
NRF2 in Viral Infection

Abstract
:1. Introduction
2. Reported Cases of NRF2 Activation during Viral Infections
3. Reported Cases of NRF2 Downregulation during Viral Infections
4. Importance of NRF2 in Viral Infections
4.1. Protective Role of NRF2
4.2. Pathogenic Role of NRF2

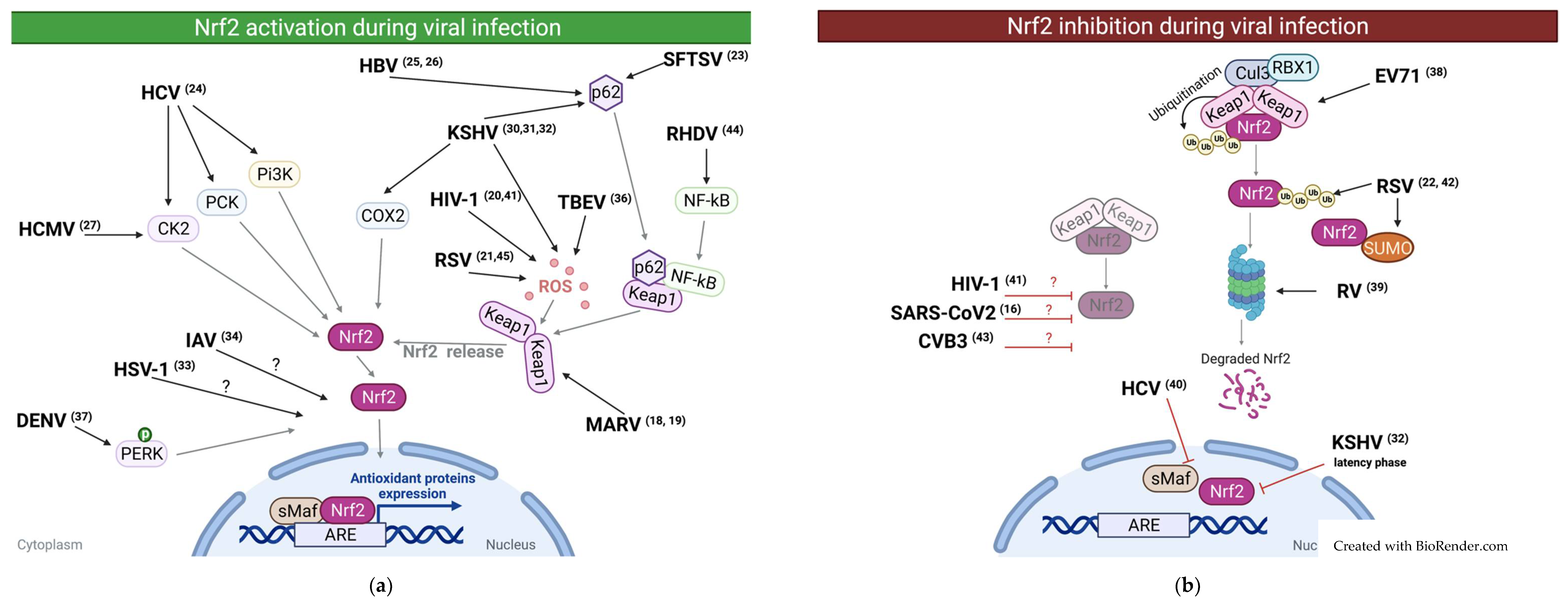{kind=link}
| Virus | Model | Effect on NRF2 | Viral Replication | Patho/Cytoprotective effect | Mechanism | Ref |
|---|---|---|---|---|---|---|
| HIV | In vitro expression of HIV protein tat in neuroblastoma cell line | ↑ | ND | Induction of ROS | [20] | |
| In vitro infection of primary human monocytes and in vivo rat | ↓ | ND | ND | [41] | ||
| RSV | In vitro with human alveolar basal epithelial cell line | ↑ | ↓ | ND | [21] | |
| In vivo with Nrf2+/+ and Nrf2−/− mice | ↑ | ↓ | ND | [45] | ||
| In vivo with mice and in vitro with human alveolar basal epithelial cell line | ↓ | ND | Proteasomal degradation of Nrf2 | [22] | ||
| In vitro with human alveolar epithelial cell line | ↓ | ND | SUMOylation and ubiquitination of Nrf2 induce its degradation | [42] | ||
| SFTSV | In vivo with mice | ↑ | ND | Pathogenic: mice infected with SFTSV mutated for this nonstructural protein showed a higher survival rate and less weight loss. | SFTSV viral protein interact with TRIM21 that normally mediated p62 ubiquitination. Hence p62 is stabilized and interact with KEAP1 leading to set free Nrf2 | [23] |
| HCV | In vitro with hepatocyte cell line | ↑ | ND | HCV core proteins activate Nrf2/ARE pathway via several independent mechanisms. 1. Nrf2 activation was triggered by protein kinase C in response to accumulation of ROS. 2. Nrf2 was also activated in ROS independent manner 3. The effect of some viral proteins was mediated through casein kinase 2 and phosphoinositide 3 kinase. | [24] | |
| In vitro with hepatocyte cell line | ↓ | ND | HCV Core and nonstructural proteins induce nuclear export of sMaf a Nrf2 partner | [40] | ||
| In vitro with hepatocellular carcinoma cell line | ND | ND | Pathogenic: Persistent activation of NRF2 makes HCV positive tumor cells resistant to oxidative damage and anticancer agent | [55] | ||
| HCMV | In vitro with primary human foreskin fibroblasts | ↑ | ↓ | Casein Kinase 2 mediated | [27] | |
| HBV | In vitro with human hepatoma derived cell lines | ↑ | ND | Triggered by HBV proteins (HBx and LHBs) | [25] | |
| In vitro with human hepatocyte cell lines | ↑ | ND | HBV protein HBx stimulate p62-Keap1 interaction leading to Nrf2 activation | [26] | ||
| MARV | In vitro with human kidney cell line | ↑ | ND | Pathogenic: Infected mice deficient for Nrf2 have a higher survival rate than infected control mice | VP24 viral protein bind to KEAP1 setting free Nrf2 | [18] |
| In vitro with lymphocyte cell line | ↑ | ↓ | Cytoprotective: Activation of antioxidant mechanisms | MARV protein Mvp24 interact directly with KEAP1 | [19] | |
| KSHV | In vitro with Human dermal microvascular endothelial cell line | ↑ | ND | Pathogenic: Nrf2 contribute to the expression of host factor VEGF-A, VEGF-D and COX-2 important for KSHV pathogenesis | Induction of ROS | [30] |
| In vitro with primary effusion lymphoma cell line and tissue | ↑ | ↑ | COX-2/PGE2 axis induces Nrf2 through prostaglandin E receptor 4 (EP4) | [32] | ||
| In vitro with TIVE and LTC cells | ↑ | ↑ | Two distinct mechanisms: (1) Ros induction. (2) ROS-independent and through P62–Keap1 interaction | [31] | ||
| IAV | In vitro with alveolar type II cells and alveolar macrophages isolated from human lungs infected | ↑ | ↓ | ND | [34] | |
| Primary human nasal epithelial cell | ND | ↓ | ND | [46] | ||
| In vitro Madin darby canine kidney cell line | ND | ↓ | ND | [47] | ||
| HSV1 | In vitro with primary normal human dermal fibroblast cell line | ↑ | ↓ | ND | [33] | |
| TBEV | In vitro with human embryonic kidney cell line | ↑ | ND | TBEV nonstructural protein induces ROS production | [35] | |
| Broad | In vitro chemical activation of NRF2 before infection inhibited replication of HSV-1/2,VACV, ZKV, SARS-CoV2 | ↑ | ↓ | ND | [16] | |
| DENV | In vitro with murine monocytic cell line | ↑ | ND | DENV infection induces ER stress that phosphorylate PERK that activate Nrf2 | [37] | |
| In vitro with monocyte isolated from human peripheral blood mononuclear cell from healthy donors | ↑ | ND | ND | [36] | ||
| RHDV | In vivo with rabbits | ↓ | ND | Cytoprotective: Upregulation of NRF2 reduce infected rabbit’s death rate | Nuclear export of Nrf2 by KEAP1-NFκB complex | [44] |
| EV71 | In vitro with human rhabdomyosarcoma cell line | ↓ | ↓ | Up regulation of Keap1 | [38] | |
| CMV | In vivo with mice Nrf2−/− and Nrf2+/+ | ND | ↓ | ND | [51] | |
| ZIKV | In vitro with primary human macrophages | ND | ↓ | ND | [49] | |
| EBOV | In vitro with human cell lines | ND | ↓ | ND | [48] | |
| RV | In vitro with human cell lines | ND | ↓ | ND | [50] | |
| In vitro with green monkey fetal kidney cell line | ↓ | ND | Proteasomal degradation dependency | [39] | ||
| CVB3 | In vitro with mice cardiomyocytes and in vivo with mice | ↓ | ND | Contribute to oxidative stress | [43] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| 4-OI | 4-octyl itaconate |
| AKT | Protein Kinase B |
| AM | Alveolar macrophages |
| AOE | Antioxidant Enzyme |
| ARE | Antioxidant response element |
| Bach-1 | BTB and CNC homology 1 |
| BAL | Bronchoalveolar lavage |
| CD36 | Cluster Determinant 36 |
| CK2 | Casein Kinase 2 |
| CUL1 | CULLIN1 |
| CUL3 | CULLIN3 |
| DENV | Dengue virus |
| DMF | Dimethyl fumarate |
| EBOV | Ebola virus |
| ERK | Extracellular signal-regulated kinase |
| EV71 | Enterovirus 71 |
| GCLC | Glutamate-Cysteine Ligase catalytic subunit |
| GCLM | Glutamate-Cysteine Ligase Modifier subunit |
| GSH-Px | glutathion peroxidase |
| GSK3 | Glycogen synthase kinase 3 |
| HBV | Human Hepatitis B Virus |
| HCMV | Human Cytomegalovirus |
| HCV | Hepatitis C Virus |
| HIV | Human Immunodeficiency Virus |
| HMOX-1 | Heme Oxygenase-1 (gene) |
| HO-1 | Heme oxygenase 1 |
| HSV1 | Herpes Simplex Virus 1 |
| HPI | Hour post infection |
| IAV | Influenza A Virus |
| IFN | Interferon |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| KSHV | Kaposi’s Sarcoma-associated Herpes Virus |
| LPS | Lipopolysaccharide |
| MARV | Marburgvirus |
| MARVS | Mitochondrial antiviral signaling |
| MCMV | Murine cytomegalovirus |
| MOI | Multiplicity of infection |
| mRNA | messenger Ribonucleic Acid |
| NAD(P)H | Nicotinamide Adenine Dinucleotide Phosphate Hydrogen |
| NFκB | Nuclear factor-kappa B |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NS | Nonstructural protein |
| ORF | Open Reading Frame |
| PI3K | Phosphatidylinositol-3-Kinase |
| PKC | Protein kinase C |
| PML-NBs | Membrane free subnuclear compartments named promyelocytic leukemia protein-nuclear bodies |
| ROS | Reactive oxygen species |
| SARS-CoV2 | Severe Acute Respiratory Syndrome-Corona Virus |
| SFTSV | Severe Fever with Thrombocytopenia Syndrome Virus |
| sMAF | Small musculo-aponeurotic fibrosarcoma |
| SOD | Superoxide dismutase |
| SOD2 | Superoxide dismutase 2 |
| STING | Stimulator of Interferon genes |
| TBB | 4,5,6,7-tetrabromobenzotriazole |
| tBHQ | tert-butylhydroquinone |
| TLR | Toll-like receptor |
| TEBV | Tick-Borne Encephalitis virus |
| TRIM21 | Tripartite motif-containing protein 21 |
| RHDV | Rabbit Hemorrhagic Disease Viruses |
| RNF4 | RING finger protein 4 |
| RSV | Respiratory Syncytial Virus |
| RV | Rotavirus |
| VACV | Vaccinia virus |
| WT | Wild type |
| ZIKV | Zika virus |
References
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, O.; Murphy, P.; Prydz, H.; Kolsto, A.B. Interaction of the CNC-bZIP factor TCF11/LCR-F1/Nrf1 with MafG: Binding-site selection and regulation of transcription. Nucleic Acids Res. 1998, 26, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Brandtoft, A.M.; Gunderstofte, C.; Villadsen, N.L.; Krapp, C.; Thielke, A.L.; Laustsen, A.; Peri, S.; Hansen, A.L.; Bonefeld, L.; et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 2018, 9, 3506. [Google Scholar] [CrossRef] [Green Version]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Ramezani, A.; Nahad, M.P.; Faghihloo, E. The role of Nrf2 transcription factor in viral infection. J. Cell. Biochem. 2018, 119, 6366–6382. [Google Scholar] [CrossRef]
- Page, A.; Volchkova, V.A.; Reid, S.P.; Mateo, M.; Bagnaud-Baule, A.; Nemirov, K.; Shurtleff, A.C.; Lawrence, P.; Reynard, O.; Ottmann, M.; et al. Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell Rep. 2014, 6, 1026–1036. [Google Scholar] [CrossRef]
- Edwards, M.R.; Johnson, B.; Mire, C.E.; Xu, W.; Shabman, R.S.; Speller, L.N.; Leung, D.W.; Geisbert, T.W.; Amarasinghe, G.K.; Basler, C.F. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep. 2014, 6, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Mastrantonio, R.; Cervelli, M.; Pietropaoli, S.; Mariottini, P.; Colasanti, M.; Persichini, T. HIV-Tat Induces the Nrf2/ARE Pathway through NMDA Receptor-Elicited Spermine Oxidase Activation in Human Neuroblastoma Cells. PLoS ONE 2016, 11, e0149802. [Google Scholar] [CrossRef]
- Sun, T.; Yu, H.Y.; Zhang, C.L.; Zhu, T.N.; Huang, S.H. Respiratory syncytial virus infection up-regulates TLR7 expression by inducing oxidative stress via the Nrf2/ARE pathway in A549 cells. Arch. Virol. 2018, 163, 1209–1217. [Google Scholar] [CrossRef]
- Komaravelli, N.; Tian, B.; Ivanciuc, T.; Mautemps, N.; Brasier, A.R.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2. Free Radic. Biol. Med. 2015, 88, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Jiang, Z.; Shin, W.J.; Jung, J.U. Severe Fever with Thrombocytopenia Syndrome Virus NSs Interacts with TRIM21 To Activate the p62-Keap1-Nrf2 Pathway. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Koh, K.; Kim, Y.E.; Ahn, J.H.; Kim, S. Upregulation of Nrf2 expression by human cytomegalovirus infection protects host cells from oxidative stress. J. Gen. Virol. 2013, 94, 1658–1668. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Song, J.D.; Chung, H.T.; Park, Y.C. Protein kinase CK2 mediates peroxynitrite-induced heme oxygenase-1 expression in articular chondrocytes. Int. J. Mol. Med. 2012, 29, 1039–1044. [Google Scholar] [CrossRef]
- Afonyushkin, T.; Oskolkova, O.V.; Binder, B.R.; Bochkov, V.N. Involvement of CK2 in activation of electrophilic genes in endothelial cells by oxidized phospholipids. J. Lipid Res. 2011, 52, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Gjyshi, O.; Bottero, V.; Veettil, M.V.; Dutta, S.; Singh, V.V.; Chikoti, L.; Chandran, B. Kaposi's sarcoma-associated herpesvirus induces Nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog. 2014, 10, e1004460. [Google Scholar] [CrossRef]
- Gjyshi, O.; Flaherty, S.; Veettil, M.V.; Johnson, K.E.; Chandran, B.; Bottero, V. Kaposi's sarcoma-associated herpesvirus induces Nrf2 activation in latently infected endothelial cells through SQSTM1 phosphorylation and interaction with polyubiquitinated Keap1. J. Virol. 2015, 89, 2268–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjyshi, O.; Roy, A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chandran, B. Activated Nrf2 Interacts with Kaposi's Sarcoma-Associated Herpesvirus Latency Protein LANA-1 and Host Protein KAP1 To Mediate Global Lytic Gene Repression. J. Virol. 2015, 89, 7874–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyler, E.; Franke, V.; Menegatti, J.; Kocks, C.; Boltengagen, A.; Praktiknjo, S.; Walch-Ruckheim, B.; Bosse, J.; Rajewsky, N.; Grasser, F.; et al. Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 2019, 10, 4878. [Google Scholar] [CrossRef] [Green Version]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir. Res. 2012, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Kuzmenko, Y.V.; Smirnova, O.A.; Ivanov, A.V.; Starodubova, E.S.; Karpov, V.L. Nonstructural Protein 1 of Tick-Borne Encephalitis Virus Induces Oxidative Stress and Activates Antioxidant Defense by the Nrf2/ARE Pathway. Intervirology 2016, 59, 111–117. [Google Scholar] [CrossRef]
- Olagnier, D.; Peri, S.; Steel, C.; van Montfoort, N.; Chiang, C.; Beljanski, V.; Slifker, M.; He, Z.; Nichols, C.N.; Lin, R.; et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.L.; Lin, Y.S.; Chen, C.L.; Tsai, T.T.; Tsai, C.C.; Wu, Y.W.; Ou, Y.D.; Chu, Y.Y.; Wang, J.M.; Yu, C.Y.; et al. Activation of Nrf2 by the dengue virus causes an increase in CLEC5A, which enhances TNF-alpha production by mononuclear phagocytes. Sci. Rep. 2016, 6, 32000. [Google Scholar] [CrossRef] [Green Version]
- Bai, Z.; Zhao, X.; Li, C.; Sheng, C.; Li, H. EV71 virus reduces Nrf2 activation to promote production of reactive oxygen species in infected cells. Gut Pathog. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Patra, U.; Mukhopadhyay, U.; Mukherjee, A.; Sarkar, R.; Chawla-Sarkar, M. Progressive Rotavirus Infection Downregulates Redox-Sensitive Transcription Factor Nrf2 and Nrf2-Driven Transcription Units. Oxid. Med. Cell. Longev. 2020, 2020, 7289120. [Google Scholar] [CrossRef]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [Green Version]
- Staitieh, B.S.; Ding, L.; Neveu, W.A.; Spearman, P.; Guidot, D.M.; Fan, X. HIV-1 decreases Nrf2/ARE activity and phagocytic function in alveolar macrophages. J. Leukoc. Biol. 2017, 102, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Komaravelli, N.; Ansar, M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces NRF2 degradation through a promyelocytic leukemia protein-ring finger protein 4 dependent pathway. Free Radic. Biol. Med. 2017, 113, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Ai, F.; Zheng, J.; Zhang, Y.; Fan, T. Inhibition of 12/15-LO ameliorates CVB3-induced myocarditis by activating Nrf2. Chem. Biol. Interact. 2017, 272, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Wei, H.; Song, Y.; Chen, M.; Fan, Z.; Qiu, R.; Zhu, W.; Xu, W.; Wang, F. NF-kappaB and Keap1 Interaction Represses Nrf2-Mediated Antioxidant Response in Rabbit Hemorrhagic Disease Virus Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Arakaki, Y.; Esumi, T.; Kohnomi, S.; Yamamoto, C.; Suzuki, Y.; Takahashi, E.; Konishi, S.; Kido, H.; Kuzuhara, T. Bakuchiol Is a Phenolic Isoprenoid with Novel Enantiomer-selective Anti-influenza A Virus Activity Involving Nrf2 Activation. J. Biol. Chem. 2015, 290, 28001–28017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Konduru, K.; Solovena, V.; Zhou, Z.H.; Kumari, N.; Takeda, K.; Nekhai, S.; Bavari, S.; Kaplan, G.G.; Yamada, K.M.; et al. Therapeutic potential of the heme oxygenase-1 inducer hemin against Ebola virus infection. Curr. Trends Immunol. 2016, 17, 117–123. [Google Scholar]
- Huang, H.; Falgout, B.; Takeda, K.; Yamada, K.M.; Dhawan, S. Nrf2-dependent induction of innate host defense via heme oxygenase-1 inhibits Zika virus replication. Virology 2017, 503, 1–5. [Google Scholar] [CrossRef]
- Patra, U.; Mukhopadhyay, U.; Sarkar, R.; Mukherjee, A.; Chawla-Sarkar, M. RA-839, a selective agonist of Nrf2/ARE pathway, exerts potent anti-rotaviral efficacy in vitro. Antivir. Res. 2019, 161, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Seelige, R.; Saddawi-Konefka, R.; Adams, N.M.; Picarda, G.; Sun, J.C.; Benedict, C.A.; Bui, J.D. Interleukin-17D and Nrf2 mediate initial innate immune cell recruitment and restrict MCMV infection. Sci. Rep. 2018, 8, 13670. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Sacca, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herengt, A.; Thyrsted, J.; Holm, C.K. NRF2 in Viral Infection. Antioxidants 2021, 10, 1491. https://doi.org/10.3390/antiox10091491
Herengt A, Thyrsted J, Holm CK. NRF2 in Viral Infection. Antioxidants. 2021; 10(9):1491. https://doi.org/10.3390/antiox10091491
Chicago/Turabian StyleHerengt, Angela, Jacob Thyrsted, and Christian K. Holm. 2021. "NRF2 in Viral Infection" Antioxidants 10, no. 9: 1491. https://doi.org/10.3390/antiox10091491
APA StyleHerengt, A., Thyrsted, J., & Holm, C. K. (2021). NRF2 in Viral Infection. Antioxidants, 10(9), 1491. https://doi.org/10.3390/antiox10091491