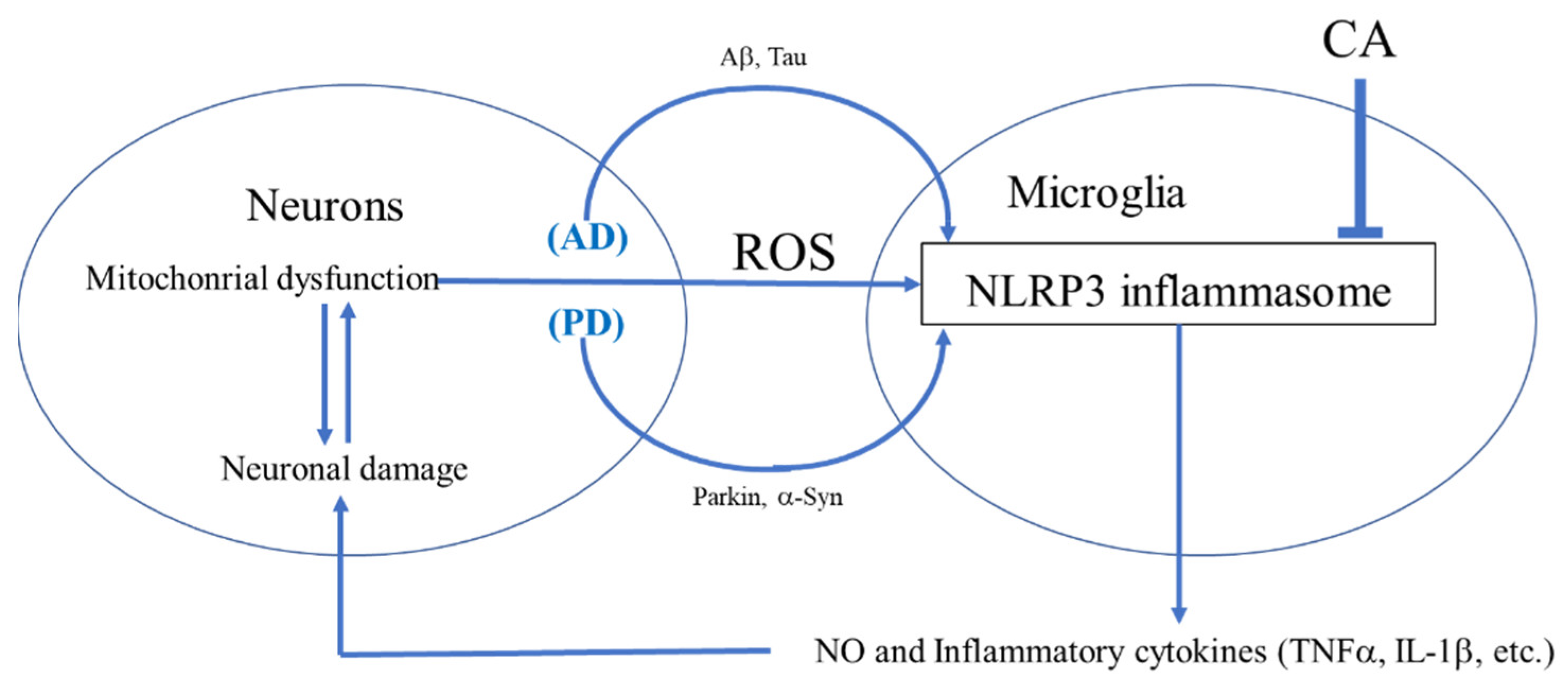
2.2. Anti-Inflammatory Effects of CA on Brain Microglia
3. Role of NLRP3 Inflammasome
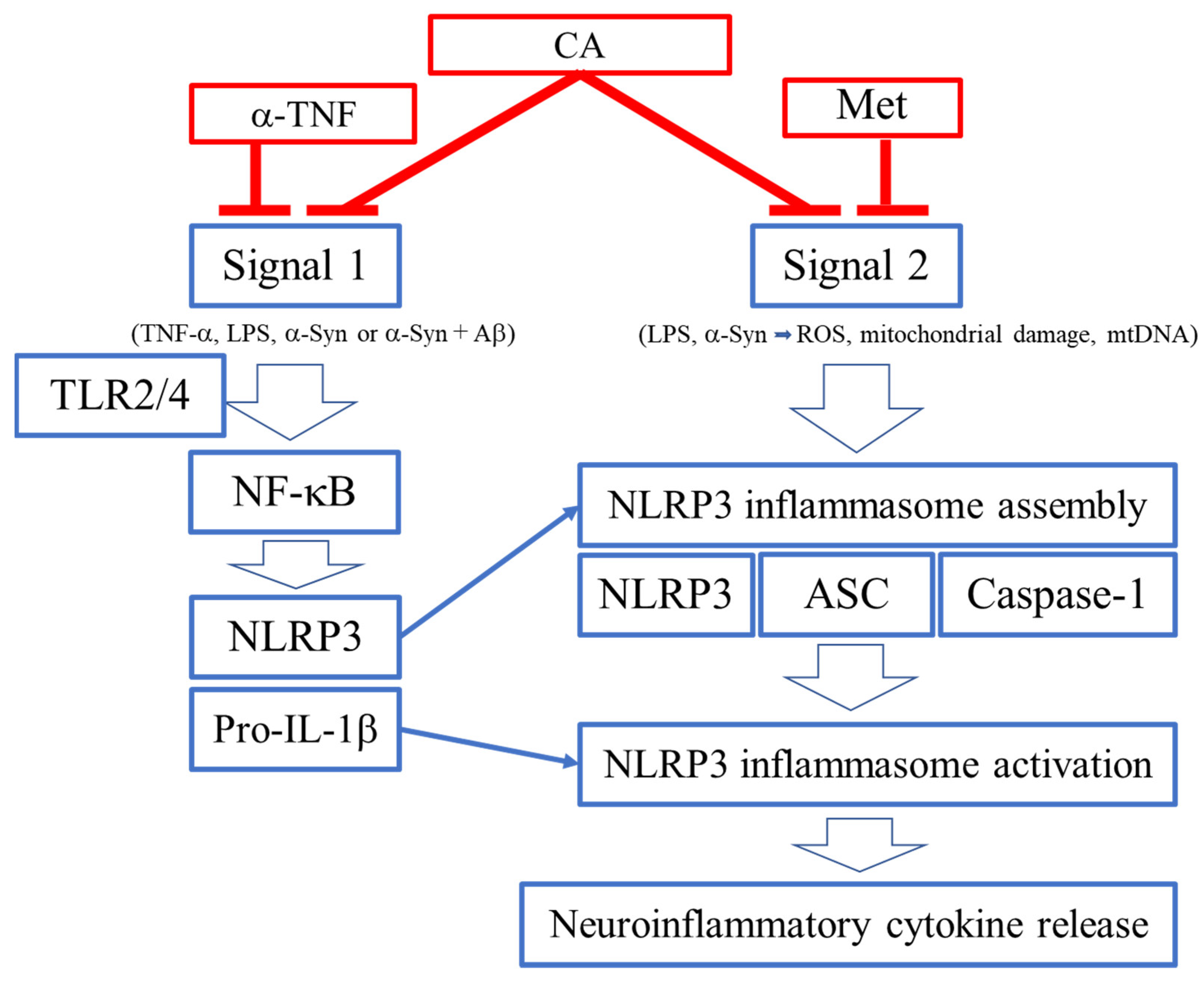
3.1. Proteins of the NLRP3 Inflammasome
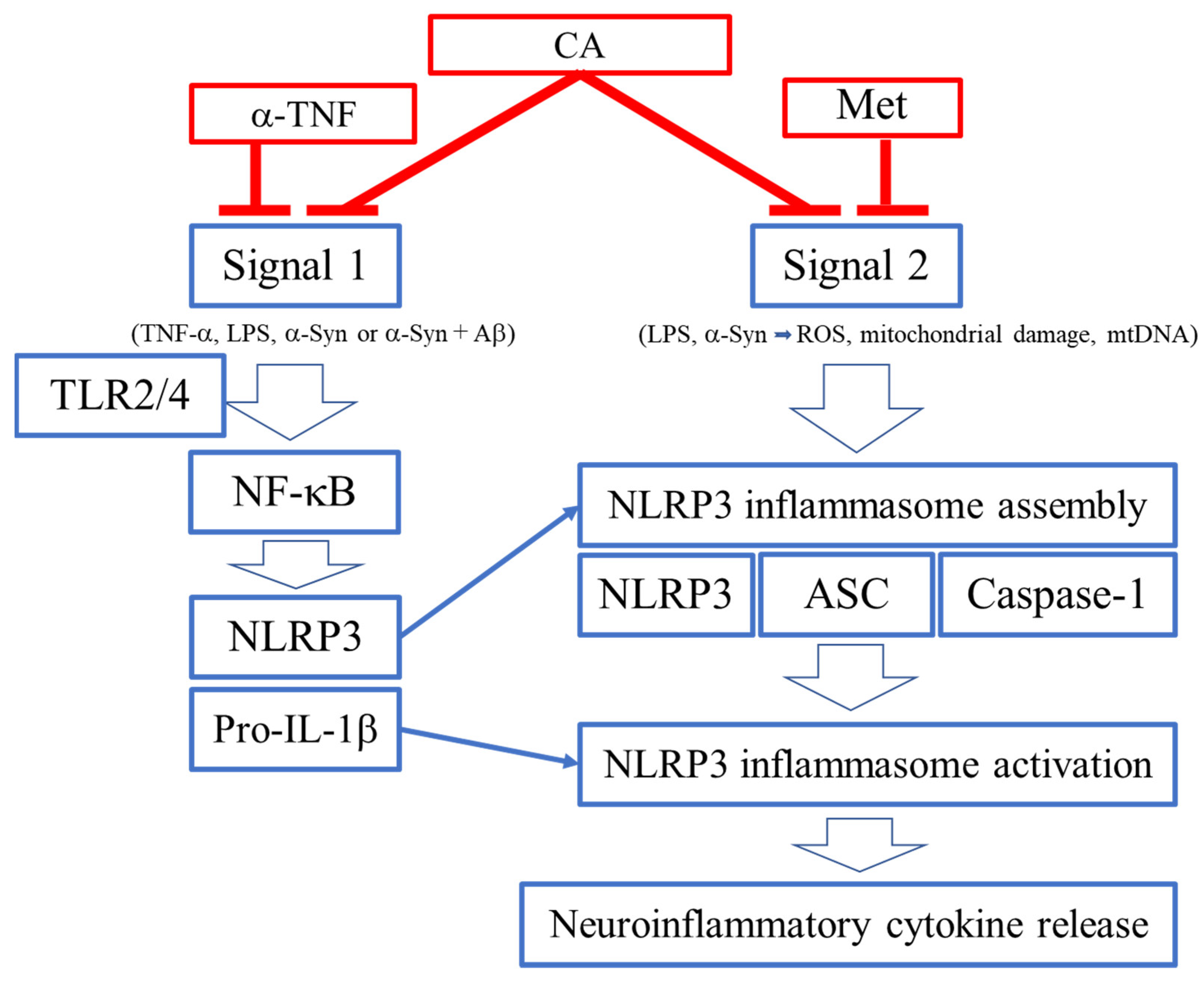
3.2. Signals 1 and 2 for Activation of NLRP3 Inflammasome
3.3. Activation of NLRP3 Inflammasome by Misfolded Proteins
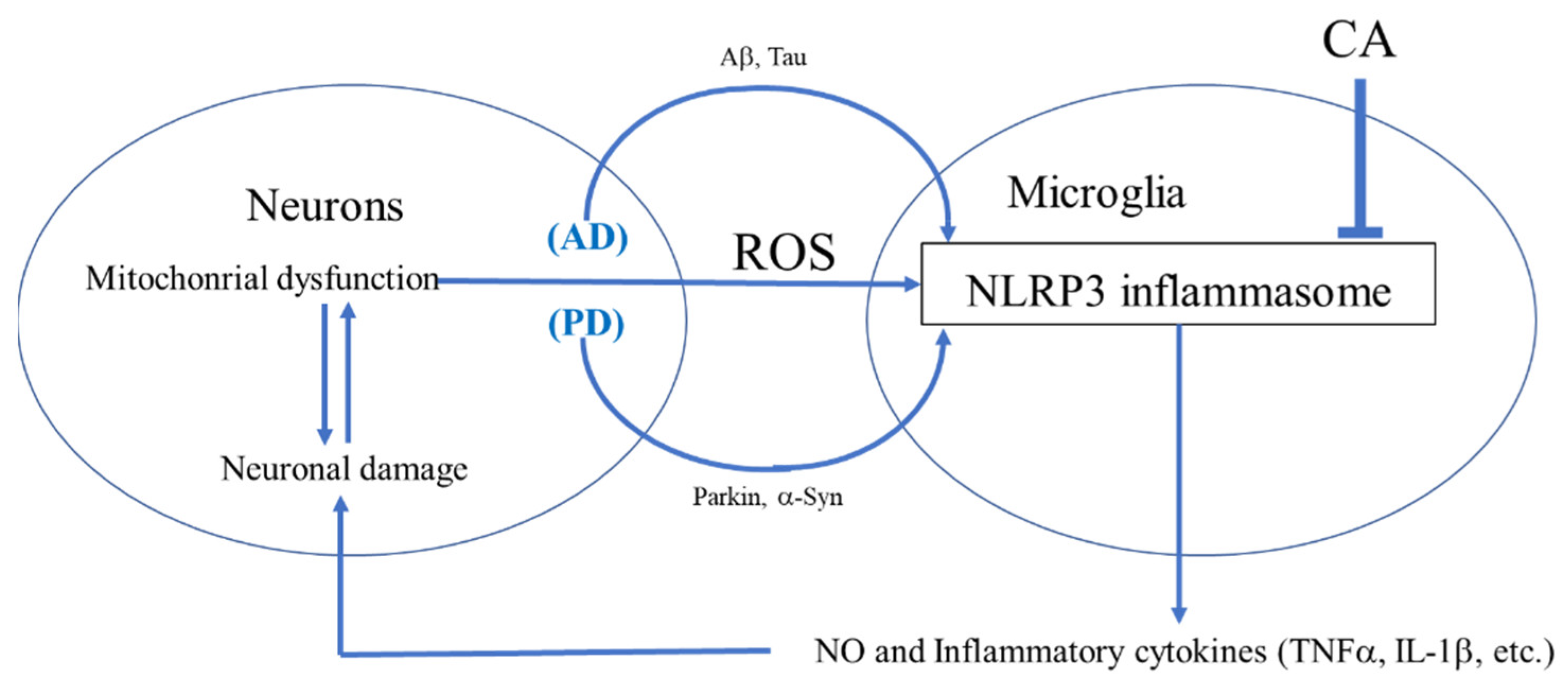
4. Inhibition of NLRP3 Activation by CA
4.1. Activation of the NLRP3 Inflammasome Complex
4.2. ROS as a Signal 2 Activator and Therapeutic Target
4.3. In Addition to α-Syn and Aβ, Possibly Other Misfolded Proteins May Contribute to Activation of the NLRP3 Inflammasome through ROS Generation
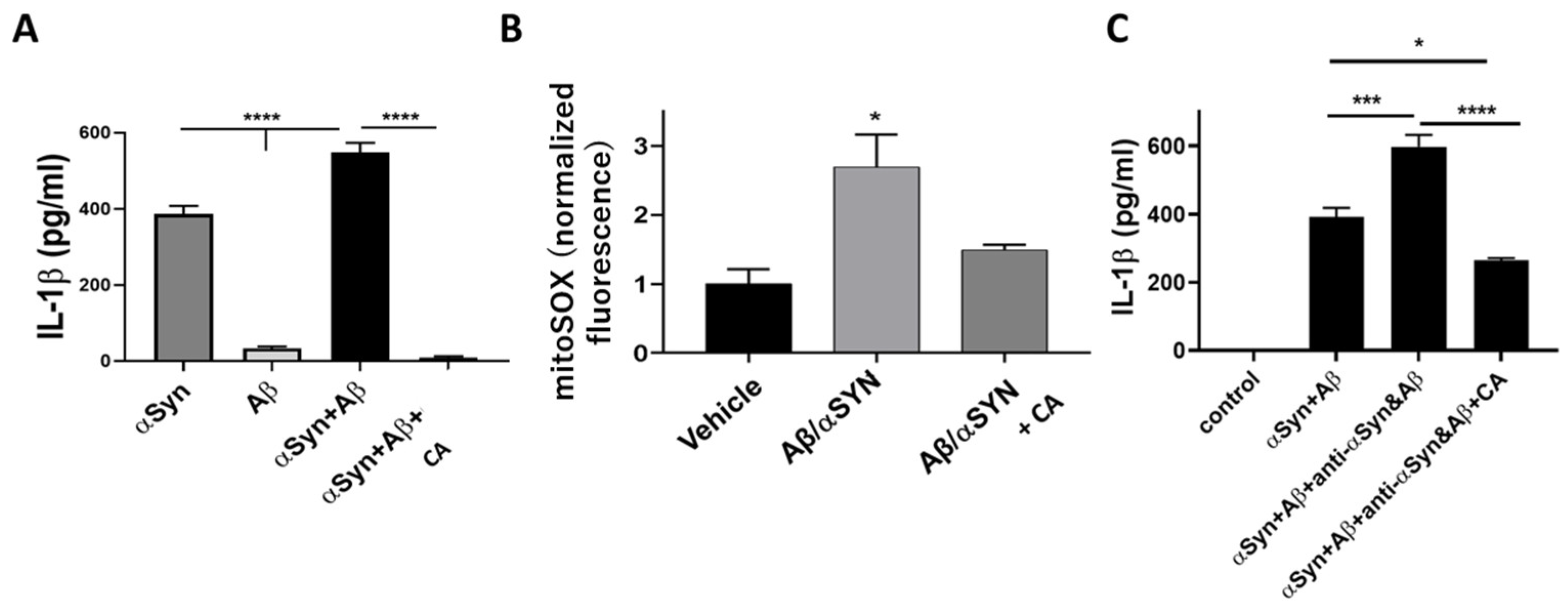
4.4. CA as an Inhibitor of NLRP3 Inflammasome Activation in PD- and AD-Related Models
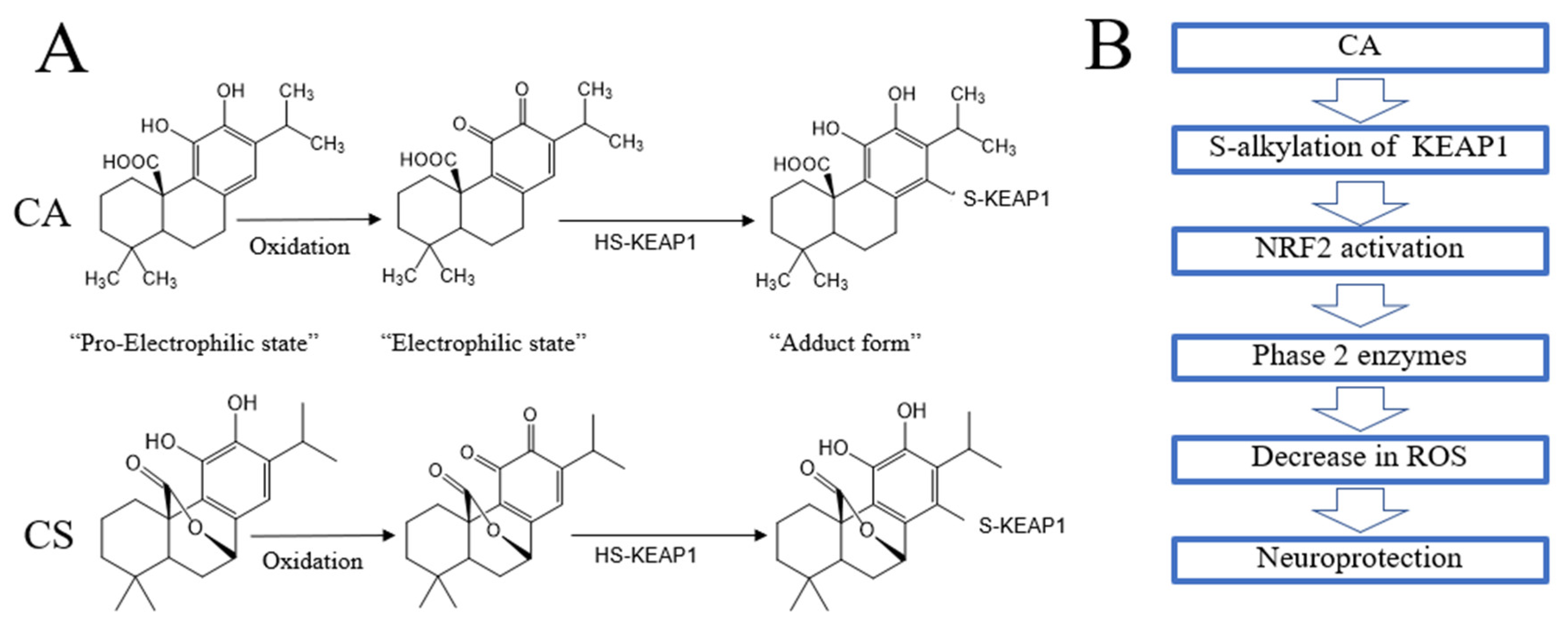
5. Direct and Indirect Antioxidant Actions of Rosemary and CA
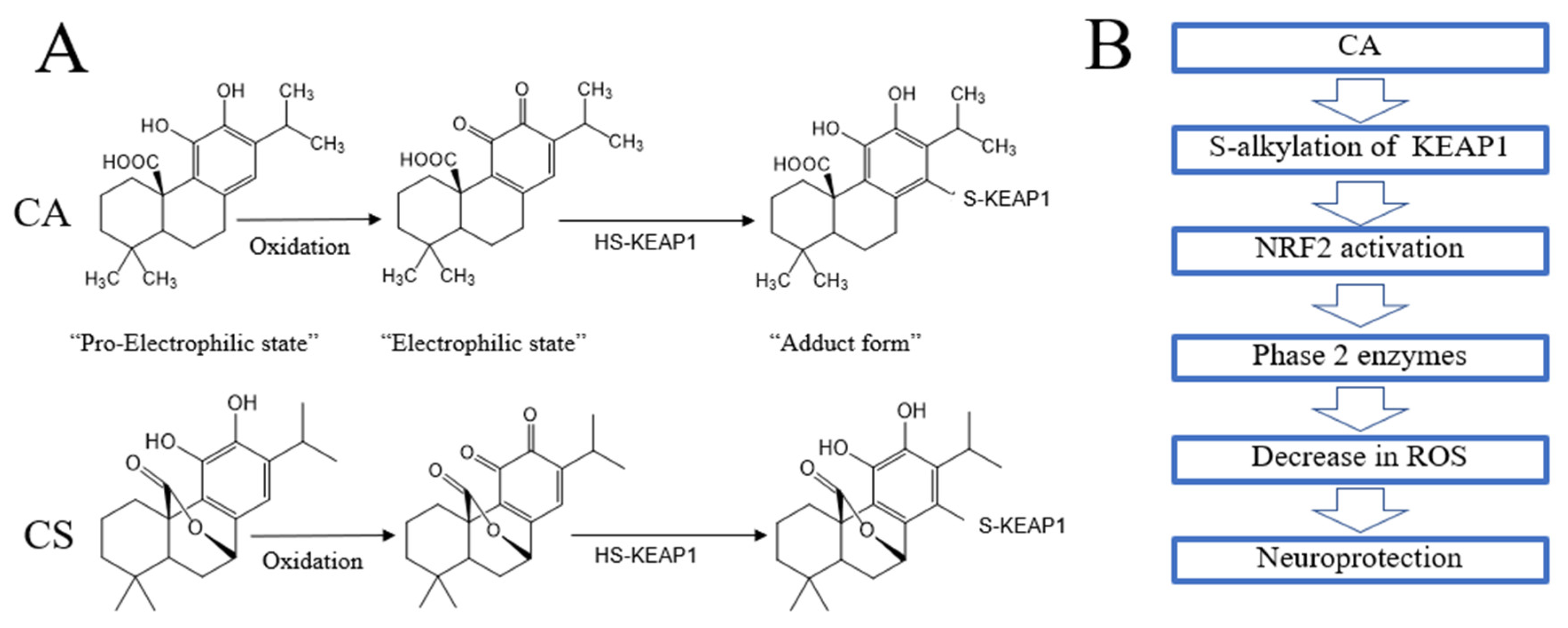
5.1. Direct Antioxidant Action
5.2. Indirect Antioxidant Action
5.3. CA-Mediated Protection of the Brain in AD and PD
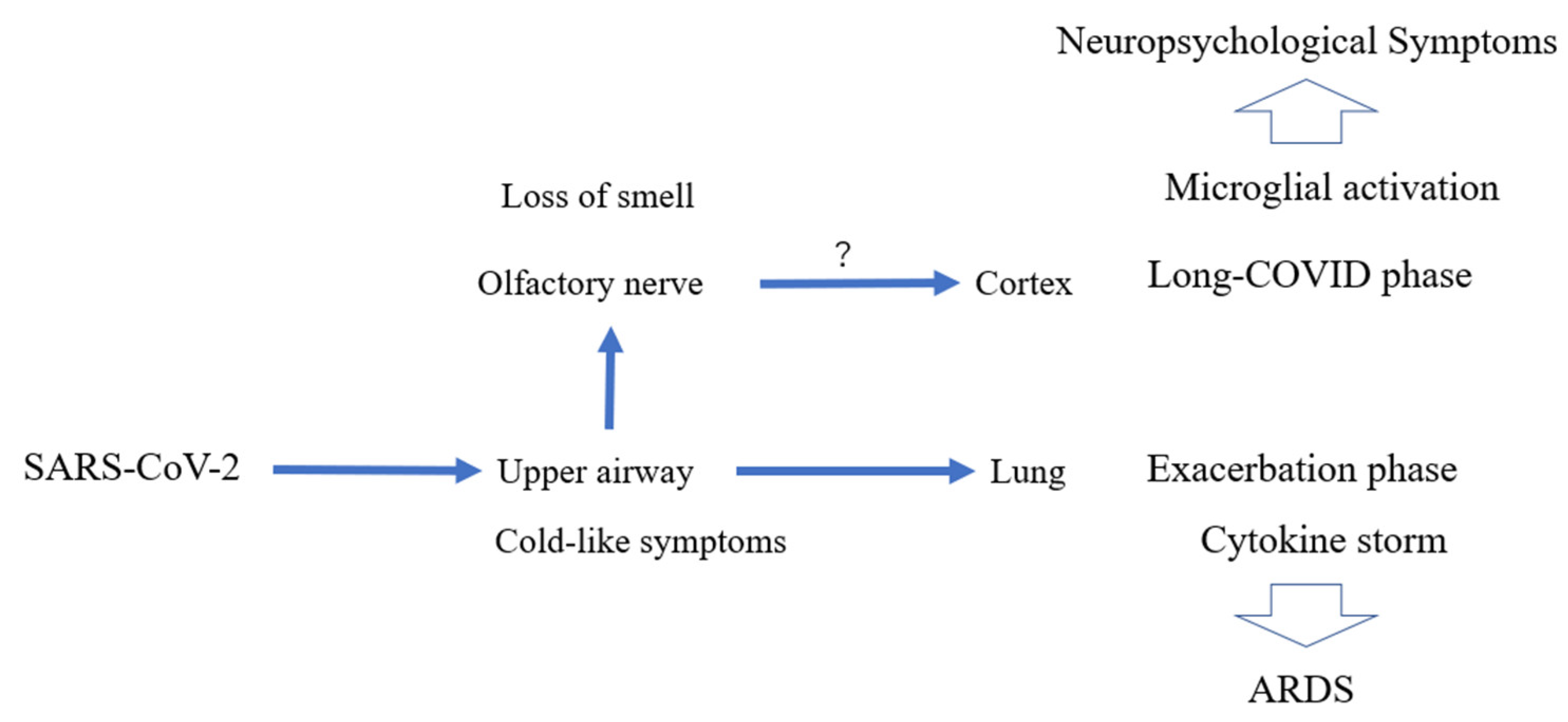
6. Complicated Manifestations of COVID-19
6.1. Manifestations
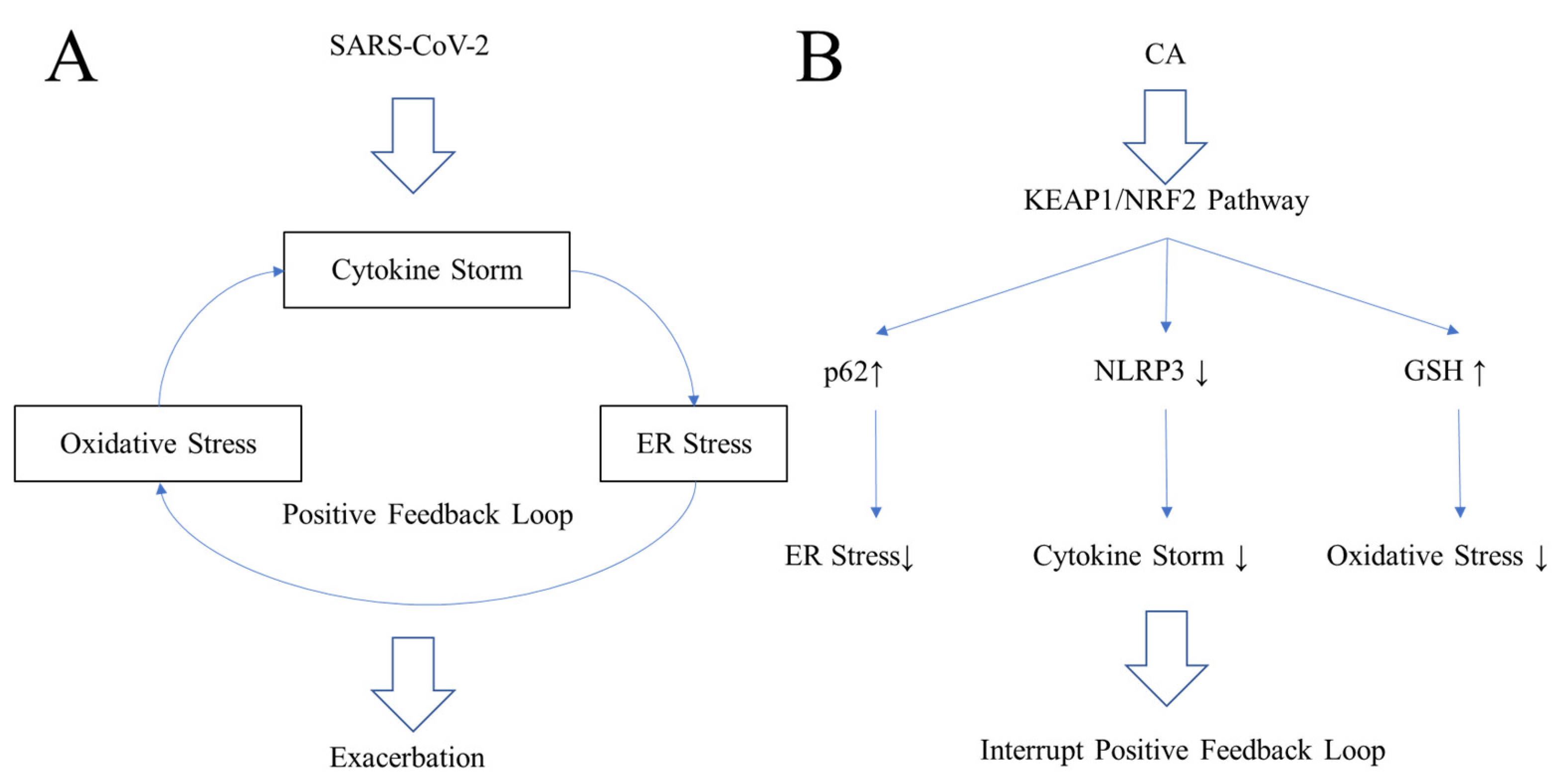
6.2. Cytokine Storm
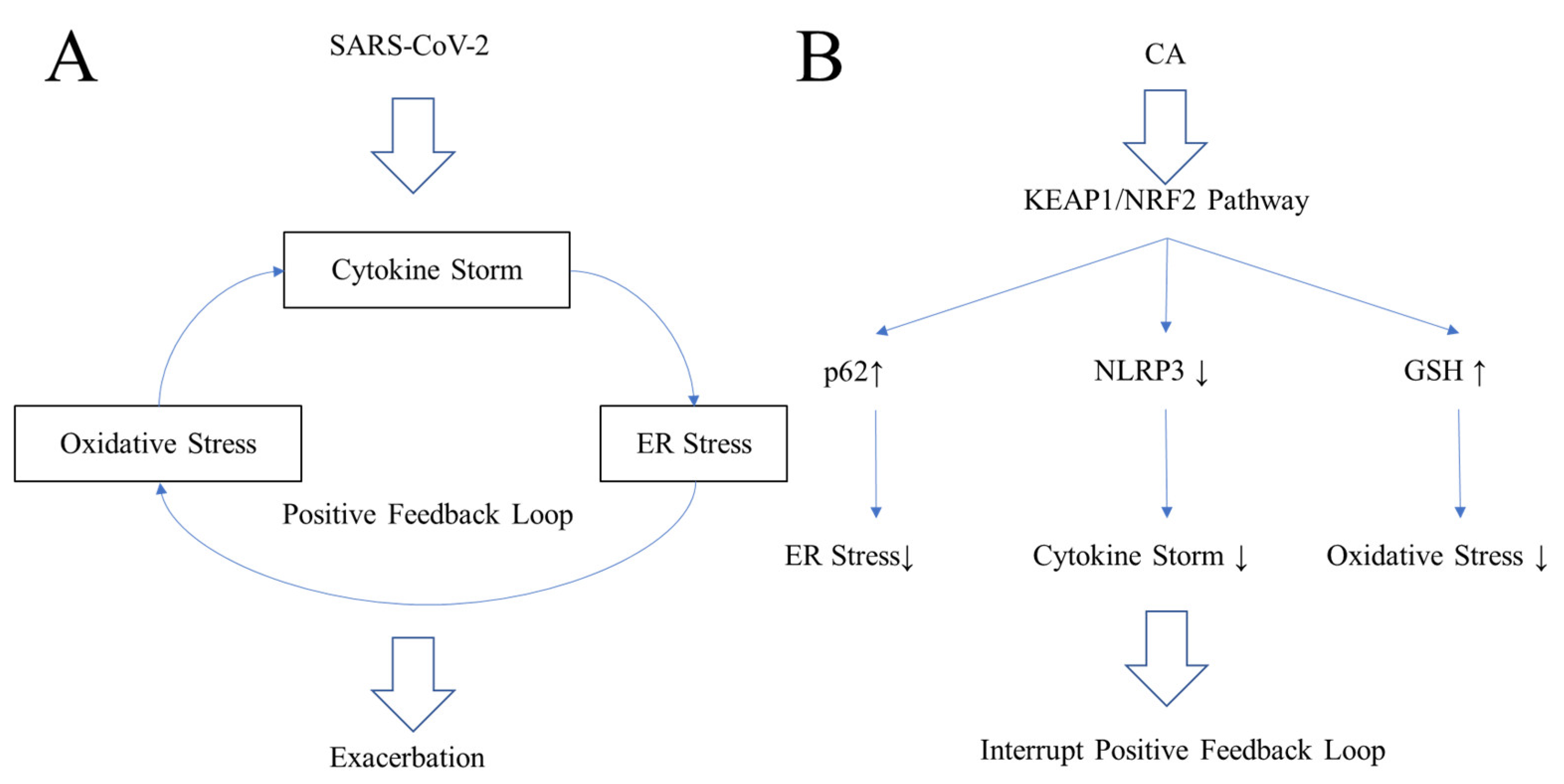
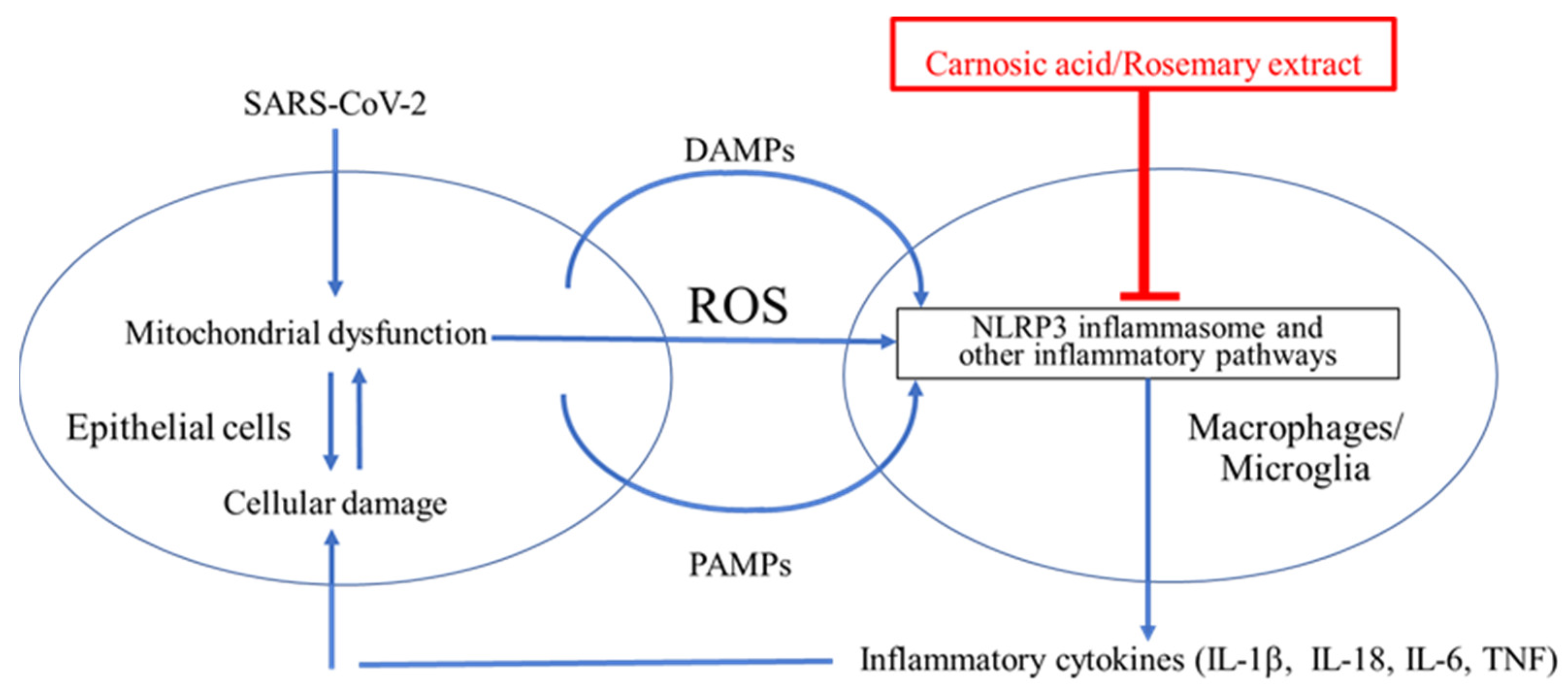
6.3. Oxidative Stress in COVID-19
6.4. Endoplasmic Reticulum (ER) Stress in COVID-19
6.5. Inhibiting the Positive Feedback Loop of COVID-19
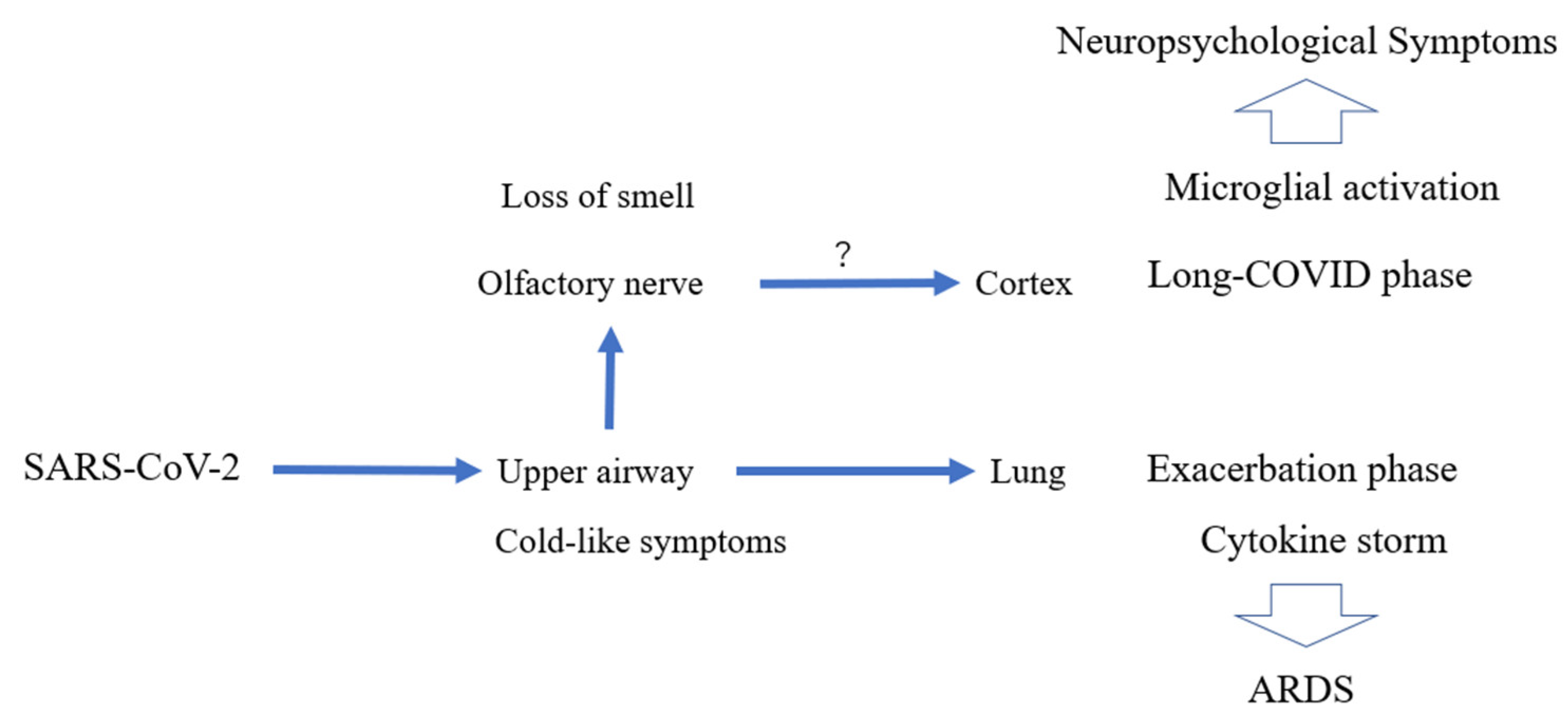
7. Potential Relationship of Neurological Sequelae in COVID-19 and Microglial Activation
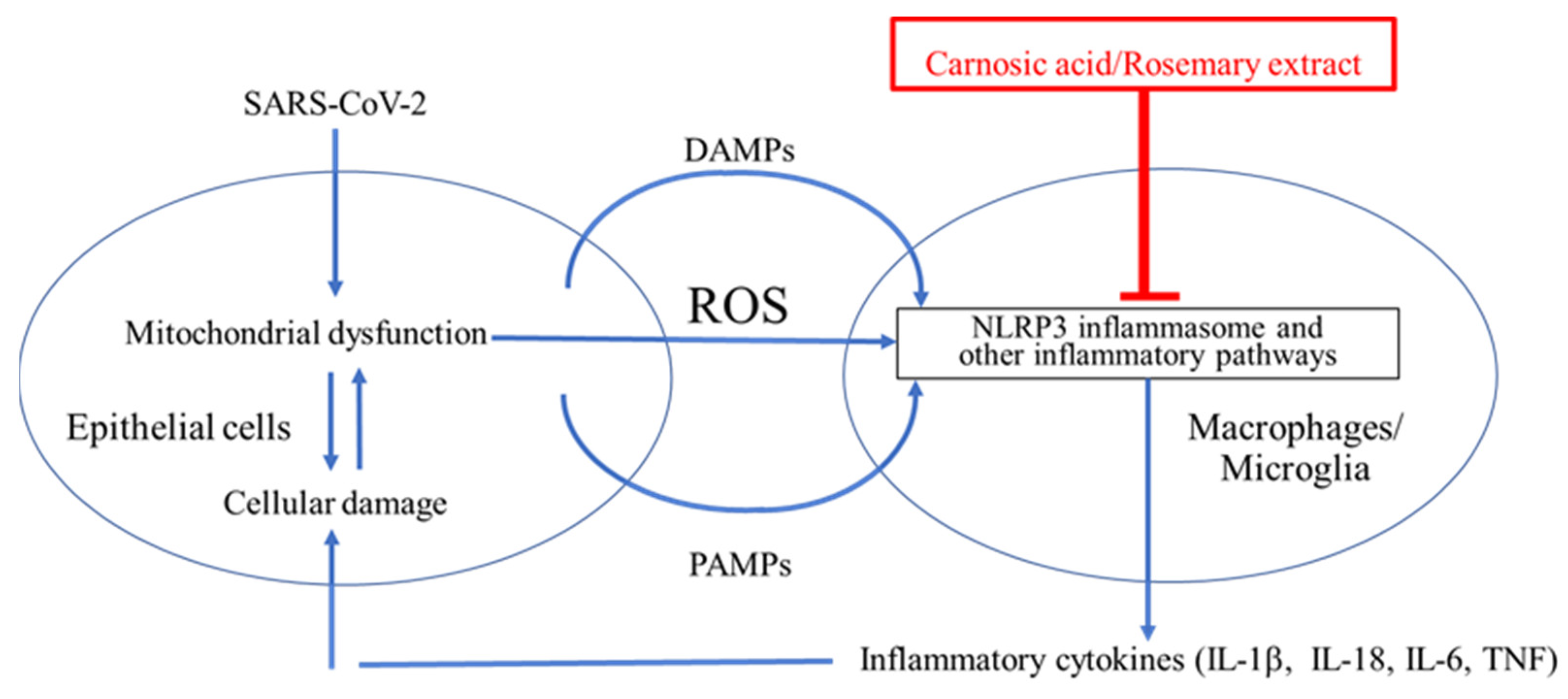
8. CA as a Potential COVID-19 Therapeutic via Inhibition of NLRP3 Inflammasome, Other Inflammatory Pathways, and SARS-CoV-2 Infection
8.1. ARDS Mediated by NLRP3 Inflammasome
8.2. CA as a Potential Treatment for COVID-19 by Suppressing Infection and Cytokine Production
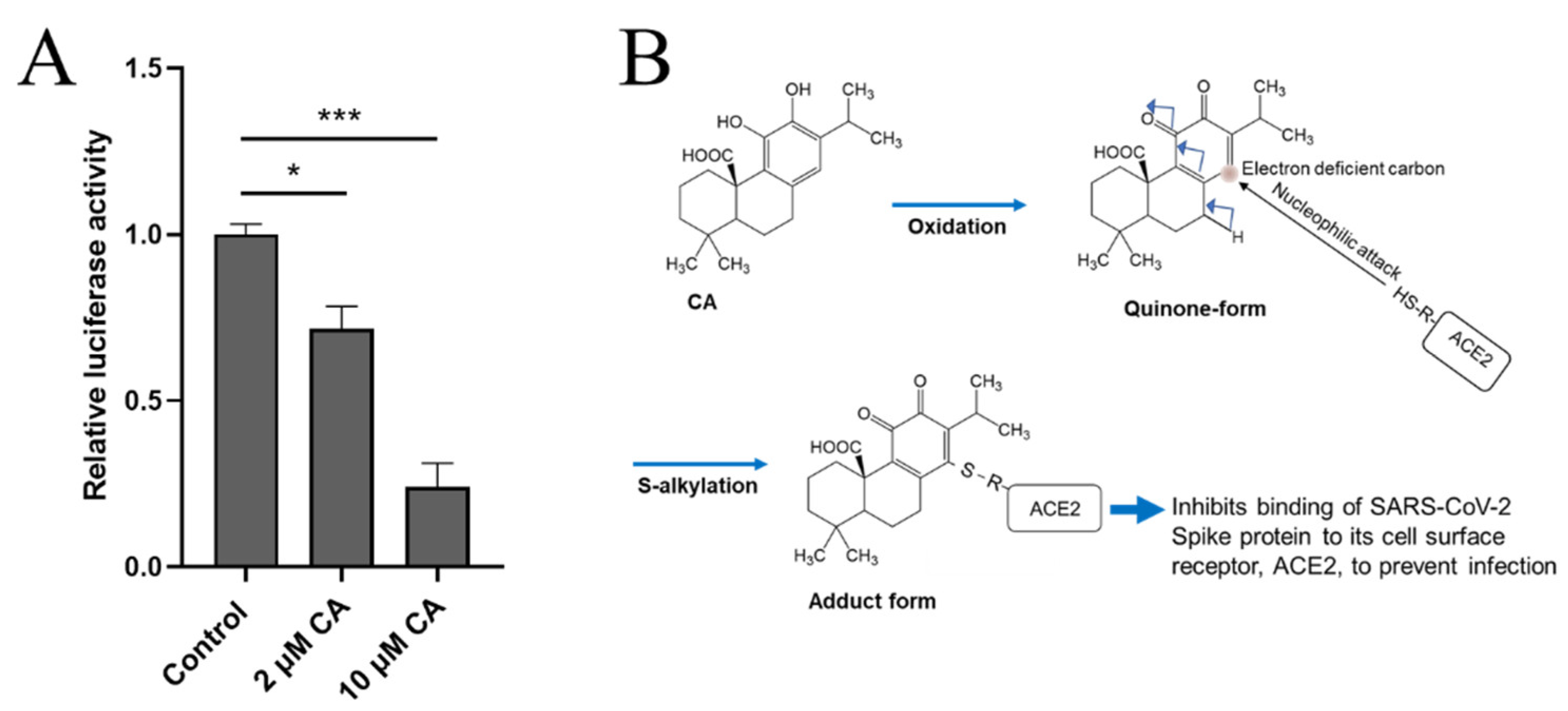
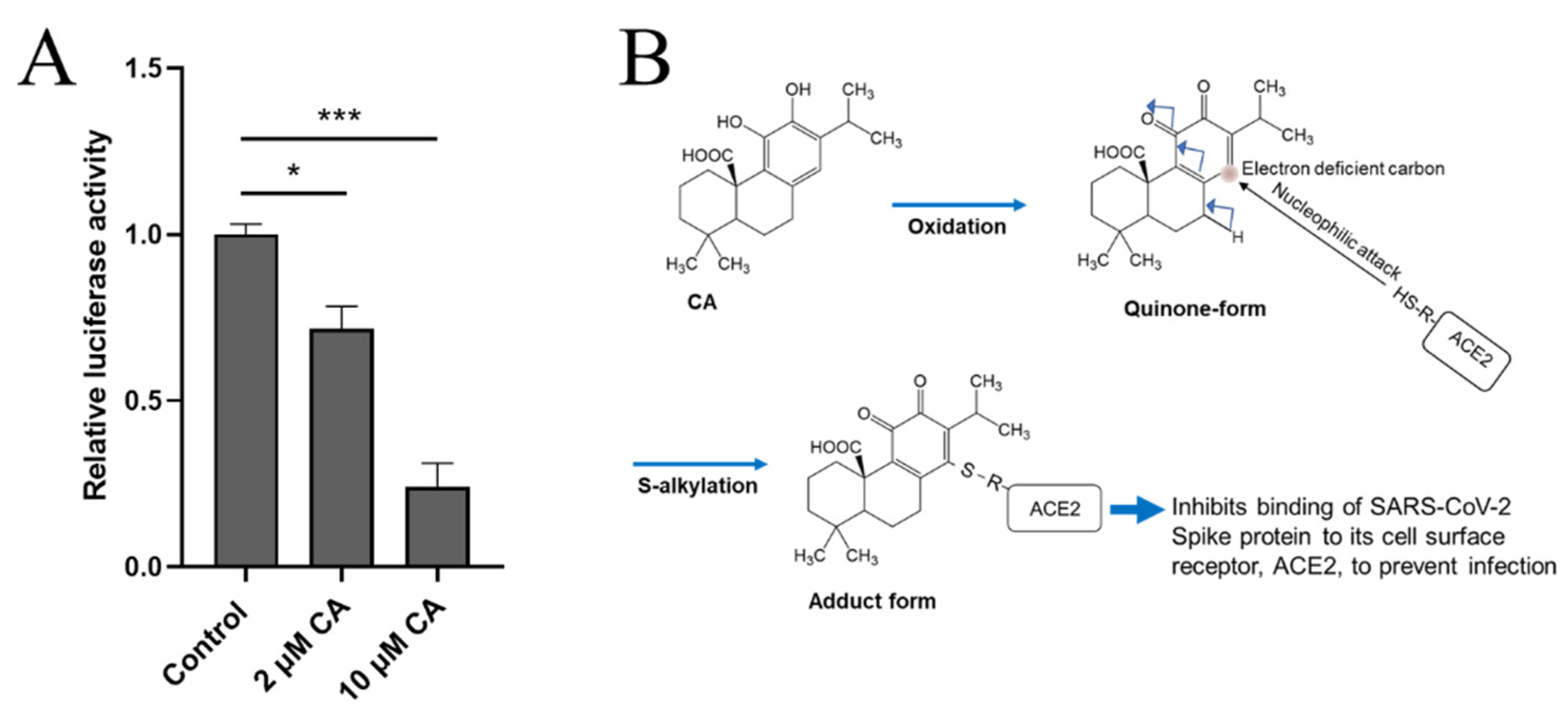
8.3. CA as a Potential Anti-Infectious Agent against SARS-CoV-2 by Inhibiting Binding to ACE2
9. Proposal for Testing Rosemary Extract or CA against SARS-CoV-2 Infection, Long- COVID, and Neurodegenerative Disorders
- CA passes through the blood–brain barrier (BBB) with good bioavailability after oral administration [54].
- CA inhibits activated microglia, in part, via inhibition of the NLRP3 inflammasome [18].
- Rosemary (containing CA and CS) is already used clinically as a therapeutic against rheumatoid arthritis in Germany and is on the FDA “generally regarded as safe” (GRAS) list in the U.S. [103,104]. Moreover, CA manifests very few side effects at very high concentrations in two-species toxicity testing.
10. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Nieto, G.; Ros, G.; Castillo, J. Antioxidant and antimicrobial properties of rosemary (Rosmarinus officinalis, L.): A review. Medicines 2018, 5, 98. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, J.R.; Camargo, S.E.A.; de Oliveira, L.D. Rosmarinus officinalis L.(rosemary) as therapeutic and prophylactic agent. J. Biomed. Sci. 2019, 26, 5. [Google Scholar] [PubMed]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer effects of rosemary (Rosmarinus officinalis L.) extract and rosemary extract polyphenols. Nutrients 2016, 8, 731. [Google Scholar]
- Generally Recognized as Safe (GRAS) in USA. Available online: https://www.fda.gov/food/food-ingredients-packaging/generally-recognized-safe-gras (accessed on 19 November 2021).
- 5. Current EU Approval and Their E Number in EU. Available online: https://ec.europa.eu/food/safety/food-improvement-agents/additives/database_en (accessed on 19 November 2021).
- Approved Food Additives by Ministry of Health, Labor and Welfare of Japan. Available online: https://www.ffcr.or.jp/tenka/list/post-12.html?OpenDocument (accessed on 19 November 2021).
- Andrade, J.M.; Faustino, C.; Garcia, C.; Ladeiras, D.; Reis, C.P.; Rijo, P. Rosmarinus officinalis L.: An update review of its phytochemistry and biological activity. Future Sci. OA 2018, 4, FSO283. [Google Scholar] [PubMed] [Green Version]
- De Oliveira, M.R. The dietary components carnosic acid and carnosol as neuroprotective agents: A mechanistic view. Mol. Neurobiol. 2016, 53, 6155–6168. [Google Scholar] [CrossRef] [PubMed]
- Loussouarn, M.; Krieger-Liszkay, A.; Svilar, L.; Bily, A.; Birtić, S.; Havaux, M. Carnosic acid and carnosol, two major antioxidants of rosemary, act through different mechanisms. Plant Physiol. 2017, 175, 1381–1394. [Google Scholar] [CrossRef] [Green Version]
- Chae, I.G.; Yu, M.H.; Im, N.K.; Jung, Y.T.; Lee, J.; Chun, K.S.; Lee, I.S. Effect of Rosmarinus officinalis L. on MMP-9, MCP-1 levels, and cell migration in RAW 264.7 and smooth muscle cells. J. Med. Food 2012, 15, 879–886. [Google Scholar]
- Kuo, C.F.; Su, J.D.; Chiu, C.H.; Peng, C.C.; Chang, C.H.; Sung, T.Y.; Huang, S.H.; Lee, W.C.; Chyau, C.C. Anti-inflammatory effects of supercritical carbon dioxide extract and its isolated carnosic acid from Rosmarinus officinalis leaves. J. Agric. Food Chem. 2011, 59, 3674–3685. [Google Scholar] [CrossRef]
- Estévez, M.; Ventanas, S.; Ramírez, R.; Cava, R. Analysis of volatiles in porcine liver pâtés with added sage and rosemary essential oils by using SPME-GC-MS. J. Agric. Food Chem. 2004, 52, 5168–5174. [Google Scholar] [CrossRef]
- Foresti, R.; Bains, S.K.; Pitchumony, T.S.; Brás, L.E.D.C.; Drago, F.; Dubois-Randé, J.-L.; Bucolo, C.; Motterlini, R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol. Res. 2013, 76, 132–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Shen, Z.; Yu, H.; Lu, G.; Yu, Y.; Liu, X.; Zheng, P. Carnosic acid protects against acetaminophen-induced hepatotoxicity by potentiating Nrf2-mediated antioxidant capacity in mice. Korean J. Physiol. Pharmacol. 2016, 20, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagitai, M.; Itoh, S.; Kitagawa, T.; Takenouchi, T.; Kitani, H.; Satoh, T. Carnosic acid, a pro-electrophilic compound, inhibits LPS-induced activation of microglia. Biochem. Biophys. Res. Commun. 2012, 418, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Hadad, N.; Levy, R. Combination of EPA with carotenoids and polyphenol synergistically attenuated the transformation of microglia to M1 phenotype via inhibition of NF-κB. Neuromol. Med. 2017, 19, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Han, J.J.; Zhang, F.; Liu, S.; Zhu, L.; Wang, Z.Z.; Zhang, G.X.; Zhang, Y. Carnosol modulates Th17 cell differentiation and microglial switch in experimental autoimmune encephalomyelitis. Front. Immunol. 2018, 9, 1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Ho, M.S. Microglia in Parkinson’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 335–353. [Google Scholar]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2015, 53, 6709–6715. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef]
- Sun, L.; Ma, W.; Gao, W.; Xing, Y.; Chen, L.; Xia, Z.; Zhang, Z.; Dai, Z. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N.Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Scott, M.J. Caspase-1 as a multifunctional inflammatory mediator: Noncytokine maturation roles. J. Leukoc. Biol. 2016, 100, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.R.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxidative Med. Cell. Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. β-Amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.-W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Wu, A.G.; Zhou, X.G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.Q.; Pan, R.; Yu, C.L.; Law, B.Y.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88 Pt B, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Lipton, S.A. Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 2007, 30, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; McKercher, S.R.; Lipton, S.A. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic. Biol Med. 2013, 65, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Research 2017, 6, 2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Okamoto, S.I.; Cui, J.; Watanabe, Y.; Furuta, K.; Suzuki, M.; Tohyama, K.; Lipton, S.A. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic phase II inducers. Proc. Natl. Acad. Sci. USA 2006, 103, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-W.; Lee, M.-S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharm. Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Han, M.; Li, S.; Li, L. Verapamil inhibits early acute liver failure through suppressing the NLRP3 inflammasome pathway. J. Cell. Mol. Med. 2021, 25, 5963–5975. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondriallLocalization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, K.; Zhang, Y.; Wang, Y.; Meng, X.; Wang, Y.; Yu, Y.; Chen, H. Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Agents Actions 2020, 69, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Wu, Y.F.; Yan, F.; Li, C.C.; Dai, Z.; You, Q.D.; Jiang, Z.Y.; Di, B. 5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), a novel Nrf2 activator targeting brain tissue, protects against MPTP-induced subacute Parkinson’s disease in mice by inhibiting the NLRP3 inflammasome and protects PC12 cells against oxidative stress. Free Radic. Biol. Med. 2019, 134, 288–303. [Google Scholar] [PubMed]
- Nagasaki, H.; Yoshimura, T.; Aoki, N. Real-time monitoring of inflammation status in 3T3-L1 adipocytes possessing a secretory Gaussia luciferase gene under the control of nuclear factor-κB response element. Biochem. Biophys. Res. Commun. 2012, 420, 623–627. [Google Scholar] [CrossRef]
- Xian, H.; Liu, Y.; Rundberg Nilsson, A.; Gatchalian, R.; Crother, T.R.; Tourtellotte, W.G.; Zhang, Y.; Aleman-Muench, G.R.; Lewis, G.; Chen, W.; et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 2021, 54, 1463–1477.e11. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-α therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Tabuchi, T.; Takahashi, T.; Kosaka, K.; Satoh, T. Activated glutathione metabolism participates in protective effects of carnosic acid against oxidative stress in neuronal HT22 cells. Planta Medica. 2009, 76, 683–688. [Google Scholar] [CrossRef]
- Satoh, T.; Izumi, M.; Inukai, Y.; Tsutsumi, Y.; Nakayama, N.; Kosaka, K.; Shimojo, Y.; Kitajima, C.; Itoh, K.; Yokoi, T.; et al. Carnosic acid protects neuronal HT22 cells through activation of the antioxidant-responsive element in free carboxylic acid- and catechol hydroxyl moieties-dependent manners. Neurosci. Lett. 2008, 434, 260–265. [Google Scholar] [CrossRef]
- Tena, M.T.; Valcárcel, M.; Hidalgo, P.J.; Ubera, J.L. Supercritical fluid extraction of natural antioxidants from rosemary: comparison with liquid solvent sonication. Anal. Chem. 1997, 69, 521–526. [Google Scholar] [CrossRef]
- Satoh, T.; Stalder, R.; McKercher, S.R.; Williamson, R.E.; Roth, G.P.; Lipton, S.A. Nrf2 and HSF-1 pathway activation via hydroquinone-based proelectrophilic small molecules is regulated by electrochemical oxidation potential. ASN Neuro. 2015, 7, 1759091415593294. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Saitoh, S.; Hosaka, M.; Kosaka, K. Simple ortho- and para-hydroquinones as compounds neuroprotective against oxidative stress in a manner associated with specific transcriptional activation. Biochem. Biophys. Res. Commun. 2009, 379, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kosaka, K.; Itoh, K.; Kobayashi, A.; Yamamoto, M.; Shimojo, Y.; Kitajima, C.; Cui, J.; Kamins, J.; Okamoto, S.; et al. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J. Neurochem. 2008, 104, 1116–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef]
- Nagar, S.; Noveral, S.M.; Trudler, D.; Lopez, K.M.; McKercher, S.R.; Han, X.; Yates, J.R., 3rd; Piña-Crespo, J.C.; Nakanishi, N.; Satoh, T.; et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc. Natl. Acad. Sci. USA 2017, 114, E4048–E4056. [Google Scholar]
- Satoh, T.; Rezaie, T.; Seki, M.; Sunico, C.R.; Tabuchi, T.; Kitagawa, T.; Yanagitai, M.; Senzaki, M.; Kosegawa, C.; Taira, H.; et al. Dual neuroprotective pathways of a pro-electrophilic compound via HSF-1-activated heat-shock proteins and Nrf2-activated phase 2 antioxidant response enzymes. J. Neurochem. 2011, 119, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Maynard, M.E.; Underwood, E.L.; Redell, J.B.; Zhao, J.; Kobori, N.; Hood, K.N.; Moore, A.N.; Dash, P.K. Carnosic acid improves outcome after repetitive mild traumatic brain injury. J. Neurotrauma. 2019, 36, 2147–2152. [Google Scholar] [CrossRef]
- Rezaie, T.; McKercher, S.R.; Kosaka, K.; Seki, M.; Wheeler, L.; Viswanath, V.; Chun, T.; Joshi, R.; Valencia, M.; Sasaki, S.; et al. Protective effect of carnosic acid, a pro-electrophilic compound, in models of oxidative stress and light-induced retinal degeneration. Invest. Ophthalmol. Vis. Sci. 2012, 53, 7847–7854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwager, J.; Richard, N.; Fowler, A.; Seifert, N.; Raederstorff, D. Carnosol and related substances modulate chemokine and cytokine production in macrophages and chondrocytes. Molecules 2016, 21, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habtemariam, S. The Therapeutic Potential of rosemary (Rosmarinus officinalis) diterpenes for Alzheimer’s disease. Evid. Based Complement. Altern. Med. 2016, 2016, 2680409. [Google Scholar] [CrossRef] [Green Version]
- Perry, N.S.L.; Menzies, R.; Hodgson, F.; Wedgewood, P.; Howes, M.R.; Brooker, H.J.; Wesnes, K.A.; Perry, E.K. A randomised double-blind placebo-controlled pilot trial of a combined extract of sage, rosemary and melissa, traditional herbal medicines, on the enhancement of memory in normal healthy subjects, including influence of age. Phytomedicine 2018, 39, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-R.; Tsai, C.-W.; Chang, S.-W.; Lin, C.-Y.; Huang, L.-C. Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: Involvement of antioxidative enzymes induction. Chem. Interact. 2015, 225, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.S.; Cho, H.S.; Lee, H.J.; Kim, S.Y.; Kim, S.; Lee, S.Y.; Chun, H.S. Carnosol, a component of rosemary (Rosmarinus officinalis L.) protects nigral dopaminergic neuronal cells. NeuroReport 2006, 17, 1729–1733. [Google Scholar] [PubMed]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of β-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef]
- Zhao, J.; Yu, S.; Zheng, Y.; Yang, H.; Zhang, J. Oxidative modification and its implications for the neurodegeneration of Parkinson’s disease. Mol. Neurobiol. 2017, 54, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, E.; Chin, T.Y.; Chen, C.P.; Wu, T.Y. Management of moderate toa severe Alzheimer’s disease: Focus on memantine. Taiwan J. Obstet Gynecol. 2011, 50, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef]
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development. J. Hepatol. 2020, 74, 168–184. [Google Scholar] [CrossRef]
- De Candia, P.; Prattichizzo, F.; Garavelli, S.; Matarese, G. T cells: Warriors of SARS-CoV-2 infection. Trends Immunol. 2021, 42, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-boosting, antioxidant and anti-inflammatory food supplements targeting pathogenesis of COVID-19. Front. Immunol. 2020, 11, 570122. [Google Scholar] [CrossRef]
- Uddin, M.; Mustafa, F.; Rizvi, T.A.; Loney, T.; Suwaidi, H.A.; Al-Marzouqi, A.H.H.; Eldin, A.K.; Alsabeeha, N.; Adrian, T.E.; Stefanini, C.; et al. SARS-CoV-2/COVID-19: Viral genomics, epidemiology, vaccines, and therapeutic interventions. Viruses 2020, 12, 526. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.; Swartz, T.H. Targeting the NLRP3 Inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm. Res. 2020, 70, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Papatheou, D.; Melita, H. COVID-19 infection: Viral macro- and micro-vascular coagulopathy and thromboembolism/prophylactic and therapeutic management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, H.; Li, L.; Liu, C.; Yan, S.; Chen, H.; Li, Y. Ferritin in the coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. J. Clin. Lab. Anal. 2020, 34, e23618. [Google Scholar] [CrossRef]
- Perricone, C.; Bartoloni, E.; Bursi, R.; Cafaro, G.; Guidelli, G.M.; Shoenfeld, Y.; Gerli, R. COVID-19 as part of the hyperferritinemic syndromes: The role of iron depletion therapy. Immunol. Res. 2020, 68, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Tamura, T. Hydrogen gas therapy: From preclinical studies to clinical trials. Curr. Pharm. Des. 2021, 27, 650–658. [Google Scholar] [CrossRef]
- Zhang, J.; Rao, X.; Li, Y.; Zhu, Y.; Liu, F.; Guo, G.; Luo, G.; Meng, Z.; De Backer, D.; Xiang, H.; et al. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann. Intensiv. Care 2021, 11, 5. [Google Scholar] [CrossRef]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between endoplasmic reticulum stress and anti-viral activities: A novel therapeutic target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef] [PubMed]
- Sureda, A.; Alizadeh, J.; Nabavi, S.F.; Berindan-Neagoe, I.; Cismaru, C.A.; Jeandet, P.; Łos, M.J.; Clementi, E.; Nabavi, S.M.; Ghavami, S. Endoplasmic reticulum as a potential therapeutic target for COVID-19 infection management? Eur. J. Pharmacol. 2020, 882, 173288. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Nouri, Z.; Moradi, S.Z.; Akkol, E.K.; Piri, S.; Sobarzo-Sánchez, E.; Farzaei, M.H.; Echeverría, J. Targeting multiple signal transduction pathways of SARS-CoV-2: Approaches to COVID-19 therapeutic candidates. Molecules 2021, 26, 2917. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jiménez-Villegas, J.; Escoll, M.; Fernández-Ginés, R.; Garcia Yagüe, A.J.; Lastra, D.; Manda, G.; Rojo, A.I.; et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharmacol. Sci. 2020, 41, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; The ARIA Group; Cristol, J.-P.; Czarlewski, W.; Anto, J.M.; Martineau, A.; Haahtela, T.; Fonseca, S.C.; Iaccarino, G.; Blain, H.; et al. Nrf2-interacting nutrients and COVID-19: Time for research to develop adaptation strategies. Clin. Transl. Allergy 2020, 10, 58. [Google Scholar]
- Jiang, T.; Harder, B.; de la Vega, M.R.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, K.; Mimura, J.; Itoh, K.; Satoh, T.; Shimojo, Y.; Kitajima, C.; Maruyama, A.; Yamamoto, M.; Shirasawa, T. Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells. J. Biochem. 2010, 147, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Cholevas, C.; Polyzoidis, K.; Politis, A. Long-COVID syndrome-associated brain fog and chemofog: Luteolin to the rescue. BioFactors 2021, 47, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Hugon, J.; Msika, E.-F.; Queneau, M.; Farid, K.; Paquet, C. Long COVID: Cognitive complaints (brain fog) and dysfunction of the cingulate cortex. J. Neurol 2021, Jun 18, 1–3. [Google Scholar] [CrossRef]
- Awogbindin, I.O.; Ben-Azu, B.; Olusola, B.A.; Akinluyi, E.T.; Adeniyi, P.A.; Di Paolo, T.; Tremblay, M.È. Microglial implications in SARS-CoV-2 infection and COVID-19: Lessons from viral RNA neurotropism and possible relevance to Parkinson’s disease. Front. Cell Neurosci. 2021, 15, 670298. [Google Scholar] [CrossRef]
- López-Reyes, A.; Martinez-Armenta, C.; Espinosa-Velázquez, R.; Vázquez-Cárdenas, P.; Cruz-Ramos, M.; Palacios-Gonzalez, B.; Gomez-Quiroz, L.E.; Martínez-Nava, G.A. NLRP3 inflammasome: The stormy link between obesity and COVID-19. Front. Immunol. 2020, 11, 570251. [Google Scholar] [CrossRef]
- Mccarty, M.F.; Iloki Assanga, S.B.; Luján, L.L.; O’Keefe, J.H.; DiNicolantonio, J.J. Nutraceutical strategies for suppressing NLRP3 inflammasome activation: Pertinence to the management of COVID-19 and beyond. Nutrients 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Mccord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Geraci, K.P.; Gao, B. Nrf2 activator PB125® as a potential therapeutic agent against COVID-19. Antioxidants 2020, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Gao, B. Nrf2 activator PB125® as a carnosic acid-based therapeutic agent against respiratory viral diseases, including COVID-19. Free Radic. Biol Med. 2021, 175, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.; McCord, J.M. Phytochemical combination PB125 activates the Nrf2 pathway and induces cellular protection against oxidative injury. Antioxidants 2019, 8, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della-Torre, E.; Criscuolo, E.; Lanzillotta, M.; Locatelli, M.; Clementi, N.; Mancini, N.; Dagna, L.; COVID-BioB Study Group. IL-1 and IL-6 inhibition affects the neutralising activity of anti-SARS-CoV-2 antibodies in patients with COVID-19. Lancet Rheumatol. 2021, 3, e829–e831. [Google Scholar]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Al-Okbi, S.Y. Nutraceuticals of anti-inflammatory activity as complementary therapy for rheumatoid arthritis. Toxicol. Ind. Heal. 2012, 30, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Lukaczer, D.; Darland, G.; Tripp, M.; Liska, D.A.; Lerman, R.H.; Schiltz, B.; Bland, J.S. A Pilot trial evaluating meta050, a proprietary combination of reduced iso-alpha acids, rosemary extract and oleanolic acid in patients with arthritis and fibromyalgia. Phytother. Res. 2005, 19, 864–869. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satoh, T.; Trudler, D.; Oh, C.-K.; Lipton, S.A. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants 2022, 11, 124. https://doi.org/10.3390/antiox11010124
Satoh T, Trudler D, Oh C-K, Lipton SA. Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants. 2022; 11(1):124. https://doi.org/10.3390/antiox11010124
Chicago/Turabian StyleSatoh, Takumi, Dorit Trudler, Chang-Ki Oh, and Stuart A. Lipton. 2022. "Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome" Antioxidants 11, no. 1: 124. https://doi.org/10.3390/antiox11010124
APA StyleSatoh, T., Trudler, D., Oh, C.-K., & Lipton, S. A. (2022). Potential Therapeutic Use of the Rosemary Diterpene Carnosic Acid for Alzheimer’s Disease, Parkinson’s Disease, and Long-COVID through NRF2 Activation to Counteract the NLRP3 Inflammasome. Antioxidants, 11(1), 124. https://doi.org/10.3390/antiox11010124