
Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders



{kind=link}
{kind=link}
Abstract
1. Introduction
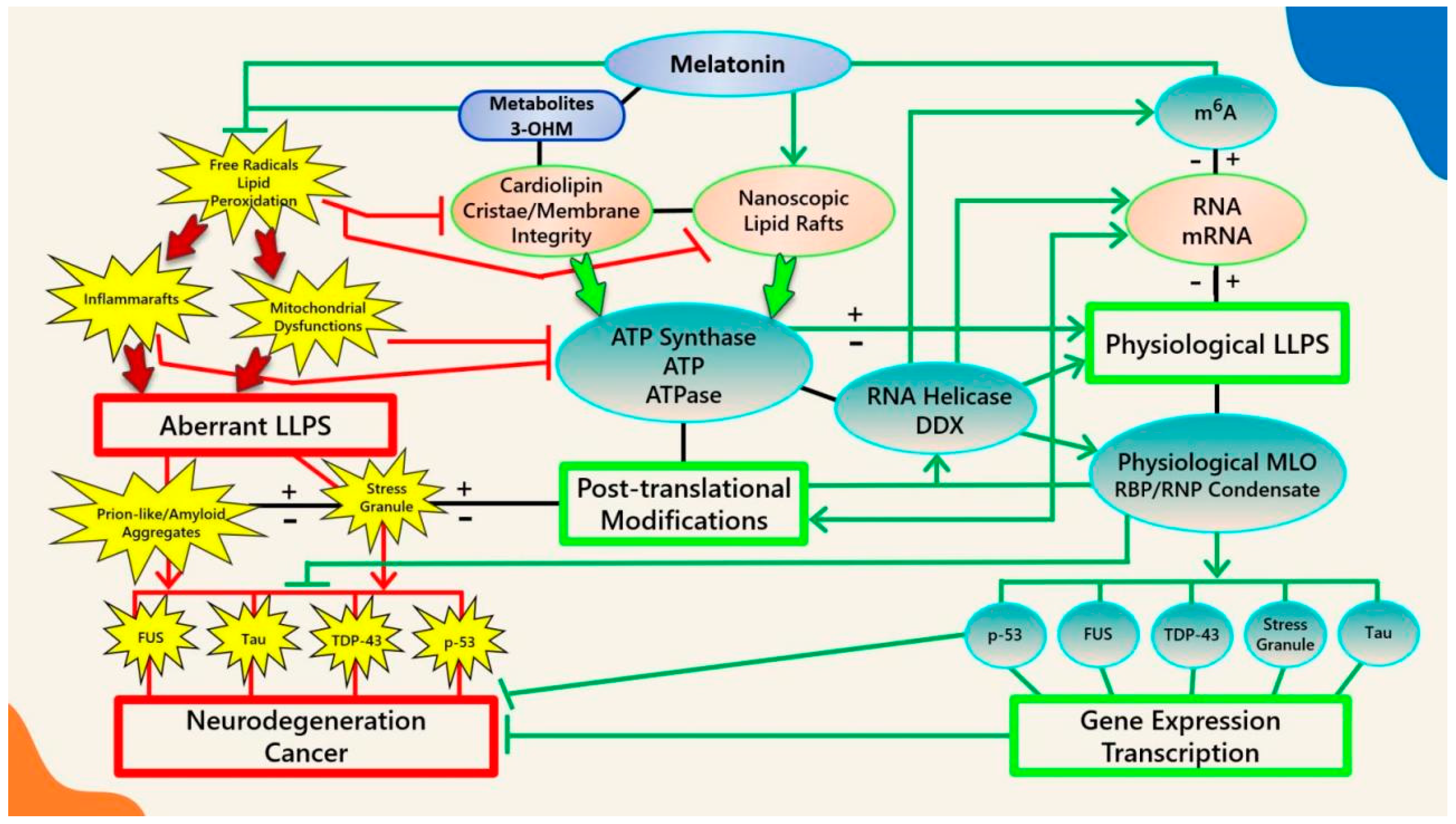
2. ATP Regulates Biomolecular Condensates
3. The Interdependence between Membranes and Membraneless Organelles
3.1. Lipid Rafts and Biomolecular Condensates in Health and Disease
3.2. Non-Mitochondrial Dimerized ATP Synthase and ATPase Are Localized in High-Curvature Lipid Rafts/Caveolae
3.2.1. Dimerized ATP Synthase/ATPase Require High-Curvature Lipid Domains
3.2.2. Translocation of ATP Dimers to Lipid Rafts Are Cellular Responses to Stress and Stimuli
3.3. Physiological Nanoscopic Lipid Raft Domains Are Stabilized by Intrinsic Negative Membrane Curvature and Reduced Line Tension
3.4. Oxidative Stress Alters Lipid Molecular Structures in Rafts and Membranes, Resulting in the Accumulation of Pathological MLOs
3.5. ROS-Externalized Cardiolipin Facilitates the Accumulation of Amyloid/Prionoid Aggregates and Activates Autophagic and Inflammatory Signaling
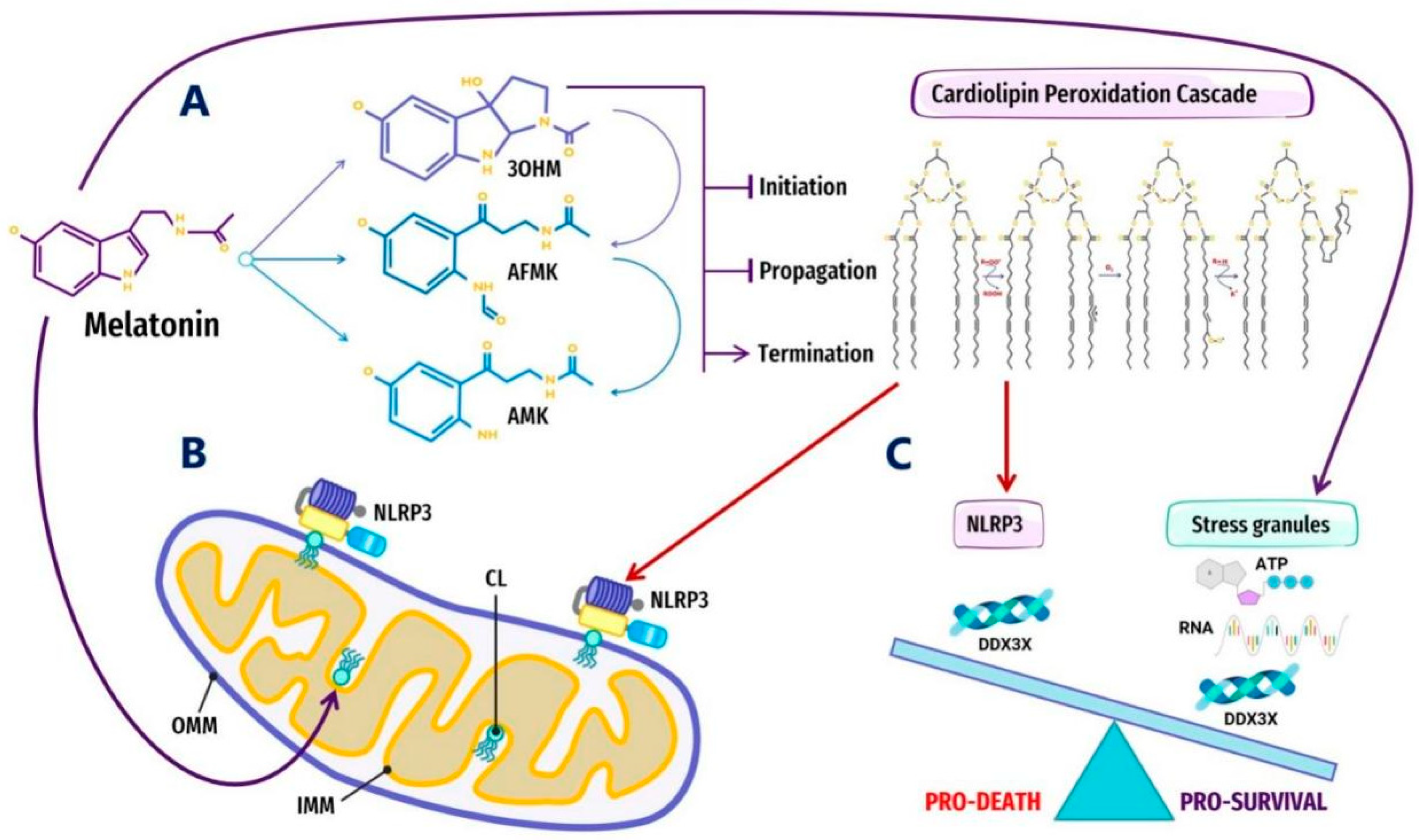
3.6. Melatonin Inhibits Cardiolipin Peroxidation to Prevent the Aggregation of Pathological MLOs at Membranes
3.7. Melatonin Regulates Membrane Lipid Dynamics and Composition via Phase Separation
3.8. Melatonin Increases Membrane Fluidity and Reduces Line Tension to Stabilize and Maintain Nanoscopic Lipid Raft Domains
3.9. Melatonin Maintains a High Cytosolic ATP:ADP Ratio through the Optimization of VDAC-CYB5R3 Redox Complexes in Lipid Rafts
4. Melatonin Is a Potent Ancient Antioxidant That Protects ATP Levels to Regulate the Formation and Dissolution of MLOs
4.1. Melatonin Metabolite 3-OHM Inhibits Lipid Peroxidation by Hydroperoxyl Radical
4.2. Melatonin Is Preferentially Located at Hydrophilic/Hydrophobic Membrane Interfaces
4.3. Melatonin Metabolite Free Radical Scavenging Cascades Rescue Cardiolipin from Hydroperoxyl Radicals (•OOH)
4.4. Melatonin May Regulate Glycolytic G Bodies by Increasing ATP
5. Melatonin May Attenuate the Stress-Induced Aggregation of Pathological MLOs via Post-Translational Modification and RNA Modification in an ATP-Dependent Manner
5.1. Cellular Stress and Mutations Drive Dysregulated LLPS to Form Pathological Aggregates in Neurodegenerative Disorders
5.2. Melatonin Inhibits/Disaggregates Pathological Tau Neurofibrillary Tangles and May Regulate the Phosphorylation of Tau in Neurodegenerative Disorders
5.3. Melatonin May Ameliorate Pathological Tau Fibrillation by Protecting Lipid Composition in Membranes and Lipid Rafts
5.4. Melatonin Regulates p53 and Other Biomolecular Condensates through the ATP-Dependent Ubiquitin–Protease System in Neurodegenerative Disorders
5.4.1. Aberrant Phase Separation/Droplet Formation May Cause Pathological Prion-like Aggregation and Inactivation of p53 in Neurodegenerative Disorders
5.4.2. The Potential Regulation of Ubiquitination/SUMOylation in MLO Assembly and Dissolution by Melatonin in an ATP-Dependent Manner
5.5. Post-Transcriptional Modifications of RNA by m6A Regulate Phase-Separated MLOs
RNA Regulation by N6-Methyladenosine (m6A) in Neurodegenerative Disorders
5.6. Potential Regulation of RNA and RNA m6A Modifications by Melatonin
5.7. The Ancient Relationships between Melatonin, ATP, RNA, and Membraneless Organelles
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-OHM | 3-hydroxymelatonin |
| Aβ | β-amyloid peptide |
| AD | Alzheimer’s disease |
| ADP | adenosine diphosphate |
| AICD | amyloid precursor protein intracellular domain |
| ALS | amyotrophic lateral sclerosis |
| ANT | adenine nucleotide translocator |
| APP | amyloid precursor protein |
| ASC | apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain |
| ATP | adenosine triphosphate |
| CL | cardiolipin |
| CYB5R3 | NADH-cytochrome b5 reductase 3 |
| Cyt | c cytochrome c |
| DDX3(X) | DEAD-box RNA helicase |
| DNA | deoxyribonucleic acid |
| eIF2a | eukaryotic translation initiation factor 2 alpha |
| ER | endoplasmic reticulum |
| FTO | frontotemporal dementia |
| FUS | fused in sarcoma |
| H+ | proton |
| H2O2 | hydrogen peroxide |
| IDP | intrinsically disordered protein |
| IDR | intrinsically disordered region |
| IMM | inner mitochondrial membrane |
| Lo | liquid-ordered |
| Lc | circular liquid-condensed |
| Ld | liquid-disordered |
| LLPS | liquid–liquid phase separation |
| m6A | N6-methyladenosine |
| mM | millimolar |
| μM | micromolar |
| MDM2 | mouse double minute 2 homolog |
| MLO | membraneless organelle |
| MAM | mitochondria-associated membrane |
| MOM | mitochondrial outer membrane |
| mPTP | mitochondrial permeability transition pore |
| mRNA | messenger RNA |
| MT | microtubule |
| NE | nuclear envelope |
| NFT | neurofibrillary tangles |
| NLRP3 | NLR pyrin domain containing 3 (inflammasome) |
| nM | nanomolar |
| NPC | nuclear pore complex |
| O2•− | superoxide radical |
| •OH | hydroxyl radical |
| •OOH | hydroperoxyl radical |
| OXPHOS | oxidative phosphorylation |
| PD | Parkinson’s disorder |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase kinase |
| Pi | inorganic phosphate |
| PLD | prion-like domain |
| PML | promyelocytic leukemia proteins |
| PTM | post-translational modification |
| RNA | ribonucleic acid |
| RBP | RNA-binding protein |
| RNP | ribonucleoprotein |
| ROS | reactive oxygen species |
| RP | ribosomal protein |
| SG | stress granule |
| SUMO | small ubiquitin-like modifier |
| TCA | tricarboxylic acid (cycle) |
| TDP-43 | TAR DNA-binding protein 43 |
| Ub | ubiquitin |
| UCP1 | uncoupling protein 1 |
| UPS | ubiquitin-protease system |
| VDAC | voltage-dependent anion channel |
| ZFP217 | zinc finger protein 217 |
References
- Feng, Z.; Chen, X.; Wu, X.; Zhang, M. Formation of Biological Condensates via Phase Separation: Characteristics, Analytical Methods, and Physiological Implications. J. Biol. Chem. 2019, 294, 14823–14835. [Google Scholar] [CrossRef]
- Oparin, A.I.; Synge, A. The Origin of Life on the Earth/Translated from the Russian by Ann Synge; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1957. [Google Scholar] [CrossRef]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-Triggered Phase Separation is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase Transitions in the Assembly of Multivalent Signalling Proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef]
- Su, X.; Ditlev, J.A.; Hui, E.; Xing, W.; Banjade, S.; Okrut, J.; King, D.S.; Taunton, J.; Rosen, M.K.; Vale, R.D. Phase Separation of Signaling Molecules Promotes T Cell Receptor Signal Transduction. Science 2016, 352, 595–599. [Google Scholar] [CrossRef]
- Laflamme, G.; Mekhail, K. Biomolecular Condensates as Arbiters of Biochemical Reactions inside the Nucleus. Commun. Biol. 2020, 3, 773. [Google Scholar] [CrossRef] [PubMed]
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. J. Mol. Biol. 2018, 430, 4666–4684. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Dormann, D. Liquid-Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192. [Google Scholar] [CrossRef]
- Zbinden, A.; Pérez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef]
- Taniue, K.; Akimitsu, N. Aberrant Phase Separation and Cancer. FEBS J. 2021. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Ahlers, J.; Adams, E.M.; Bader, V.; Pezzotti, S.; Winklhofer, K.F.; Tatzelt, J.; Havenith, M. The Key Role of Solvent in Condensation: Mapping Water in Liquid-Liquid Phase-Separated FUS. Biophys. J. 2021, 120, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Lawrence Zipursky, S.; Matsudaira, P.; Baltimore, D.; Darnell, J. Biochemical Energetics; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Manchester, K.L. Free Energy ATP Hydrolysis and Phosphorylation Potential. Biochem. Educ. 1980, 8, 70–72. [Google Scholar] [CrossRef]
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-Dependent Steps in the Binding of Ubiquitin Conjugates to the 26S Proteasome That Commit to Degradation. Mol. Cell 2010, 40, 671–681. [Google Scholar] [CrossRef]
- Callis, J. The Ubiquitination Machinery of the Ubiquitin System. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef]
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A Manually Curated Network of the PML Nuclear Body Interactome Reveals an Important Role for PML-NBs in SUMOylation Dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread Occurrence of the Droplet State of Proteins in the Human Proteome. Proc. Natl. Acad. Sci. USA 2020, 117, 33254–33262. [Google Scholar] [CrossRef]
- Hondele, M.; Heinrich, S.; De Los Rios, P.; Weis, K. Membraneless Organelles: Phasing out of Equilibrium. Emerg. Top Life Sci. 2020, 4, 331–342. [Google Scholar] [CrossRef]
- Garcia-Jove Navarro, M.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA Is a Critical Element for the Sizing and the Composition of Phase-Separated RNA-Protein Condensates. Nat. Commun. 2019, 10, 3230. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-Binding Proteins: Modular Design for Efficient Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Niaki, A.G.; Sarkar, J.; Cai, X.; Rhine, K.; Vidaurre, V.; Guy, B.; Hurst, M.; Lee, J.C.; Koh, H.R.; Guo, L.; et al. Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol. Cell 2020, 77, 82–94.e4. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid-Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Hatzopoulos, M.H.; Eastoe, J.; Dowding, P.J.; Rogers, S.E.; Heenan, R.; Dyer, R. Are Hydrotropes Distinct from Surfactants? Langmuir 2011, 27, 12346–12353. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active Liquid-like Behavior of Nucleoli Determines Their Size and Shape in Xenopus Laevis Oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef]
- Pal, S.; Paul, S. ATP Controls the Aggregation of Aβ16-22 Peptides. J. Phys. Chem. B 2020, 124, 210–223. [Google Scholar] [CrossRef]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP Alternations in an Alzheimer’s Disease Transgenic Mouse Model. J. Alzheimers. Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef]
- Salis, A.; Ninham, B.W. Models and Mechanisms of Hofmeister Effects in Electrolyte Solutions, and Colloid and Protein Systems Revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef] [PubMed]
- Mandl, I.; Grauer, A.; Neuberg, C. Solubilization of Insoluble Matter in Nature; I. The Part Played by Salts of Adenosinetriphosphate. Biochim. Biophys. Acta 1952, 8, 654–663. [Google Scholar] [CrossRef]
- Mehringer, J.; Do, T.-M.; Touraud, D.; Hohenschutz, M.; Khoshsima, A.; Horinek, D.; Kunz, W. Hofmeister versus Neuberg: Is ATP Really a Biological Hydrotrope? Cell Rep. Phys. Science 2021, 2, 100343. [Google Scholar] [CrossRef]
- Schwenke, W.D.; Soboll, S.; Seitz, H.J.; Sies, H. Mitochondrial and Cytosolic ATP/ADP Ratios in Rat Liver in Vivo. Biochem. J 1981, 200, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP Levels inside Single Living Cells with Fluorescence Resonance Energy Transfer-Based Genetically Encoded Indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef]
- Fang, D.; Maldonado, E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018, 138, 41–69. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e15. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.J. Adenine Nucleotide Translocase, Mitochondrial Stress, and Degenerative Cell Death. Oxid. Med. Cell. Longev. 2013, 2013, 146860. [Google Scholar] [CrossRef] [PubMed]
- Depaoli, M.R.; Karsten, F.; Madreiter-Sokolowski, C.T.; Klec, C.; Gottschalk, B.; Bischof, H.; Eroglu, E.; Waldeck-Weiermair, M.; Simmen, T.; Graier, W.F.; et al. Real-Time Imaging of Mitochondrial ATP Dynamics Reveals the Metabolic Setting of Single Cells. Cell Rep. 2018, 25, 501–512.e3. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Adenosine Triphosphate Energy-Independently Controls Protein Homeostasis with Unique Structure and Diverse Mechanisms. Protein Sci. 2021, 30, 1277–1293. [Google Scholar] [CrossRef] [PubMed]
- Takaine, M.; Imamura, H.; Yoshida, S. High and Stable ATP Levels Prevent Aberrant Intracellular Protein Aggregation. bioRxiv 2021, 2021, 801738. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS Assembles into Stress Granules and Is a Prosurvival Factor during Hyperosmolar Stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic Stress Granules: Dynamic Modulators of Cell Signaling and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Hilliker, A. Analysis of RNA Helicases in P-Bodies and Stress Granules. Methods Enzymol. 2012, 511, 323–346. [Google Scholar] [CrossRef]
- Sathyanarayanan, U.; Musa, M.; Bou Dib, P.; Raimundo, N.; Milosevic, I.; Krisko, A. ATP Hydrolysis by Yeast Hsp104 Determines Protein Aggregate Dissolution and Size in Vivo. Nat. Commun. 2020, 11, 5226. [Google Scholar] [CrossRef]
- Kang, J.; Lim, L.; Song, J. ATP Enhances at Low Concentrations but Dissolves at High Concentrations Liquid-Liquid Phase Separation (LLPS) of ALS/FTD-Causing FUS. Biochem. Biophys. Res. Commun. 2018, 504, 545–551. [Google Scholar] [CrossRef]
- Kang, J.; Lim, L.; Song, J. ATP Binds and Inhibits the Neurodegeneration-Associated Fibrillization of the FUS RRM Domain. Commun. Biol. 2019, 2, 223. [Google Scholar] [CrossRef]
- Dang, M.; Lim, L.; Kang, J.; Song, J. ATP Biphasically Modulates LLPS of TDP-43 PLD by Specifically Binding Arginine Residues. Commun. Biol. 2021, 4, 714. [Google Scholar] [CrossRef]
- Heo, C.E.; Han, J.Y.; Lim, S.; Lee, J.; Im, D.; Lee, M.J.; Kim, Y.K.; Kim, H.I. ATP Kinetically Modulates Pathogenic Tau Fibrillations. ACS Chem. Neurosci. 2020, 11, 3144–3152. [Google Scholar] [CrossRef]
- Farid, M.; Corbo, C.P.; Alonso, A.D.C. Tau Binds ATP and Induces Its Aggregation. Microsc. Res. Tech. 2014, 77, 133–137. [Google Scholar] [CrossRef]
- Newby, G.A.; Lindquist, S. Blessings in Disguise: Biological Benefits of Prion-like Mechanisms. Trends Cell Biol. 2013, 23, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; McGinnis, J.P.; Si, K. Translational Control by Prion-like Proteins. Trends Cell Biol. 2018, 28, 494–505. [Google Scholar] [CrossRef]
- Schuster, B.S.; Dignon, G.L.; Tang, W.S.; Kelley, F.M.; Ranganath, A.K.; Jahnke, C.N.; Simpkins, A.G.; Regy, R.M.; Hammer, D.A.; Good, M.C.; et al. Identifying Sequence Perturbations to an Intrinsically Disordered Protein That Determine Its Phase-Separation Behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 11421–11431. [Google Scholar] [CrossRef] [PubMed]
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically Disordered Linkers Determine the Interplay between Phase Separation and Gelation in Multivalent Proteins. Elife 2017, 6, e30294. [Google Scholar] [CrossRef] [PubMed]
- Owen, I.; Shewmaker, F. The Role of Post-Translational Modifications in the Phase Transitions of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Küffner, A.M.; Linsenmeier, M.; Grigolato, F.; Prodan, M.; Zuccarini, R.; Capasso Palmiero, U.; Faltova, L.; Arosio, P. Sequestration within Biomolecular Condensates Inhibits Aβ-42 Amyloid Formation. Chem. Sci. 2021, 12, 4373–4382. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, G.; Kedersha, N. Relationship of GW/P-Bodies with Stress Granules. Adv. Exp. Med. Biol. 2013, 768, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress Granules and Processing Bodies Are Dynamically Linked Sites of mRNP Remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef]
- Nostramo, R.; Xing, S.; Zhang, B.; Herman, P.K. Insights into the Role of P-Bodies and Stress Granules in Protein Quality Control. Genetics 2019, 213, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, D.; Sheth, U.; Valencia-Sanchez, M.A.; Brengues, M.; Parker, R. Processing Bodies Require RNA for Assembly and Contain Nontranslating mRNAs. RNA 2005, 11, 371–382. [Google Scholar] [CrossRef]
- Loll-Krippleber, R.; Brown, G.W. P-Body Proteins Regulate Transcriptional Rewiring to Promote DNA Replication Stress Resistance. Nat. Commun. 2017, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Mugler, C.F.; Hondele, M.; Heinrich, S.; Sachdev, R.; Vallotton, P.; Koek, A.Y.; Chan, L.Y.; Weis, K. ATPase Activity of the DEAD-Box Protein Dhh1 Controls Processing Body Formation. Elife 2016, 5, e18746. [Google Scholar] [CrossRef]
- Wang, C.; Schmich, F.; Srivatsa, S.; Weidner, J.; Beerenwinkel, N.; Spang, A. Context-Dependent Deposition and Regulation of mRNAs in P-Bodies. Elife 2018, 7, e29815. [Google Scholar] [CrossRef]
- Hermesh, O.; Jansen, R.-P. Take the (RN)A-Train: Localization of mRNA to the Endoplasmic Reticulum. Biochim. Biophys. Acta 2013, 1833, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.A.; Palazzo, A.F. Localization of mRNAs to the Endoplasmic Reticulum. Wiley Interdiscip. Rev. RNA 2014, 5, 481–492. [Google Scholar] [CrossRef]
- Reid, D.W.; Nicchitta, C.V. Primary Role for Endoplasmic Reticulum-Bound Ribosomes in Cellular Translation Identified by Ribosome Profiling. J. Biol. Chem. 2012, 287, 5518–5527. [Google Scholar] [CrossRef]
- Majewski, J.; Jones, E.M.; Vander Zanden, C.M.; Biernat, J.; Mandelkow, E.; Chi, E.Y. Lipid Membrane Templated Misfolding and Self-Assembly of Intrinsically Disordered Tau Protein. Sci. Rep. 2020, 10, 13324. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tuominen, E.K.J.; Kinnunen, P.K.J. Formation of Amyloid Fibers Triggered by Phosphatidylserine-Containing Membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar] [CrossRef]
- Goñi, F.; Martá-Ariza, M.; Herline, K.; Peyser, D.; Boutajangout, A.; Mehta, P.; Drummond, E.; Prelli, F.; Wisniewski, T. Anti-β-Sheet Conformation Monoclonal Antibody Reduces Tau and Aβ Oligomer Pathology in an Alzheimer’s Disease Model. Alzheimers. Res. Ther. 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Dregni, A.J.; Mandala, V.S.; Wu, H.; Elkins, M.R.; Wang, H.K.; Hung, I.; DeGrado, W.F.; Hong, M. In Vitro 0N4R Tau Fibrils Contain a Monomorphic β-Sheet Core Enclosed by Dynamically Heterogeneous Fuzzy Coat Segments. Proc. Natl. Acad. Sci. USA 2019, 116, 16357–16366. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; Özkan, N.; Farías, G.G. Complex Interactions between Membrane-Bound Organelles, Biomolecular Condensates and the Cytoskeleton. Front. Cell Dev. Biol. 2020, 8, 618733. [Google Scholar] [CrossRef] [PubMed]
- Snead, W.T.; Gladfelter, A.S. The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell 2019, 76, 295–305. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Compartmentalization of Cells; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Mizuuchi, R.; Ichihashi, N. Primitive Compartmentalization for the Sustainable Replication of Genetic Molecules. Life 2021, 11, 191. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Pucadyil, T.J. Chapter 2—Dynamic Remodeling of Membranes Catalyzed by Dynamin. In Current Topics in Membranes; Chernomordik, L.V., Kozlov, M.M., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 68. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming Together to Define Membrane Contact Sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Sallese, M.; Pulvirenti, T.; Luini, A. The Physiology of Membrane Transport and Endomembrane-Based Signalling. EMBO J. 2006, 25, 2663–2673. [Google Scholar] [CrossRef]
- Jaqaman, K.; Ditlev, J.A. Biomolecular Condensates in Membrane Receptor Signaling. Curr. Opin. Cell Biol. 2021, 69, 48–54. [Google Scholar] [CrossRef]
- Case, L.B.; Zhang, X.; Ditlev, J.A.; Rosen, M.K. Stoichiometry Controls Activity of Phase-Separated Clusters of Actin Signaling Proteins. Science 2019, 363, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.C.; Alvarez, S.; Kondo, Y.; Lee, Y.K.; Chung, J.K.; Lam, H.Y.M.; Biswas, K.H.; Kuriyan, J.; Groves, J.T. A Molecular Assembly Phase Transition and Kinetic Proofreading Modulate Ras Activation by SOS. Science 2019, 363, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the Cell Membrane Interface in Modulating Production and Uptake of Alzheimer’s Beta Amyloid Protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic Processing of the Alzheimer Beta-Amyloid Precursor Protein Depends on Lipid Rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low Cholesterol Stimulates the Nonamyloidogenic Pathway by Its Effect on the Alpha -Secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bagnat, M.; Keränen, S.; Shevchenko, A.; Shevchenko, A.; Simons, K. Lipid Rafts Function in Biosynthetic Delivery of Proteins to the Cell Surface in Yeast. Proc. Natl. Acad. Sci. USA 2000, 97, 3254–3259. [Google Scholar] [CrossRef]
- Ikonen, E. Roles of Lipid Rafts in Membrane Transport. Curr. Opin. Cell Biol. 2001, 13, 470–477. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. Lipid Rafts in Health and Disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.-L. Lipid Rafts: Keys to Neurodegeneration. Brain Res. Bull. 2010, 82, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Maguy, A.; Hebert, T.E.; Nattel, S. Involvement of Lipid Rafts and Caveolae in Cardiac Ion Channel Function. Cardiovasc. Res. 2006, 69, 798–807. [Google Scholar] [CrossRef]
- Vey, M.; Pilkuhn, S.; Wille, H.; Nixon, R.; DeArmond, S.J.; Smart, E.J.; Anderson, R.G.; Taraboulos, A.; Prusiner, S.B. Subcellular Colocalization of the Cellular and Scrapie Prion Proteins in Caveolae-like Membranous Domains. Proc. Natl. Acad. Sci. USA 1996, 93, 14945–14949. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The Prion Protein and Lipid Rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Jury, E.C.; Kabouridis, P.S.; Flores-Borja, F.; Mageed, R.A.; Isenberg, D.A. Altered Lipid Raft-Associated Signaling and Ganglioside Expression in T Lymphocytes from Patients with Systemic Lupus Erythematosus. J. Clin. Investig. 2004, 113, 1176–1187. [Google Scholar] [CrossRef]
- Chazal, N.; Gerlier, D. Virus Entry, Assembly, Budding, and Membrane Rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237. [Google Scholar] [CrossRef]
- Murai, T. The Role of Lipid Rafts in Cancer Cell Adhesion and Migration. Int. J. Cell Biol. 2012, 2012, 763283. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid Rafts as Signaling Hubs in Cancer Cell Survival/death and Invasion: Implications in Tumor Progression and Therapy: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef]
- Greenlee, J.D.; Subramanian, T.; Liu, K.; King, M.R. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2021, 81, 5–17. [Google Scholar] [CrossRef]
- Codini, M.; Garcia-Gil, M.; Albi, E. Cholesterol and Sphingolipid Enriched Lipid Rafts as Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2021, 22, 726. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.-T.; Chang, Y.-C.; Shimobayashi, S.F.; Shin, Y.; Strom, A.R.; Brangwynne, C.P. Nucleated Transcriptional Condensates Amplify Gene Expression. Nat. Cell Biol. 2020, 22, 1187–1196. [Google Scholar] [CrossRef]
- Shrinivas, K.; Sabari, B.R.; Coffey, E.L.; Klein, I.A.; Boija, A.; Zamudio, A.V.; Schuijers, J.; Hannett, N.M.; Sharp, P.A.; Young, R.A.; et al. Enhancer Features That Drive Formation of Transcriptional Condensates. Mol. Cell 2019, 75, 549–561.e7. [Google Scholar] [CrossRef] [PubMed]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Fagman, J.B.; Chen, C.; Alberti, S.; Liu, B. Protein Phase Separation and Its Role in Tumorigenesis. Elife 2020, 9, e60264. [Google Scholar] [CrossRef]
- Vernon, R.M.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi Contacts Are an Overlooked Protein Feature Relevant to Phase Separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef]
- Ditlev, J.A. Membrane-Associated Phase Separation: Organization and Function Emerge from a Two-Dimensional Milieu. J. Mol. Cell Biol. 2021, 13, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabouille, C. Membrane-Bound Meet Membraneless in Health and Disease. Cells 2019, 8, 1000. [Google Scholar] [CrossRef]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-Ras, K-Ras, and Inner Plasma Membrane Raft Proteins Operate in Nanoclusters with Differential Dependence on the Actin Cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [PubMed]
- Banjade, S.; Rosen, M.K. Phase Transitions of Multivalent Proteins Can Promote Clustering of Membrane Receptors. Elife 2014, 3, e04123. [Google Scholar] [CrossRef]
- Lee, I.-H.; Imanaka, M.Y.; Modahl, E.H.; Torres-Ocampo, A.P. Lipid Raft Phase Modulation by Membrane-Anchored Proteins with Inherent Phase Separation Properties. ACS Omega. 2019, 4, 6551–6559. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.K.; Huang, W.Y.C.; Carbone, C.B.; Nocka, L.M.; Parikh, A.N.; Vale, R.D.; Groves, J.T. Coupled Membrane Lipid Miscibility and Phosphotyrosine-Driven Protein Condensation Phase Transitions. Biophys. J. 2021, 120, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef]
- Pullman, M.E.; Penefsky, H.S.; Datta, A.; Racker, E. Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation. I. Purification and Properties of Soluble Dinitrophenol-Stimulated Adenosine Triphosphatase. J. Biol. Chem. 1960, 235, 3322–3329. [Google Scholar] [CrossRef]
- Ernster, L.; Schatz, G. Mitochondria: A Historical Review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A Resolution of F1-ATPase from Bovine Heart Mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Grüber, G.; Wieczorek, H.; Harvey, W.R.; Müller, V. Structure–function Relationships of A-, F- and V-ATPases. J. Exp. Biol. 2001, 204, 2597–2605. [Google Scholar] [CrossRef]
- Mitchell, P. Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. Biol. Rev. Camb. Philos. Soc. 1966, 41, 445–502. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP Synthase--a Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biology, Structure and Mechanism of P-Type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Beyenbach, K.W.; Wieczorek, H. The V-Type H+ ATPase: Molecular Structure and Function, Physiological Roles and Regulation. J. Exp. Biol. 2006, 209 Pt 4, 577–589. [Google Scholar] [CrossRef]
- Gaudet, R.; Wiley, D.C. Structure of the ABC ATPase Domain of Human TAP1, the Transporter Associated with Antigen Processing. EMBO J. 2001, 20, 4964–4972. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Oiwa, K.; Nishizaka, T.; Furuike, S.; Noji, H.; Itoh, H.; Yoshida, M.; Kinosita Jr, K. Coupling of Rotation and Catalysis in F(1)-ATPase Revealed by Single-Molecule Imaging and Manipulation. Cell 2007, 130, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Okuno, D.; Iino, R.; Noji, H. Rotation and Structure of FoF1-ATP Synthase. J. Biochem. 2011, 149, 655–664. [Google Scholar] [CrossRef]
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef]
- Das, B.; Mondragon, M.O.; Sadeghian, M.; Hatcher, V.B.; Norin, A.J. A Novel Ligand in Lymphocyte-Mediated Cytotoxicity: Expression of the Beta Subunit of H+ Transporting ATP Synthase on the Surface of Tumor Cell Lines. J. Exp. Med. 1994, 180, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wang, X.; Li, Y.; Cao, Y.; Chen, X. Extracellular ATP a New Player in Cancer Metabolism: NSCLC Cells Internalize ATP In Vitro and In Vivo Using Multiple Endocytic Mechanisms. Mol. Cancer Res. 2016, 14, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.L.; Kenan, D.J.; Ashley, T.A.; Roy, J.A.; Goodman, M.D.; Misra, U.K.; Cheek, D.J.; Pizzo, S.V. Endothelial Cell Surface F1-F0 ATP Synthase Is Active in ATP Synthesis and Is Inhibited by Angiostatin. Proc. Natl. Acad. Sci. USA 2001, 98, 6656–6661. [Google Scholar] [CrossRef]
- Arakaki, N.; Nagao, T.; Niki, R.; Toyofuku, A.; Tanaka, H.; Kuramoto, Y.; Emoto, Y.; Shibata, H.; Magota, K.; Higuti, T. Possible Role of Cell Surface H+ -ATP Synthase in the Extracellular ATP Synthesis and Proliferation of Human Umbilical Vein Endothelial Cells. Mol. Cancer Res. 2003, 1, 931–939. [Google Scholar] [PubMed]
- Bae, T.-J.; Kim, M.-S.; Kim, J.-W.; Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, K.-B.; Lee, C.S.; Kim, J.-H.; Chang, S.Y.; et al. Lipid Raft Proteome Reveals ATP Synthase Complex in the Cell Surface. Proteomics 2004, 4, 3536–3548. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Kamanna, V.S.; Zhang, M.C.; Kashyap, M.L. Niacin Inhibits Surface Expression of ATP Synthase Beta Chain in HepG2 Cells: Implications for Raising HDL. J. Lipid Res. 2008, 49, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.O.; Jacquet, S.; Esteve, J.-P.; Rolland, C.; Cabezón, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Tercé, F.; et al. Ectopic Beta-Chain of ATP Synthase Is an Apolipoprotein A-I Receptor in Hepatic HDL Endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Burrell, H.E.; Wlodarski, B.; Foster, B.J.; Buckley, K.A.; Sharpe, G.R.; Quayle, J.M.; Simpson, A.W.M.; Gallagher, J.A. Human Keratinocytes Release ATP and Utilize Three Mechanisms for Nucleotide Interconversion at the Cell Surface. J. Biol. Chem. 2005, 280, 29667–29676. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Wattez, A.; Galván-valencia, M.; Ghestem, A.; David, J.-P.; Lemoine, J.; Sautiére, P.-E.; Dachary, J.; Mazat, J.-P.; Michalski, J.-C.; et al. Association of ATP Synthase Alpha-Chain with Neurofibrillary Degeneration in Alzheimer’s Disease. Neuroscience 2003, 117, 293–303. [Google Scholar] [CrossRef]
- Chi, S.L.; Pizzo, S.V. Cell Surface F1Fo ATP Synthase: A New Paradigm? Ann. Med. 2006, 38, 429–438. [Google Scholar] [CrossRef]
- Abrams, A. Structure and Function of Membrane-Bound ATPase in Bacteria. In The Enzymes of Biological Membranes: Volume 3 Membrane Transport (FIRST EDITION); Martonosi, A., Ed.; Springer: Boston, MA, USA, 1976. [Google Scholar] [CrossRef]
- Guo, H.; Suzuki, T.; Rubinstein, J.L. Structure of a Bacterial ATP Synthase. Elife 2019, 8, e43128. [Google Scholar] [CrossRef]
- Krulwich, T.A.; Sachs, G.; Padan, E. Molecular Aspects of Bacterial pH Sensing and Homeostasis. Nat. Rev. Microbiol. 2011, 9, 330–343. [Google Scholar] [CrossRef]
- Xing, S.-L.; Yan, J.; Yu, Z.-H.; Zhu, C.-Q. Neuronal Cell Surface ATP Synthase Mediates Synthesis of Extracellular ATP and Regulation of Intracellular pH. Cell Biol. Int. 2011, 35, 81–86. [Google Scholar] [CrossRef]
- Murakami, T.; Shibuya, I.; Ise, T.; Chen, Z.S.; Akiyama, S.; Nakagawa, M.; Izumi, H.; Nakamura, T.; Matsuo, K.; Yamada, Y.; et al. Elevated Expression of Vacuolar Proton Pump Genes and Cellular PH in Cisplatin Resistance. Int. J. Cancer 2001, 93, 869–874. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Ruff, K.M.; Roberts, S.; Chilkoti, A.; Pappu, R.V. Advances in Understanding Stimulus-Responsive Phase Behavior of Intrinsically Disordered Protein Polymers. J. Mol. Biol. 2018, 430, 4619–4635. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Chidlow Jr, J.H.; Sessa, W.C. Caveolae, Caveolins, and Cavins: Complex Control of Cellular Signalling and Inflammation. Cardiovasc. Res. 2010, 86, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Mangiullo, R.; Gnoni, A.; Leone, A.; Gnoni, G.V.; Papa, S.; Zanotti, F. Structural and Functional Characterization of FoF1-ATP Synthase on the Extracellular Surface of Rat Hepatocytes. BBA—Bioenerg. 2008, 1777, 1326–1335. [Google Scholar] [CrossRef]
- Kim, B.-W.; Choo, H.-J.; Lee, J.-W.; Kim, J.-H.; Ko, Y.-G. Extracellular ATP Is Generated by ATP Synthase Complex in Adipocyte Lipid Rafts. Exp. Mol. Med. 2004, 36, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, M.R.; Berrocal-Carrillo, M.; Gasset, M.; Mata, A.M. The Plasma Membrane Ca2+-ATPase Isoform 4 Is Localized in Lipid Rafts of Cerebellum Synaptic Plasma Membranes. J. Biol. Chem. 2006, 281, 447–453. [Google Scholar] [CrossRef]
- Noble, K.; Zhang, J.; Wray, S. Lipid Rafts, the Sarcoplasmic Reticulum and Uterine Calcium Signalling: An Integrated Approach. J. Physiol. 2006, 570 Pt 1, 29–35. [Google Scholar] [CrossRef]
- Nagata, J.; Guerra, M.T.; Shugrue, C.A.; Gomes, D.A.; Nagata, N.; Nathanson, M.H. Lipid Rafts Establish Calcium Waves in Hepatocytes. Gastroenterology 2007, 133, 256–267. [Google Scholar] [CrossRef]
- Krishnan, G.; Chatterjee, N. Detergent Resistant Membrane Fractions Are Involved in Calcium Signaling in Müller Glial Cells of Retina. Int. J. Biochem. Cell Biol. 2013, 45, 1758–1766. [Google Scholar] [CrossRef]
- Tsutsumi, Y.M.; Horikawa, Y.T.; Jennings, M.M.; Kidd, M.W.; Niesman, I.R.; Yokoyama, U.; Head, B.P.; Hagiwara, Y.; Ishikawa, Y.; Miyanohara, A.; et al. Cardiac-Specific Overexpression of Caveolin-3 Induces Endogenous Cardiac Protection by Mimicking Ischemic Preconditioning. Circulation 2008, 118, 1979–1988. [Google Scholar] [CrossRef]
- Shaul, P.W.; Anderson, R.G. Role of Plasmalemmal Caveolae in Signal Transduction. Am. J. Physiol. 1998, 275, L843–L851. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as Plasma Membrane Sensors, Protectors and Organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Razani, B.; Lisanti, M.P. Emerging Themes in Lipid Rafts and Caveolae. Cell 2001, 106, 403–411. [Google Scholar] [CrossRef]
- Chen, Z.; Rand, R.P. The Influence of Cholesterol on Phospholipid Membrane Curvature and Bending Elasticity. Biophys. J. 1997, 73, 267–276. [Google Scholar] [CrossRef]
- Wang, W.; Yang, L.; Huang, H.W. Evidence of Cholesterol Accumulated in High Curvature Regions: Implication to the Curvature Elastic Energy for Lipid Mixtures. Biophys. J. 2007, 92, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Rivel, T.; Ramseyer, C. The Influence of Curvature on the Properties of the Plasma Membrane. Insights from Atomistic Molecular Dynamics Simulations. Sci. Rep. 2017, 7, 16078. [Google Scholar] [CrossRef] [PubMed]
- Krishna, A.; Sengupta, D. Interplay between Membrane Curvature and Cholesterol: Role of Palmitoylated Caveolin-1. Biophys. J. 2019, 116, 69–78. [Google Scholar] [CrossRef]
- Smart, E.J.; Graf, G.A.; McNiven, M.A.; Sessa, W.C.; Engelman, J.A.; Scherer, P.E.; Okamoto, T.; Lisanti, M.P. Caveolins, Liquid-Ordered Domains, and Signal Transduction. Mol. Cell Biol. 1999, 19, 7289–7304. [Google Scholar] [CrossRef]
- Murata, M.; Peränen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. VIP21/caveolin Is a Cholesterol-Binding Protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef]
- Esparza-Perusquía, M.; Olvera-Sánchez, S.; Pardo, J.P.; Mendoza-Hernández, G.; Martínez, F.; Flores-Herrera, O. Structural and Kinetics Characterization of the F1F0-ATP Synthase Dimer. New Repercussion of Monomer-Monomer Contact. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 975–981. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Interface Mobility between Monomers in Dimeric Bovine ATP Synthase Participates in the Ultrastructure of Inner Mitochondrial Membranes. Proc. Natl. Acad. Sci. USA 2021, 118, e2021012118. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F1Fo-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [PubMed]
- Habersetzer, J.; Larrieu, I.; Priault, M.; Salin, B.; Rossignol, R.; Brèthes, D.; Paumard, P. Human F1F0 ATP Synthase, Mitochondrial Ultrastructure and OXPHOS Impairment: A (super-)complex Matter? PLoS ONE 2013, 8, e75429. [Google Scholar] [CrossRef]
- Mannella, C.A. The Relevance of Mitochondrial Membrane Topology to Mitochondrial Function. Biochim. Biophys. Acta 2006, 1762, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kühlbrandt, W. Dimers of Mitochondrial ATP Synthase Induce Membrane Curvature and Self-Assemble into Rows. Proc. Natl. Acad. Sci. USA 2019, 116, 4250–4255. [Google Scholar] [CrossRef]
- Allen-Worthington, K.; Xie, J.; Brown, J.L.; Edmunson, A.M.; Dowling, A.; Navratil, A.M.; Scavelli, K.; Yoon, H.; Kim, D.-G.; Bynoe, M.S.; et al. The F0F1 ATP Synthase Complex Localizes to Membrane Rafts in Gonadotrope Cells. Mol. Endocrinol. 2016, 30, 996–1011. [Google Scholar] [CrossRef][Green Version]
- Kim, B.-W.; Lee, J.-W.; Choo, H.-J.; Lee, C.S.; Jung, S.-Y.; Yi, J.-S.; Ham, Y.-M.; Lee, J.-H.; Hong, J.; Kang, M.-J.; et al. Mitochondrial Oxidative Phosphorylation System Is Recruited to Detergent-Resistant Lipid Rafts during Myogenesis. Proteomics 2010, 10, 2498–2515. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.H.; Peuchen, E.H.; Dovichi, N.J.; Weeks, D.L. Dual Roles for ATP in the Regulation of Phase Separated Protein Aggregates in Xenopus Oocyte Nucleoli. Elife 2018, 7, e35224. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Kurzawa, N.; Werner, T.; Günthner, I.; Helm, D.; Huber, W.; Bantscheff, M.; Savitski, M.M. Proteome-Wide Solubility and Thermal Stability Profiling Reveals Distinct Regulatory Roles for ATP. Nat. Commun. 2019, 10, 1155. [Google Scholar] [CrossRef]
- Ma, Z.; Cao, M.; Liu, Y.; He, Y.; Wang, Y.; Yang, C.; Wang, W.; Du, Y.; Zhou, M.; Gao, F. Mitochondrial F1Fo-ATP Synthase Translocates to Cell Surface in Hepatocytes and Has High Activity in Tumor-like Acidic and Hypoxic Environment. Acta Biochim. Biophys. Sin. 2010, 42, 530–537. [Google Scholar] [CrossRef]
- Villa-Pulgarín, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marbán, Á.; Müller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and Lipid Raft-Located FOF1-ATP Synthase as Major Therapeutic Targets in the Antileishmanial and Anticancer Activities of Ether Lipid Edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Jin, S.; Zhou, F.; Katirai, F.; Li, P.-L. Lipid Raft Redox Signaling: Molecular Mechanisms in Health and Disease. Antioxid. Redox Signal. 2011, 15, 1043–1083. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Insel, P.A. Lipid Rafts and Caveolae and Their Role in Compartmentation of Redox Signaling. Antioxid. Redox Signal. 2009, 11, 1357–1372. [Google Scholar] [CrossRef]
- Koneru, S.; Penumathsa, S.V.; Thirunavukkarasu, M.; Samuel, S.M.; Zhan, L.; Han, Z.; Maulik, G.; Das, D.K.; Maulik, N. Redox Regulation of Ischemic Preconditioning Is Mediated by the Differential Activation of Caveolins and Their Association with eNOS and GLUT-4. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2060–H2072. [Google Scholar] [CrossRef]
- Kuzmin, P.I.; Akimov, S.A.; Chizmadzhev, Y.A.; Zimmerberg, J.; Cohen, F.S. Line Tension and Interaction Energies of Membrane Rafts Calculated from Lipid Splay and Tilt. Biophys. J. 2005, 88, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, D.J.; Zak, T.J.; Robia, S.L. Cardiac Calcium ATPase Dimerization Measured by Cross-Linking and Fluorescence Energy Transfer. Biophys. J. 2016, 111, 1192–1202. [Google Scholar] [CrossRef]
- Simons, K.; Sampaio, J.L. Membrane Organization and Lipid Rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Kinnun, J.J.; Bolmatov, D.; Lavrentovich, M.O.; Katsaras, J. Lateral Heterogeneity and Domain Formation in Cellular Membranes. Chem. Phys. Lipids 2020, 232, 104976. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts Defined: A Report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Ouweneel, A.B.; Thomas, M.J.; Sorci-Thomas, M.G. The Ins and Outs of Lipid Rafts: Functions in Intracellular Cholesterol Homeostasis, Microparticles, and Cell Membranes: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 676–686. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Marquardt, D.; Dies, H.; Kučerka, N.; Yamani, Z.; Harroun, T.A.; Katsaras, J.; Shi, A.-C.; Rheinstädter, M.C. The Observation of Highly Ordered Domains in Membranes with Cholesterol. PLoS ONE 2013, 8, e66162. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.F.F.; Pokorny, A.; Hinderliter, A. Thermodynamics of Membrane Domains. Biochim. Biophys. Acta 2005, 1720, 1–13. [Google Scholar] [CrossRef]
- Igarashi, M.; Honda, A.; Kawasaki, A.; Nozumi, M. Neuronal Signaling Involved in Neuronal Polarization and Growth: Lipid Rafts and Phosphorylation. Front. Mol. Neurosci. 2020, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Bolmatov, D.; Soloviov, D.; Zhernenkov, M.; Zav’yalov, D.; Mamontov, E.; Suvorov, A.; Cai, Y.Q.; Katsaras, J. Molecular Picture of the Transient Nature of Lipid Rafts. Langmuir 2020, 36, 4887–4896. [Google Scholar] [CrossRef]
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid Rafts in Glial Cells: Role in Neuroinflammation and Pain Processing: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 655–666. [Google Scholar] [CrossRef]
- Sviridov, D.; Mukhamedova, N.; Miller, Y.I. Lipid Rafts as a Therapeutic Target: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 687–695. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.-K.; Kim, C.-W.; Kim, Y.-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated with Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am. J. Pathol. 2006, 168, 1107–1118; quiz 1404–1405. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid Rafts and Clusters of Apoptotic Signaling Molecule-Enriched Rafts in Cancer Therapy. Future Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.M.; Hooper, N.M.; Turner, A.J. The Involvement of Lipid Rafts in Alzheimer’s Disease. Mol. Membr. Biol. 2006, 23, 111–122. [Google Scholar] [CrossRef]
- Rushworth, J.V.; Hooper, N.M. Lipid Rafts: Linking Alzheimer’s Amyloid-β Production, Aggregation, and Toxicity at Neuronal Membranes. Int. J. Alzheimers. Dis. 2011, 2011, 603052. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid Rafts and Alzheimer’s Disease: Protein-Lipid Interactions and Perturbation of Signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Mesa-Herrera, F.; Taoro-González, L.; Valdés-Baizabal, C.; Diaz, M.; Marín, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. The Challenge of Lipid Rafts. J. Lipid Res. 2009, 50, S323–S328. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging Coexisting Fluid Domains in Biomembrane Models Coupling Curvature and Line Tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef]
- Meinhardt, S.; Vink, R.L.C.; Schmid, F. Monolayer Curvature Stabilizes Nanoscale Raft Domains in Mixed Lipid Bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 4476–4481. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Feigenson, G.W. Lowering Line Tension with High Cholesterol Content Induces a Transition from Macroscopic to Nanoscopic Phase Domains in Model Biomembranes. BBA—Biomembr. 2019, 1861, 478–485. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Z.; Wang, X.; Shyy, J.Y.-J.; Zhu, Y. Cholesterol Loading Increases the Translocation of ATP Synthase Beta Chain into Membrane Caveolae in Vascular Endothelial Cells. Biochim. Biophys. Acta 2006, 1761, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Leyva, J.A.; Bianchet, M.A.; Amzel, L.M. Understanding ATP Synthesis: Structure and Mechanism of the F1-ATPase (Review). Mol. Membr. Biol. 2003, 20, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the Heart of Mitochondrial Metabolism. Cell. Mol. Life Sci. 2008, 65, 2493–2506. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Vähäheikkilä, M.; Peltomaa, T.; Róg, T.; Vazdar, M.; Pöyry, S.; Vattulainen, I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane? Chem. Phys. Lipids 2018, 214, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sankhagowit, S.; Lee, E.Y.; Wong, G.C.L.; Malmstadt, N. Oxidation of Membrane Curvature-Regulating Phosphatidylethanolamine Lipid Results in Formation of Bilayer and Cubic Structures. Langmuir 2016, 32, 2450–2457. [Google Scholar] [CrossRef]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef]
- Garcia, A.; Pochinda, S.; Elgaard-Jørgensen, P.N.; Khandelia, H.; Clarke, R.J. Evidence for ATP Interaction with Phosphatidylcholine Bilayers. Langmuir 2019, 35, 9944–9953. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Coban, O.; Popov, J.; Burger, M.; Vobornik, D.; Johnston, L.J. Transition from Nanodomains to Microdomains Induced by Exposure of Lipid Monolayers to Air. Biophys. J. 2007, 92, 2842–2853. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.W.; Visser, N.V.; Kouptsova, O.; Visser, A.J. Oxidation of Unsaturated Phospholipids in Membrane Bilayer Mixtures Is Accompanied by Membrane Fluidity Changes. Biochim. Biophys. Acta 2000, 1487, 61–73. [Google Scholar] [CrossRef]
- Runas, K.A.; Malmstadt, N. Low Levels of Lipid Oxidation Radically Increase the Passive Permeability of Lipid Bilayers. Soft Matter. 2015, 11, 499–505. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae Membrane Dynamics—A Paradigm Change. Trends Cell Biol. 2020, 30, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Agrawal, A.; Ramachandran, R. Exploring the Links between Lipid Geometry and Mitochondrial Fission: Emerging Concepts. Mitochondrion 2019, 49, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Vetica, F.; Sansone, A.; Meliota, C.; Batani, G.; Roberti, M.; Chatgilialoglu, C.; Ferreri, C. Free-Radical-Mediated Formation of Trans-Cardiolipin Isomers, Analytical Approaches for Lipidomics and Consequences of the Structural Organization of Membranes. Biomolecules 2020, 10, 1189. [Google Scholar] [CrossRef]
- Elías-Wolff, F.; Lindén, M.; Lyubartsev, A.P.; Brandt, E.G. Curvature Sensing by Cardiolipin in Simulated Buckled Membranes. Soft Matter 2019, 15, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.; Ramanathan, A.; Lopez, C.F. Cardiolipin-Dependent Properties of Model Mitochondrial Membranes from Molecular Simulations. Biophys. J. 2019, 117, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Cardiolipin and Mitochondrial Cristae Organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef]
- Beltrán-Heredia, E.; Tsai, F.-C.; Salinas-Almaguer, S.; Cao, F.J.; Bassereau, P.; Monroy, F. Membrane Curvature Induces Cardiolipin Sorting. Commun Biol 2019, 2, 225. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin Membrane Domains in Prokaryotes and Eukaryotes. Biochim. Biophys. Acta 2009, 1788, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Huang, K.C.; Wingreen, N.S. Lipid Localization in Bacterial Cells through Curvature-Mediated Microphase Separation. Biophys. J. 2008, 95, 1034–1049. [Google Scholar] [CrossRef]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding Structure, Function, and Mutations in the Mitochondrial ATP Synthase. Microb. Cell Fact. 2015, 2, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Gao, J.; Siira, S.J.; Shearwood, A.-M.; Ermer, J.A.; Hofferek, V.; Mathews, J.C.; Zheng, M.; Reid, G.E.; Rackham, O.; et al. Cardiolipin is Required for Membrane Docking of Mitochondrial Ribosomes and Protein Synthesis. J. Cell Sci. 2020, 133, jcs240374. [Google Scholar] [CrossRef]
- Basu Ball, W.; Neff, J.K.; Gohil, V.M. The Role of Nonbilayer Phospholipids in Mitochondrial Structure and Function. FEBS Lett. 2018, 592, 1273–1290. [Google Scholar] [CrossRef]
- Eble, K.S.; Coleman, W.B.; Hantgan, R.R.; Cunningham, C.C. Tightly Associated Cardiolipin in the Bovine Heart Mitochondrial ATP Synthase as Analyzed by 31P Nuclear Magnetic Resonance Spectroscopy. J. Biol. Chem. 1990, 265, 19434–19440. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Yaguzhinsky, L.S.; Dagda, R.K. Non-Bilayer Structures in Mitochondrial Membranes Regulate ATP Synthase Activity. Biochim. Biophys. Acta Biomembr. 2018, 1860, 586–599. [Google Scholar] [CrossRef]
- Prola, A.; Blondelle, J.; Vandestienne, A.; Piquereau, J.; Denis, R.G.P.; Guyot, S.; Chauvin, H.; Mourier, A.; Maurer, M.; Henry, C.; et al. Cardiolipin Content Controls Mitochondrial Coupling and Energetic Efficiency in Muscle. Sci. Adv. 2021, 7, eabd6322. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional Role of Cardiolipin in Mitochondrial Bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of Cardiolipin Alterations in Mitochondrial Dysfunction and Disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Slabe, T.J.; Stoll, M.S.; Minkler, P.E.; Hoppel, C.L. Myocardial Ischemia Selectively Depletes Cardiolipin in Rabbit Heart Subsarcolemmal Mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2770–H2778. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative Stress, Cardiolipin and Mitochondrial Dysfunction in Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; López Torres, M.; Portero-Otín, M.; Pamplona, R.; Barja, G. Influence of Hyper- and Hypothyroidism on Lipid Peroxidation, Unsaturation of Phospholipids, Glutathione System and Oxidative Damage to Nuclear and Mitochondrial DNA in Mice Skeletal Muscle. Mol. Cell Biochem. 2001, 221, 41–48. [Google Scholar] [CrossRef]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Emerging Roles of Cardiolipin Remodeling in Mitochondrial Dysfunction Associated with Diabetes, Obesity, and Cardiovascular Diseases. J. Biomed. Res. 2010, 24, 6–15. [Google Scholar] [CrossRef]
- Ahmadpour, S.T.; Mahéo, K.; Servais, S.; Brisson, L.; Dumas, J.-F. Cardiolipin, the Mitochondrial Signature Lipid: Implication in Cancer. Int. J. Mol. Sci. 2020, 21, 8031. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.M.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin Profile Changes Are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease. J. Alzheimers. Dis. 2015, 43, 1375–1392. [Google Scholar] [CrossRef]
- Gaudioso, A.; Garcia-Rozas, P.; Casarejos, M.J.; Pastor, O.; Rodriguez-Navarro, J.A. Lipidomic Alterations in the Mitochondria of Aged Parkin Null Mice Relevant to Autophagy. Front. Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Polimova, A.M.; Maciel, E.; Tyurin, V.A.; Kapralova, V.I.; Winnica, D.E.; Vikulina, A.S.; Domingues, M.R.M.; McCoy, J.; Sanders, L.H.; et al. LC/MS Analysis of Cardiolipins in Substantia Nigra and Plasma of Rotenone-Treated Rats: Implication for Mitochondrial Dysfunction in Parkinson’s Disease. Free Radic. Res. 2015, 49, 681–691. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef] [PubMed]
- Zegallai, H.M.; Hatch, G.M. Barth Syndrome: Cardiolipin, Cellular Pathophysiology, Management, and Novel Therapeutic Targets. Mol. Cell Biochem. 2021, 476, 1605–1629. [Google Scholar] [CrossRef]
- Cole, L.K.; Kim, J.H.; Amoscato, A.A.; Tyurina, Y.Y.; Bay, R.H.; Karimi, B.; Siddiqui, T.J.; Kagan, V.E.; Hatch, G.M.; Kauppinen, T.M. Aberrant Cardiolipin Metabolism is Associated with Cognitive Deficiency and Hippocampal Alteration in Tafazzin Knockdown Mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3353–3367. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Amoscato, A.A.; Fink, A.B.; Anthonymuthu, T.; New, L.A.; Kochanek, P.M.; Watkins, S.; Kagan, V.E.; Bayır, H. Imaging Mass Spectrometry Reveals Loss of Polyunsaturated Cardiolipins in the Cortical Contusion, Hippocampus, and Thalamus after Traumatic Brain Injury. J. Neurochem. 2016, 139, 659–675. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Hier, Z.E.; Clark, R.S.B.; Kochanek, P.M.; Kagan, V.E.; Bayır, H. Detection of Brain Specific Cardiolipins in Plasma after Experimental Pediatric Head Injury. Exp. Neurol. 2019, 316, 63–73. [Google Scholar] [CrossRef]
- Helmer, P.O.; Wienken, C.M.; Korf, A.; Hayen, H. Mass Spectrometric Investigation of Cardiolipins and Their Oxidation Products after Two-Dimensional Heart-Cut Liquid Chromatography. J. Chromatogr. A 2020, 1619, 460918. [Google Scholar] [CrossRef]
- Helmer, P.O.; Nicolai, M.M.; Schwantes, V.; Bornhorst, J.; Hayen, H. Investigation of Cardiolipin Oxidation Products as a New Endpoint for Oxidative Stress in C. Elegans by Means of Online Two-Dimensional Liquid Chromatography and High-Resolution Mass Spectrometry. Free Radic. Biol. Med. 2021, 162, 216–224. [Google Scholar] [CrossRef]
- Jacob, R.F.; Mason, R.P. Lipid Peroxidation Induces Cholesterol Domain Formation in Model Membranes. J. Biol. Chem. 2005, 280, 39380–39387. [Google Scholar] [CrossRef] [PubMed]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a Method to Reveal Structures and Membrane-Interactions of Amyloidogenic Proteins. Biochim. Biophys. Acta 2007, 1768, 1862–1885. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Tyurina, Y.Y.; Jiang, J.; Tokarska-Schlattner, M.; Boissan, M.; Lacombe, M.-L.; Epand, R.; Schlattner, U.; Epand, R.M.; Kagan, V.E. Externalization of Cardiolipin as an “Eat-Me” Mitophageal Signal Is Facilitated by NDPK-D. Biophys. J. 2014, 106, 184a. [Google Scholar] [CrossRef]
- Manganelli, V.; Capozzi, A.; Recalchi, S.; Riitano, G.; Mattei, V.; Longo, A.; Misasi, R.; Garofalo, T.; Sorice, M. The Role of Cardiolipin as a Scaffold Mitochondrial Phospholipid in Autophagosome Formation: In Vitro Evidence. Biomolecules 2021, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of Peroxidized Cardiolipin with Rat-Heart Mitochondrial Membranes: Induction of Permeability Transition and Cytochrome c Release. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.-L.; et al. Crystal Structure and Biochemical Analyses Reveal Beclin 1 as a Novel Membrane Binding Protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Pariselli, F.; Dupaigne, P.; Budihardjo, I.; Lutter, M.; Antonsson, B.; Diolez, P.; Manon, S.; Martinou, J.-C.; Goubern, M.; et al. tBid Interaction with Cardiolipin Primarily Orchestrates Mitochondrial Dysfunctions and Subsequently Activates Bax and Bak. Cell Death Differ. 2005, 12, 614–626. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Schug, Z.T.; Houtkooper, R.H.; MacKenzie, E.D.; Brooks, D.G.; Wanders, R.J.A.; Petit, P.X.; Vaz, F.M.; Gottlieb, E. Cardiolipin Provides an Essential Activating Platform for Caspase-8 on Mitochondria. J. Cell Biol. 2008, 183, 681–696. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.V.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural Mechanism for NEK7-Licensed Activation of NLRP3 Inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Seoane, P.I.; Lee, B.; Hoyle, C.; Yu, S.; Lopez-Castejon, G.; Lowe, M.; Brough, D. The NLRP3-Inflammasome as a Sensor of Organelle Dysfunction. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Yeon, S.H.; Yang, G.; Lee, H.E.; Lee, J.Y. Oxidized Phosphatidylcholine Induces the Activation of NLRP3 Inflammasome in Macrophages. J. Leukoc. Biol. 2017, 101, 205–215. [Google Scholar] [CrossRef]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the Endoplasmic Reticulum and the Mitochondrion Tie the Knot: The Mitochondria-Associated Membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Sha, B. The ER Stress Sensor PERK Luminal Domain Functions as a Molecular Chaperone to Interact with Misfolded Proteins. Acta Cryst. D Struct. Biol. 2016, 72 Pt 12, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Maity, S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules 2021, 11, 173. [Google Scholar] [CrossRef]
- Gomes, E.; Shorter, J. The Molecular Language of Membraneless Organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef]
- Gilmozzi, V.; Gentile, G.; Castelo Rueda, M.P.; Hicks, A.A.; Pramstaller, P.P.; Zanon, A.; Lévesque, M.; Pichler, I. Interaction of Alpha-Synuclein with Lipids: Mitochondrial Cardiolipin as a Critical Player in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 2020, 14, 578993. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein Aggregation Nucleates through Liquid-Liquid Phase Separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The Close Encounter between Alpha-Synuclein and Mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.-I.; Anthony, M.D.; Edwards, R.H. Lipid Rafts Mediate the Synaptic Localization of Alpha-Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Ludtmann, M.; Angelova, P.; Ninkina, N.; Gandhi, S.; Buchman, V.; Abramov, A. A Physiological Role for Alpha-Synuclein in the Regulation of ATP Synthesis. Biophys. J. 2016, 110, 471a. [Google Scholar] [CrossRef]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic Alpha-Synuclein Aggregates Preferentially Bind to Mitochondria and Affect Cellular Respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef]
- Ghio, S.; Camilleri, A.; Caruana, M.; Ruf, V.C.; Schmidt, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Cauchi, R.J.; Kamp, F.; et al. Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem. Neurosci. 2019, 10, 3815–3829. [Google Scholar] [CrossRef]
- Annunziata, I.; Sano, R.; d’Azzo, A. Mitochondria-Associated ER Membranes (MAMs) and Lysosomal Storage Diseases. Cell Death Dis. 2018, 9, 328. [Google Scholar] [CrossRef]
- Faustino Mollinedo, C.G. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics 2021, 13, 763. [Google Scholar] [CrossRef] [PubMed]
- Dart, C. Lipid Microdomains and the Regulation of Ion Channel Function. J. Physiol. 2010, 588 Pt 17, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; London, E. Measurement of Lipid Nanodomain (raft) Formation and Size in sphingomyelin/POPC/cholesterol Vesicles Shows TX-100 and Transmembrane Helices Increase Domain Size by Coalescing Preexisting Nanodomains but Do Not Induce Domain Formation. Biophys. J. 2011, 101, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Varma, R.; Sarasij, R.C.; Gousset, K.; Krishnamoorthy, G.; Rao, M.; Mayor, S. Nanoscale Organization of Multiple GPI-Anchored Proteins in Living Cell Membranes. Cell 2004, 116, 577–589. [Google Scholar] [CrossRef]
- Ma, Y.; Hinde, E.; Gaus, K. Nanodomains in Biological Membranes. Essays Biochem. 2015, 57, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, R.M. Reactive Oxygen Species at Phospholipid Bilayers: Distribution, Mobility and Permeation. Biochim. Biophys. Acta 2014, 1838, 438–444. [Google Scholar] [CrossRef]
- Ayuyan, A.G.; Cohen, F.S. Lipid Peroxides Promote Large Rafts: Effects of Excitation of Probes in Fluorescence Microscopy and Electrochemical Reactions during Vesicle Formation. Biophys. J. 2006, 91, 2172–2183. [Google Scholar] [CrossRef]
- Squecco, R.; Tani, A.; Zecchi-Orlandini, S.; Formigli, L.; Francini, F. Melatonin Affects Voltage-Dependent Calcium and Potassium Currents in MCF-7 Cell Line Cultured Either in Growth or Differentiation Medium. Eur. J. Pharmacol. 2015, 758, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, M.; Suzuki, Y.; Yamamura, H. Inhibitory Effect of Melatonin on Voltage-Dependent Potassium (Kv4.2) Channels. Proc. Annu. Meet. Jpn. Pharmacol. Soc. 2020, 93, 1. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D.; Escames, G.; Rodriguez, M.I.; Lopez, L.C. Melatonin Role in the Mitochondrial Function. Front. Biosci. 2007, 12, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhao, C.; Xiang, H.; Zhao, X.; Zhong, R. Melatonin Inhibits Formation of Mitochondrial Permeability Transition Pores and Improves Oxidative Phosphorylation of Frozen-Thawed Ram Sperm. Front. Endocrinol. 2019, 10, 896. [Google Scholar] [CrossRef]
- Petrosillo, G.; Moro, N.; Ruggiero, F.M.; Paradies, G. Melatonin Inhibits Cardiolipin Peroxidation in Mitochondria and Prevents the Mitochondrial Permeability Transition and Cytochrome c Release. Free Radic. Biol. Med. 2009, 47, 969–974. [Google Scholar] [CrossRef]
- Ono, K.; Mochizuki, H.; Ikeda, T.; Nihira, T.; Takasaki, J.-I.; Teplow, D.B.; Yamada, M. Effect of Melatonin on α-Synuclein Self-Assembly and Cytotoxicity. Neurobiol. Aging. 2012, 33, 2172–2185. [Google Scholar] [CrossRef]
- Zampol, M.A.; Barros, M.H. Melatonin Improves Survival and Respiratory Activity of Yeast Cells Challenged by Alpha-Synuclein and Menadione. Yeast 2018, 35, 281–290. [Google Scholar] [CrossRef]
- Samir, P.; Kanneganti, T.-D. DDX3X Sits at the Crossroads of Liquid-Liquid and Prionoid Phase Transitions Arbitrating Life and Death Cell Fate Decisions in Stressed Cells. DNA Cell Biol. 2020, 39, 1091–1095. [Google Scholar] [CrossRef]
- Compan, V.; Martín-Sánchez, F.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrín, P. Apoptosis-Associated Speck-like Protein Containing a CARD Forms Specks but Does Not Activate Caspase-1 in the Absence of NLRP3 during Macrophage Swelling. J. Immunol. 2015, 194, 1261–1273. [Google Scholar] [CrossRef]
- Stehlik, C.; Lee, S.H.; Dorfleutner, A.; Stassinopoulos, A.; Sagara, J.; Reed, J.C. Apoptosis-Associated Speck-like Protein Containing a Caspase Recruitment Domain Is a Regulator of Procaspase-1 Activation. J. Immunol. 2003, 171, 6154–6163. [Google Scholar] [CrossRef]
- Ozgur, S.; Buchwald, G.; Falk, S.; Chakrabarti, S.; Prabu, J.R.; Conti, E. The Conformational Plasticity of Eukaryotic RNA-Dependent ATPases. FEBS J. 2015, 282, 850–863. [Google Scholar] [CrossRef]
- Owttrim, G.W. RNA Helicases: Diverse Roles in Prokaryotic Response to Abiotic Stress. RNA Biol. 2013, 10, 96–110. [Google Scholar] [CrossRef]
- Hondele, M.; Sachdev, R.; Heinrich, S.; Wang, J.; Vallotton, P.; Fontoura, B.M.A.; Weis, K. DEAD-Box ATPases Are Global Regulators of Phase-Separated Organelles. Nature 2019, 573, 144–148. [Google Scholar] [CrossRef]
- Fox, D.; Man, S.M. DDX3X: Stressing the NLRP3 Inflammasome. Cell Res. 2019, 29, 969–970. [Google Scholar] [CrossRef]
- Guan, Y.; Han, F. Key Mechanisms and Potential Targets of the NLRP3 Inflammasome in Neurodegenerative Diseases. Front. Integr. Neurosci. 2020, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, X.; Liu, M.; Fan, H.; Zheng, H.; Zhang, S.; Rahman, N.; Wołczyński, S.; Kretowski, A.; Li, X. Melatonin Inhibits Inflammasome-Associated Activation of Endothelium and Macrophages Attenuating Pulmonary Arterial Hypertension. Cardiovasc. Res. 2020, 116, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin Alleviates Intervertebral Disc Degeneration by Disrupting the IL-1β/NF-κB-NLRP3 Inflammasome Positive Feedback Loop. Bone Res. 2020, 8, 10. [Google Scholar] [CrossRef]
- Heerklotz, H. Triton Promotes Domain Formation in Lipid Raft Mixtures. Biophys. J. 2002, 83, 2693–2701. [Google Scholar] [CrossRef]
- Aponte-Santamaría, C.; Brunken, J.; Gräter, F. Stress Propagation through Biological Lipid Bilayers in Silico. J. Am. Chem. Soc. 2017, 139, 13588–13591. [Google Scholar] [CrossRef]
- Reiter, R.J. Melatonin: Lowering the High Price of Free Radicals. News Physiol. Sci. 2000, 15, 246–250. [Google Scholar] [CrossRef]
- Severcan, F.; Sahin, I.; Kazanci, N. Melatonin Strongly Interacts with Zwitterionic Model Membranes--Evidence from Fourier Transform Infrared Spectroscopy and Differential Scanning Calorimetry. Biochim. Biophys. Acta 2005, 1668, 215–222. [Google Scholar] [CrossRef]
- Mei, N.; Robinson, M.; Davis, J.H.; Leonenko, Z. Melatonin Alters Fluid Phase Co-Existence in POPC/DPPC/cholesterol Membranes. Biophys. J. 2020, 119, 2391–2402. [Google Scholar] [CrossRef]
- Bongiorno, D.; Ceraulo, L.; Ferrugia, M.; Filizzola, F.; Giordano, C.; Ruggirello, A.; Liveri, V.T. H-NMR and FT-IR Study of the State of Melatonin Confined in Membrane Models: Location and Interactions of Melatonin in Water Free Lecithin and AOT Reversed Micelles. Arkivoc 2004, 2004, 251–262. [Google Scholar] [CrossRef]
- Choi, Y.; Attwood, S.J.; Hoopes, M.I.; Drolle, E.; Karttunen, M.; Leonenko, Z. Melatonin Directly Interacts with Cholesterol and Alleviates Cholesterol Effects in Dipalmitoylphosphatidylcholine Monolayers. Soft Matter 2014, 10, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Del Mar Martínez-Senac, M.; Villalaín, J.; Gómez-Fernández, J.C. Structure of the Alzheimer Beta-Amyloid Peptide (25-35) and Its Interaction with Negatively Charged Phospholipid Vesicles. Eur. J. Biochem. 1999, 265, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.-A.; Gehman, J.D.; Separovic, F. Lipid Matrix Plays a Role in Abeta Fibril Kinetics and Morphology. FEBS Lett. 2011, 585, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-β Aggregation on Model Lipid Membranes: An Atomic Force Microscopy Study. J. Alzheimers. Dis. 2011, 26, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Ahyayauch, H.; Raab, M.; Busto, J.V.; Andraka, N.; Arrondo, J.-L.R.; Masserini, M.; Tvaroska, I.; Goñi, F.M. Binding of β-Amyloid (1-42) Peptide to Negatively Charged Phospholipid Membranes in the Liquid-Ordered State: Modeling and Experimental Studies. Biophys. J. 2012, 103, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Schauerte, J.A.; Steel, D.G.; Gafni, A. β-Amyloid (1-40) Peptide Interactions with Supported Phospholipid Membranes: A Single-Molecule Study. Biophys. J. 2012, 103, 1500–1509. [Google Scholar] [CrossRef]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef]
- Drolle, E.; Gaikwad, R.M.; Leonenko, Z. Nanoscale Electrostatic Domains in Cholesterol-Laden Lipid Membranes Create a Target for Amyloid Binding. Biophys. J. 2012, 103, L27–L29. [Google Scholar] [CrossRef]
- Finot, E.; Leonenko, Y.; Moores, B.; Eng, L.; Amrein, M.; Leonenko, Z. Effect of Cholesterol on Electrostatics in Lipid-Protein Films of a Pulmonary Surfactant. Langmuir 2010, 26, 1929–1935. [Google Scholar] [CrossRef]
- Eckert, G.P.; Kirsch, C.; Leutz, S.; Wood, W.G.; Müller, W.E. Cholesterol Modulates Amyloid Beta-Peptide’s Membrane Interactions. Pharmacopsychiatry 2003, 36 (Suppl. 2), S136–S143. [Google Scholar] [CrossRef] [PubMed]
- Gibson Wood, W.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Amyloid Beta-Protein Interactions with Membranes and Cholesterol: Causes or Casualties of Alzheimer’s Disease. Biochim. Biophys. Acta 2003, 1610, 281–290. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.-J.; Smith, M.A.; Perry, G.; et al. Melatonin Increases Survival and Inhibits Oxidative and Amyloid Pathology in a Transgenic Model of Alzheimer’s Disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Z.; Wang, Z.-F. Role of Melatonin in Alzheimer-like Neurodegeneration. Acta Pharmacol. Sin. 2006, 27, 41–49. [Google Scholar] [CrossRef]
- Vincent, B. Protective Roles of Melatonin against the Amyloid-Dependent Development of Alzheimer’s Disease: A Critical Review. Pharmacol. Res. 2018, 134, 223–237. [Google Scholar] [CrossRef]
- Chinchalongporn, V.; Shukla, M.; Govitrapong, P. Melatonin Ameliorates Aβ42 -Induced Alteration of βAPP-Processing Secretases via the Melatonin Receptor through the Pin1/GSK3β/NF-κB Pathway in SH-SY5Y Cells. J. Pineal Res. 2018, 64, e12470. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin Regulates Aβ Production/clearance Balance and Aβ Neurotoxicity: A Potential Therapeutic Molecule for Alzheimer’s Disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer Beta-Fibrillogenesis by Melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [PubMed]
- Dies, H.; Toppozini, L.; Rheinstädter, M.C. The Interaction between Amyloid-β Peptides and Anionic Lipid Membranes Containing Cholesterol and Melatonin. PLoS ONE 2014, 9, e99124. [Google Scholar] [CrossRef]
- Lu, H.; Martí, J. Binding and Dynamics of Melatonin at the Interface of Phosphatidylcholine-Cholesterol Membranes. PLoS ONE 2019, 14, e0224624. [Google Scholar] [CrossRef] [PubMed]
- Akkas, S.B.; Inci, S.; Zorlu, F.; Severcan, F. Melatonin Affects the Order, Dynamics and Hydration of Brain Membrane Lipids. J. Mol. Struct. 2007, 834–836, 207–215. [Google Scholar] [CrossRef]
- Dies, H.; Cheung, B.; Tang, J.; Rheinstädter, M.C. The Organization of Melatonin in Lipid Membranes. Biochim. Biophys. Acta 2015, 1848, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Los, D.A.; Murata, N. Membrane Fluidity and Its Roles in the Perception of Environmental Signals. Biochim. Biophys. Acta 2004, 1666, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.; Jackson, C.; Yu, B.P. Lipid Peroxidation Contributes to Age-Related Membrane Rigidity. Free Radic. Biol. Med. 1995, 18, 977–984. [Google Scholar] [CrossRef]
- De la Haba, C.; Palacio, J.R.; Martínez, P.; Morros, A. Effect of Oxidative Stress on Plasma Membrane Fluidity of THP-1 Induced Macrophages. Biochim. Biophys. Acta 2013, 1828, 357–364. [Google Scholar] [CrossRef]
- Tenchov, B.; Koynova, R. Cubic Phases in Membrane Lipids. Eur. Biophys. J. 2012, 41, 841–850. [Google Scholar] [CrossRef]
- Landh, T. From Entangled Membranes to Eclectic Morphologies: Cubic Membranes as Subcellular Space Organizers. FEBS Lett. 1995, 369, 13–17. [Google Scholar] [CrossRef]
- Catalá, Á. Lipid Peroxidation Modifies the Assembly of Biological Membranes “The Lipid Whisker Model”. Front. Physiol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Volinsky, R.; Paananen, R.; Kinnunen, P.K.J. Oxidized Phosphatidylcholines Promote Phase Separation of Cholesterol-Sphingomyelin Domains. Biophys. J. 2012, 103, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, M.; Kenworthy, A.K. Lipid Peroxidation Enhances LO/LD Domain Phase Separation in Giant Plasma Membrane Vesicles. Biophys. J. 2021, 120, 324a. [Google Scholar] [CrossRef]
- Bolmatov, D.; McClintic, W.T.; Taylor, G.; Stanley, C.B.; Do, C.; Collier, C.P.; Leonenko, Z.; Lavrentovich, M.O.; Katsaras, J. Deciphering Melatonin-Stabilized Phase Separation in Phospholipid Bilayers. Langmuir 2019, 35, 12236–12245. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Burgos, I.; Espinosa, J.R.; Joseph, J.A.; Collepardo-Guevara, R. Valency and Binding Affinity Variations Can Regulate the Multilayered Organization of Protein Condensates with Many Components. Biomolecules 2021, 11, 278. [Google Scholar] [CrossRef]
- Balmik, A.A.; Das, R.; Dangi, A.; Gorantla, N.V.; Marelli, U.K.; Chinnathambi, S. Melatonin Interacts with Repeat Domain of Tau to Mediate Disaggregation of Paired Helical Filaments. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129467. [Google Scholar] [CrossRef]
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef]
- Kaplán, P.; Racay, P.; Lehotský, J.; Mézesová, V. Change in Fluidity of Brain Endoplasmic Reticulum Membranes by Oxygen Free Radicals: A Protective Effect of Stobadine, Alpha-Tocopherol Acetate, and Butylated Hydroxytoluene. Neurochem. Res. 1995, 20, 815–820. [Google Scholar] [CrossRef]
- Yu, B.P.; Suescun, E.A.; Yang, S.Y. Effect of Age-Related Lipid Peroxidation on Membrane Fluidity and Phospholipase A2: Modulation by Dietary Restriction. Mech. Ageing. Dev. 1992, 65, 17–33. [Google Scholar] [CrossRef]
- Rudzite, V.; Jurika, E.; Jirgensons, J. Changes in Membrane Fluidity Induced by Tryptophan and Its Metabolites. In Tryptophan, Serotonin, and Melatonin: Basic Aspects and Applications; Huether, G., Kochen, W., Simat, T.J., Steinhart, H., Eds.; Springer: Boston, MA, USA, 1999. [Google Scholar] [CrossRef]
- Alexandre, H.; Mathieu, B.; Charpentier, C. Alteration in Membrane Fluidity and Lipid Composition, and Modulation of H+-ATPase Activity in Saccharomyces Cerevisiae Caused by Decanoic Acid. Available online: https://www.microbiologyresearch.org/docserver/fulltext/micro/142/3/mic-142-3-469.pdf?expires=1616871084&id=id&accname=guest&checksum=6688BACF19B736B01B5100ACDEA617F6 (accessed on 27 March 2021).
- Keeffe, E.B.; Blankenship, N.M.; Scharschmidt, B.F. Alteration of Rat Liver Plasma Membrane Fluidity and ATPase Activity by Chlorpromazine Hydrochloride and Its Metabolites. Gastroenterology 1980, 79, 222–231. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Alterations in Mitochondrial Membrane Fluidity by Lipid Peroxidation Products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Kholodenko, B.N.; Hoek, J.B.; Westerhoff, H.V. Why Cytoplasmic Signalling Proteins Should Be Recruited to Cell Membranes. Trends Cell Biol. 2000, 10, 173–178. [Google Scholar] [CrossRef]
- Botterbusch, S.; Baumgart, T. Interactions between Phase-Separated Liquids and Membrane Surfaces. NATO Adv. Sci. Inst. Ser. E Appl. Sci. 2021, 11, 1288. [Google Scholar] [CrossRef]
- Alimohamadi, H.; Rangamani, P. Modeling Membrane Curvature Generation due to Membrane−Protein Interactions. Biomolecules 2018, 8, 120. [Google Scholar] [CrossRef]
- Prévost, C.; Zhao, H.; Manzi, J.; Lemichez, E.; Lappalainen, P.; Callan-Jones, A.; Bassereau, P. IRSp53 Senses Negative Membrane Curvature and Phase Separates along Membrane Tubules. Nat. Commun. 2015, 6, 8529. [Google Scholar] [CrossRef]
- Gallop, J.L.; McMahon, H.T. BAR Domains and Membrane Curvature: Bringing Your Curves to the BAR. Biochem. Soc. Symp. 2005, 72, 223–231. [Google Scholar] [CrossRef]
- Lu, S.; Deng, R.; Jiang, H.; Song, H.; Li, S.; Shen, Q.; Huang, W.; Nussinov, R.; Yu, J.; Zhang, J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23, 1725–1734. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH Oxidase-Mediated Redox Signaling: Roles in Cellular Stress Response, Stress Tolerance, and Tissue Repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Garofalo, T.; Manganelli, V.; Grasso, M.; Mattei, V.; Ferri, A.; Misasi, R.; Sorice, M. Role of Mitochondrial Raft-like Microdomains in the Regulation of Cell Apoptosis. Apoptosis 2015, 20, 621–634. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Garcia-Bereguiain, M.A.; Martin-Romero, F.J.; Gutierrez-Merino, C. Clustering of Plasma Membrane-Bound Cytochrome b5 Reductase within “Lipid Raft” Microdomains of the Neuronal Plasma Membrane. Mol. Cell. Neurosci. 2009, 40, 14–26. [Google Scholar] [CrossRef]
- Gostincar, C.; Turk, M.; Gunde-Cimerman, N. The Evolution of Fatty Acid Desaturases and Cytochrome b5 in Eukaryotes. J. Membr. Biol. 2010, 233, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hayashi, S.; Yoshida, T. Participation of a Cytochrome b5-like Hemoprotein of Outer Mitochondrial Membrane (OM Cytochrome B) in NADH-Semidehydroascorbic Acid Reductase Activity of Rat Liver. Biochem. Biophys. Res. Commun. 1981, 101, 591–598. [Google Scholar] [CrossRef]
- Navarro, F.; Villalba, J.M.; Crane, F.L.; Mackellar, W.C.; Navas, P. A Phospholipid-Dependent NADH-Coenzyme Q Reductase from Liver Plasma Membrane. Biochem. Biophys. Res. Commun. 1995, 212, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.J.; Lappin, T.R. Recessive Congenital Methaemoglobinaemia: Cytochrome b(5) Reductase Deficiency. Br. J. Haematol. 2008, 141, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Siendones, E.; SantaCruz-Calvo, S.; Martín-Montalvo, A.; Cascajo, M.V.; Ariza, J.; López-Lluch, G.; Villalba, J.M.; Acquaviva-Bourdain, C.; Roze, E.; Bernier, M.; et al. Membrane-Bound CYB5R3 Is a Common Effector of Nutritional and Oxidative Stress Response through FOXO3a and Nrf2. Antioxid. Redox Signal. 2014, 21, 1708–1725. [Google Scholar] [CrossRef] [PubMed]
- Marques-da-Silva, D.; Samhan-Arias, A.K.; Tiago, T.; Gutierrez-Merino, C. L-Type Calcium Channels and Cytochrome b5 Reductase Are Components of Protein Complexes Tightly Associated with Lipid Rafts Microdomains of the Neuronal Plasma Membrane. J. Proteom. 2010, 73, 1502–1510. [Google Scholar] [CrossRef]
- Nikiforova, A.B.; Saris, N.-E.L.; Kruglov, A.G. External Mitochondrial NADH-Dependent Reductase of Redox Cyclers: VDAC1 or Cyb5R3? Free Radic. Biol. Med. 2014, 74, 74–84. [Google Scholar] [CrossRef]
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New Potential Biomarker for Stratification of Patients for Pharmacological Vitamin C in Adjuvant Settings of Cancer Therapy. Redox Biol. 2020, 28, 101357. [Google Scholar] [CrossRef]
- Mihara, K.; Sato, R. Molecular Cloning and Sequencing of cDNA for Yeast Porin, an Outer Mitochondrial Membrane Protein: A Search for Targeting Signal in the Primary Structure. EMBO J. 1985, 4, 769–774. [Google Scholar] [CrossRef]
- Thinnes, F.P.; Götz, H.; Kayser, H.; Benz, R.; Schmidt, W.E.; Kratzin, H.D.; Hilschmann, N. Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell. Biol. Chem. Hoppe Seyler 1989, 370, 1253–1264. [Google Scholar]
- Bàthori, G.; Parolini, I.; Tombola, F.; Szabò, I.; Messina, A.; Oliva, M.; De Pinto, V.; Lisanti, M.; Sargiacomo, M.; Zoratti, M. Porin Is Present in the Plasma Membrane Where It Is Concentrated in Caveolae and Caveolae-Related Domains. J. Biol. Chem. 1999, 274, 29607–29612. [Google Scholar] [CrossRef]
- Herrera, J.L.; Diaz, M.; Hernández-Fernaud, J.R.; Salido, E.; Alonso, R.; Fernández, C.; Morales, A.; Marin, R. Voltage-Dependent Anion Channel as a Resident Protein of Lipid Rafts: Post-Transductional Regulation by Estrogens and Involvement in Neuronal Preservation against Alzheimer’s Disease. J. Neurochem. 2011, 116, 820–827. [Google Scholar] [CrossRef]
- De Pinto, V.; Messina, A.; Lane, D.J.R.; Lawen, A. Voltage-Dependent Anion-Selective Channel (VDAC) in the Plasma Membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar] [CrossRef]
- Anishkin, A.; Loukin, S.H.; Teng, J.; Kung, C. Feeling the Hidden Mechanical Forces in Lipid Bilayer Is an Original Sense. Proc. Natl. Acad. Sci. USA 2014, 111, 7898–7905. [Google Scholar] [CrossRef] [PubMed]
- Anishkin, A.; Kung, C. Stiffened Lipid Platforms at Molecular Force Foci. Proc. Natl. Acad. Sci. USA 2013, 110, 4886–4892. [Google Scholar] [CrossRef]
- Martinac, B.; Adler, J.; Kung, C. Mechanosensitive Ion Channels of E. Coli Activated by Amphipaths. Nature 1990, 348, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Markin, V.S.; Martinac, B. Mechanosensitive Ion Channels as Reporters of Bilayer Expansion. A Theoretical Model. Biophys. J. 1991, 60, 1120–1127. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Marques-da-Silva, D.; Yanamala, N.; Gutierrez-Merino, C. Stimulation and Clustering of Cytochrome b5 Reductase in Caveolin-Rich Lipid Microdomains Is an Early Event in Oxidative Stress-Mediated Apoptosis of Cerebellar Granule Neurons. J. Proteom. 2012, 75, 2934–2949. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Fortalezas, S.; Cordas, C.M.; Moura, I.; Moura, J.J.G.; Gutierrez-Merino, C. Cytochrome b5 Reductase Is the Component from Neuronal Synaptic Plasma Membrane Vesicles That Generates Superoxide Anion upon Stimulation by Cytochrome c. Redox Biol. 2018, 15, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Gutierrez-Merino, C. Purified NADH-Cytochrome b5 Reductase Is a Novel Superoxide Anion Source Inhibited by Apocynin: Sensitivity to Nitric Oxide and Peroxynitrite. Free Radic. Biol. Med. 2014, 73, 174–189. [Google Scholar] [CrossRef]
- La Piana, G.; Marzulli, D.; Gorgoglione, V.; Lofrumento, N.E. Porin and Cytochrome Oxidase Containing Contact Sites Involved in the Oxidation of Cytosolic NADH. Arch. Biochem. Biophys. 2005, 436, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Martín-Romero, F.J.; Gutiérrez-Martín, Y.; Henao, F.; Gutiérrez-Merino, C. The NADH Oxidase Activity of the Plasma Membrane of Synaptosomes Is a Major Source of Superoxide Anion and Is Inhibited by Peroxynitrite. J. Neurochem. 2002, 82, 604–614. [Google Scholar] [CrossRef]
- Zizi, M.; Forte, M.; Blachly-Dyson, E.; Colombini, M. NADH Regulates the Gating of VDAC, the Mitochondrial Outer Membrane Channel. J. Biol. Chem. 1994, 269, 1614–1616. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Evolution of Voltage-Dependent Anion Channel Function: From Molecular Sieve to Governator to Actuator of Ferroptosis. Front. Oncol. 2017, 7, 303. [Google Scholar] [CrossRef]
- Elinder, F.; Akanda, N.; Tofighi, R.; Shimizu, S.; Tsujimoto, Y.; Orrenius, S.; Ceccatelli, S. Opening of Plasma Membrane Voltage-Dependent Anion Channels (VDAC) Precedes Caspase Activation in Neuronal Apoptosis Induced by Toxic Stimuli. Cell Death Differ. 2005, 12, 1134–1140. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. VDAC Channels Mediate and Gate the Flow of ATP: Implications for the Regulation of Mitochondrial Function. Biophys. J. 1997, 72, 1954–1962. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Tan, W.; Colombini, M. On the Role of VDAC in Apoptosis: Fact and Fiction. J. Bioenerg. Biomembr. 2005, 37, 129–142. [Google Scholar] [CrossRef]
- McCommis, K.S.; Baines, C.P. The Role of VDAC in Cell Death: Friend or Foe? Biochim. Biophys. Acta 2012, 1818, 1444–1450. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the Crossroads of Cell Metabolism, Apoptosis and Cell Stress. Cell Stress Chaperones 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-Enriched Raft-like Microdomains Are Essential Activating Platforms for Apoptotic Signals on Mitochondria. FEBS Lett. 2009, 583, 2447–2450. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; et al. Dynamics of Mitochondrial Raft-like Microdomains in Cell Life and Death. Commun. Integr. Biol. 2012, 5, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Giammarioli, A.M.; Garofalo, T.; Sorice, M. Dynamics of Lipid Raft Components during Lymphocyte Apoptosis: The Paradigmatic Role of GD3. Apoptosis 2007, 12, 941–949. [Google Scholar] [CrossRef]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Kim, J.; Minkler, P.E.; Salomon, R.G.; Anderson, V.E.; Hoppel, C.L. Cardiolipin: Characterization of Distinct Oxidized Molecular Species. J. Lipid Res. 2011, 52, 125–135. [Google Scholar] [CrossRef]
- Schneider, C. An Update on Products and Mechanisms of Lipid Peroxidation. Mol. Nutr. Food Res. 2009, 53, 315–321. [Google Scholar] [CrossRef]
- Ting, H.-C.; Chen, L.-T.; Chen, J.-Y.; Huang, Y.-L.; Xin, R.-C.; Chan, J.-F.; Hsu, Y.-H.H. Double Bonds of Unsaturated Fatty Acids Differentially Regulate Mitochondrial Cardiolipin Remodeling. Lipids Health Dis. 2019, 18, 53. [Google Scholar] [CrossRef]
- Musatov, A. Contribution of Peroxidized Cardiolipin to Inactivation of Bovine Heart Cytochrome c Oxidase. Free Radic. Biol. Med. 2006, 41, 238–246. [Google Scholar] [CrossRef]
- Afzal, N.; Lederer, W.J.; Jafri, M.S.; Mannella, C.A. Effect of Crista Morphology on Mitochondrial ATP Output: A Computational Study. Curr. Res. Physiol. 2021, 4, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Römsing, S. Development and Validation of Bioanalytical Methods: Application to Melatonin and Selected Anti-Infective Drugs; Acta Universitatis Upsaliensis: Uppsala, Sweden, 2010. [Google Scholar]
- Ceraulo, L.; Ferrugia, M.; Tesoriere, L.; Segreto, S.; Livrea, M.A.; Turco Liveri, V. Interactions of Melatonin with Membrane Models: Portioning of Melatonin in AOT and Lecithin Reversed Micelles. J. Pineal Res. 1999, 26, 108–112. [Google Scholar] [CrossRef]
- Yu, H.; Dickson, E.J.; Jung, S.-R.; Koh, D.-S.; Hille, B. High Membrane Permeability for Melatonin. J. Gen. Physiol. 2016, 147, 63–76. [Google Scholar] [CrossRef]
- Venegas, C.; García, J.A.; Escames, G.; Ortiz, F.; López, A.; Doerrier, C.; García-Corzo, L.; López, L.C.; Reiter, R.J.; Acuña-Castroviejo, D. Extrapineal Melatonin: Analysis of Its Subcellular Distribution and Daily Fluctuations. J. Pineal Res. 2012, 52, 217–227. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a Mitochondria-Targeted Antioxidant: One of Evolution’s Best Ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Coon, S.L.; Klein, D.C. Evolution of Arylalkylamine N-Acetyltransferase: Emergence and Divergence. Mol. Cell. Endocrinol. 2006, 252, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.C. Arylalkylamine N-Acetyltransferase: The Timezyme. J. Biol. Chem. 2007, 282, 4233–4237. [Google Scholar] [CrossRef]
- Ganguly, S.; Weller, J.L.; Ho, A.; Chemineau, P.; Malpaux, B.; Klein, D.C. Melatonin Synthesis: 14-3-3-Dependent Activation and Inhibition of Arylalkylamine N-Acetyltransferase Mediated by Phosphoserine-205. Proc. Natl. Acad. Sci. USA 2005, 102, 1222–1227. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but Not Vitamins C and E Maintains Glutathione Homeostasis in T-Butyl Hydroperoxide-Induced Mitochondrial Oxidative Stress. FASEB J. 2000, 14, 1677–1679. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual Role of Mitochondria in Producing Melatonin and Driving GPCR Signaling to Block Cytochrome c Release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.H.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.-X.; Reiter, R.J. Melatonin: An Ancient Molecule That Makes Oxygen Metabolically Tolerable. J. Pineal. Res. 2015, 59, 403–419. [Google Scholar] [CrossRef]
- Byeon, Y.; Lee, K.; Park, Y.-I.; Park, S.; Back, K. Molecular Cloning and Functional Analysis of Serotonin N-Acetyltransferase from the Cyanobacterium Synechocystis Sp. PCC 6803. J. Pineal. Res. 2013, 55, 371–376. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and Chloroplasts as the Original Sites of Melatonin Synthesis: A Hypothesis Related to Melatonin’s Primary Function and Evolution in Eukaryotes. J. Pineal. Res. 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Abhishek, A.; Bavishi, A.; Bavishi, A.; Choudhary, M. Bacterial Genome Chimaerism and the Origin of Mitochondria. Can. J. Microbiol. 2011, 57, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.A.; Allen, J.F. Genomics and Chloroplast Evolution: What Did Cyanobacteria Do for Plants? Genome Biol. 2003, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One Molecule, Many Derivatives: A Never-Ending Interaction of Melatonin with Reactive Oxygen and Nitrogen Species? J. Pineal. Res. 2007, 42, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-N. Distribution and Dynamics of Electron Transport Complexes in Cyanobacterial Thylakoid Membranes. Biochim. Biophys. Acta 2016, 1857, 256–265. [Google Scholar] [CrossRef]
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The Emergence of Phase Separation as an Organizing Principle in Bacteria. Biophys. J. 2021, 120, 1123–1138. [Google Scholar] [CrossRef]
- Guilhas, B.; Walter, J.-C.; Rech, J.; David, G.; Walliser, N.O.; Palmeri, J.; Mathieu-Demaziere, C.; Parmeggiani, A.; Bouet, J.-Y.; Le Gall, A.; et al. ATP-Driven Separation of Liquid Phase Condensates in Bacteria. Mol. Cell 2020, 79, 293–303.e4. [Google Scholar] [CrossRef] [PubMed]
- Muthunayake, N.S.; Tomares, D.T.; Childers, W.S.; Schrader, J.M. Phase-Separated Bacterial Ribonucleoprotein Bodies Organize mRNA Decay. Wiley Interdiscip. Rev. RNA 2020, 11, e1599. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.; Sánchez-Baracaldo, P.; Larkum, A.W.; Rutherford, A.W.; Cardona, T. Time-Resolved Comparative Molecular Evolution of Oxygenic Photosynthesis. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148400. [Google Scholar] [CrossRef]
- Pattanayak, G.K.; Liao, Y.; Wallace, E.W.J.; Budnik, B.; Drummond, D.A.; Rust, M.J. Daily Cycles of Reversible Protein Condensation in Cyanobacteria. Cell Rep. 2020, 32, 108032. [Google Scholar] [CrossRef]
- Bar Eyal, L.; Ranjbar Choubeh, R.; Cohen, E.; Eisenberg, I.; Tamburu, C.; Dorogi, M.; Ünnep, R.; Appavou, M.-S.; Nevo, R.; Raviv, U.; et al. Changes in Aggregation States of Light-Harvesting Complexes as a Mechanism for Modulating Energy Transfer in Desert Crust Cyanobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 9481–9486. [Google Scholar] [CrossRef]
- Wang, H.; Yan, X.; Aigner, H.; Bracher, A.; Nguyen, N.D.; Hee, W.Y.; Long, B.M.; Price, G.D.; Hartl, F.U.; Hayer-Hartl, M. Rubisco Condensate Formation by CcmM in β-Carboxysome Biogenesis. Nature 2019, 566, 131–135. [Google Scholar] [CrossRef]
- McKinney, D.W.; Buchanan, B.B.; Wolosiuk, R.A. Activation of Chloroplast ATPase by Reduced Thioredoxin. Phytochemistry 1978, 17, 794–795. [Google Scholar] [CrossRef]
- Curtis, S.E. Structure, Organization and Expression of Cyanobacterial ATP Synthase Genes. Photosynth. Res. 1988, 18, 223–244. [Google Scholar] [CrossRef]
- Pogoryelov, D.; Reichen, C.; Klyszejko, A.L.; Brunisholz, R.; Muller, D.J.; Dimroth, P.; Meier, T. The Oligomeric State of c Rings from Cyanobacterial F-ATP Synthases Varies from 13 to 15. J. Bacteriol. 2007, 189, 5895–5902. [Google Scholar] [CrossRef]
- Walraven, H.S.; Bakels, R.H.A. Function, Structure and Regulation of Cyanobacterial and Chloroplast ATP Synthase. Physiol. Plant 1996, 96, 526–532. [Google Scholar] [CrossRef]
- Buchert, F.E. Chapter Three—Chloroplast ATP Synthase from Green Microalgae. In Advances in Botanical Research; Hisabori, T., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 96, pp. 75–118. [Google Scholar] [CrossRef]
- Carman, G.M. An Unusual Phosphatidylethanolamine-Utilizing Cardiolipin Synthase Is Discovered in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 16402–16403. [Google Scholar] [CrossRef]
- Kobayashi, K.; Endo, K.; Wada, H. Specific Distribution of Phosphatidylglycerol to Photosystem Complexes in the Thylakoid Membrane. Front. Plant Sci. 2017, 8, 1991. [Google Scholar] [CrossRef]
- Shadyro, O.I.; Yurkova, I.L.; Kisel, M.A. Radiation-Induced Peroxidation and Fragmentation of Lipids in a Model Membrane. Int. J. Radiat. Biol. 2002, 78, 211–217. [Google Scholar] [CrossRef]
- Althoff, T.; Mills, D.J.; Popot, J.-L.; Kühlbrandt, W. Arrangement of Electron Transport Chain Components in Bovine Mitochondrial Supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef]
- Ostrander, D.B.; Zhang, M.; Mileykovskaya, E.; Rho, M.; Dowhan, W. Lack of Mitochondrial Anionic Phospholipids Causes an Inhibition of Translation of Protein Components of the Electron Transport Chain. A Yeast Genetic Model System for the Study of Anionic Phospholipid Function in Mitochondria. J. Biol. Chem. 2001, 276, 25262–25272. [Google Scholar] [CrossRef]
- Yoshioka-Nishimura, M. Close Relationships Between the PSII Repair Cycle and Thylakoid Membrane Dynamics. Plant Cell Physiol. 2016, 57, 1115–1122. [Google Scholar] [CrossRef]
- Megiatto, J.D.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Gervaldo, M.; Moore, T.A.; Moore, A.L.; Gust, D. Mimicking the Electron Transfer Chain in Photosystem II with a Molecular Triad Thermodynamically Capable of Water Oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15578–15583. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and Its Metabolites: New Findings Regarding Their Production and Their Radical Scavenging Actions. Acta Biochim. Pol. 2007, 54, 1–9. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-Hydroxymelatonin: A Melatonin Metabolite Generated as a Result of Hydroxyl Radical Scavenging. Biol. Signals Recept. 1999, 8, 70–74. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, E.A.; Martinez, G.R.; Klitzke, C.F.; de Medeiros, M.H.G.; Di Mascio, P. Oxidation of Melatonin by Singlet Molecular Oxygen (O2(1deltag)) Produces N1-Acetyl-N2-Formyl-5-Methoxykynurenine. J. Pineal. Res. 2003, 35, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, Z.; Bilska, M.A.; Reszka, K.J.; Chignell, C.F.; Bilski, P. Interaction of Singlet Molecular Oxygen with Melatonin and Related Indoles. Photochem. Photobiol. 2003, 78, 449–455. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin Directly Scavenges Hydrogen Peroxide: A Potentially New Metabolic Pathway of Melatonin Biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185. [Google Scholar] [CrossRef]
- Noda, Y.; Mori, A.; Liburdy, R.; Packer, L. Melatonin and Its Precursors Scavenge Nitric Oxide. J. Pineal. Res. 1999, 27, 159–163. [Google Scholar] [CrossRef]
- Aydogan, S.; Yerer, M.B.; Goktas, A. Melatonin and Nitric Oxide. J. Endocrinol. Investig. 2006, 29, 281–287. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin, Its Metabolites and Their Interference with Reactive Nitrogen Compounds. Molecules 2021, 26, 4105. [Google Scholar] [CrossRef] [PubMed]
- Gilad, E.; Cuzzocrea, S.; Zingarelli, B.; Salzman, A.L.; Szabó, C. Melatonin Is a Scavenger of Peroxynitrite. Life Sci. 1997, 60, PL169–PL174. [Google Scholar] [CrossRef]
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs. Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal Res. 2018, 65, e12514. [Google Scholar] [CrossRef]
- Purushothaman, A.; Sheeja, A.A.; Janardanan, D. Hydroxyl Radical Scavenging Activity of Melatonin and Its Related Indolamines. Free Radic. Res. 2020, 54, 373–383. [Google Scholar] [CrossRef]
- Galano, A. On the Direct Scavenging Activity of Melatonin towards Hydroxyl and a Series of Peroxyl Radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Cyclic 3-Hydroxymelatonin, a Key Metabolite Enhancing the Peroxyl Radical Scavenging Activity of Melatonin. RSC Adv. 2014, 4, 5220. [Google Scholar] [CrossRef]
- Galano, A.; Medina, M.E.; Tan, D.X.; Reiter, R.J. Melatonin and Its Metabolites as Copper Chelating Agents and Their Role in Inhibiting Oxidative Stress: A Physicochemical Analysis. J. Pineal. Res. 2015, 58, 107–116. [Google Scholar] [CrossRef]
- Lúcio, M.; Nunes, C.; Gaspar, D.; Ferreira, H.; Lima, J.L.F.C.; Reis, S. Antioxidant Activity of Vitamin E and Trolox: Understanding of the Factors That Govern Lipid Peroxidation Studies In Vitro. Food Biophys. 2009, 4, 312–320. [Google Scholar] [CrossRef]
- Watson, H. Biological Membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, X.; Tian, Y.; Li, W.; Wang, H.; Li, Q.; Li, Y.; Li, Z.; Wu, T. Synthesis of a New Water-Soluble Melatonin Derivative with Low Toxicity and a Strong Effect on Sleep Aid. ACS Omega. 2020, 5, 6494–6499. [Google Scholar] [CrossRef]
- Shida, C.S.; Castrucci, A.M.; Lamy-Freund, M.T. High Melatonin Solubility in Aqueous Medium. J. Pineal. Res. 1994, 16, 198–201. [Google Scholar] [CrossRef]
- Aikens, J.; Dix, T.A. Perhydroxyl Radical (HOO.) Initiated Lipid Peroxidation. The Role of Fatty Acid Hydroperoxides. J. Biol. Chem. 1991, 266, 15091–15098. [Google Scholar] [CrossRef]
- Bielski, B.H.; Arudi, R.L.; Sutherland, M.W. A Study of the Reactivity of HO2/O2- with Unsaturated Fatty Acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar] [CrossRef]
- Repetto, M.; Semprine, J.; Boveris, A. Lipid Peroxidation: Chemical Mechanism, Biological Implications and Analytical Determination. In Lipid Peroxidation; Catala, A., Ed.; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Lipid Peroxidation: Physiological Levels and Dual Biological Effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Kanner, J.; German, J.B.; Kinsella, J.E. Initiation of Lipid Peroxidation in Biological Systems. Crit. Rev. Food Sci. Nutr. 1987, 25, 317–364. [Google Scholar] [CrossRef] [PubMed]
- Southorn, P.A.; Powis, G. Free Radicals in Medicine. I. Chemical Nature and Biologic Reactions. Mayo Clin. Proc. 1988, 63, 381–389. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, F.B.; Segal, A.W. NADPH Oxidase and the Respiratory Burst. Semin. Cell Biol. 1995, 6, 357–365. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O−2 Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Gebicki, J.M.; Bielski, B.H.J. Comparison of the Capacities of the Perhydroxyl and the Superoxide Radicals to Initiate Chain Oxidation of Linoleic Acid. J. Am. Chem. Soc. 1981, 103, 7020–7022. [Google Scholar] [CrossRef]
- De Grey, A.D.N.J. HO2*: The Forgotten Radical. DNA Cell Biol. 2002, 21, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Oxygen Toxicity, Oxygen Radicals, Transition Metals and Disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, M.; Wende, K.; Kupsch, S.; Neyts, E.C.; Reuter, S.; Bogaerts, A. Effect of Head Group and Lipid Tail Oxidation in the Cell Membrane Revealed through Integrated Simulations and Experiments. Sci. Rep. 2017, 7, 5761. [Google Scholar] [CrossRef]
- Sathappa, M.; Alder, N.N. The Ionization Properties of Cardiolipin and Its Variants in Model Bilayers. Biochim. Biophys. Acta 2016, 1858, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Haines, T.H.; Dencher, N.A. Cardiolipin: A Proton Trap for Oxidative Phosphorylation. FEBS Lett. 2002, 528, 35–39. [Google Scholar] [CrossRef]
- Van den Brink-van der Laan, E.; Killian, J.A.; de Kruijff, B. Nonbilayer Lipids Affect Peripheral and Integral Membrane Proteins via Changes in the Lateral Pressure Profile. Biochim. Biophys. Acta 2004, 1666, 275–288. [Google Scholar] [CrossRef]
- Khalifat, N.; Puff, N.; Bonneau, S.; Fournier, J.-B.; Angelova, M.I. Membrane Deformation under Local pH Gradient: Mimicking Mitochondrial Cristae Dynamics. Biophys. J. 2008, 95, 4924–4933. [Google Scholar] [CrossRef]
- Parui, P.P.; Sarakar, Y.; Majumder, R.; Das, S.; Yang, H.; Yasuhara, K.; Hirota, S. Determination of Proton Concentration at Cardiolipin-Containing Membrane Interfaces and Its Relation with the Peroxidase Activity of Cytochrome c. Chem. Sci. 2019, 10, 9140–9151. [Google Scholar] [CrossRef]
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. pH Difference across the Outer Mitochondrial Membrane Measured with a Green Fluorescent Protein Mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-Dependent Decline in the Cytochrome c Oxidase Activity in Rat Heart Mitochondria: Role of Cardiolipin. FEBS Lett. 1997, 406, 136–138. [Google Scholar] [CrossRef]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative Damage to Cardiac Mitochondria: Cytochrome Oxidase and Cardiolipin Alterations. FEBS Lett. 1998, 424, 155–158. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Generated by the Mitochondrial Respiratory Chain Affect the Complex III Activity via Cardiolipin Peroxidation in Beef-Heart Submitochondrial Particles. Mitochondrion 2001, 1, 151–159. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive Oxygen Species Generated from the Mitochondrial Electron Transport Chain Induce Cytochrome c Dissociation from Beef-Heart Submitochondrial Particles via Cardiolipin Peroxidation. Possible Role in the Apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Affect Mitochondrial Electron Transport Complex I Activity through Oxidative Cardiolipin Damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of Cardiolipin Peroxidation and Ca2+ in Mitochondrial Dysfunction and Disease. Cell Calcium. 2009, 45, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Arnarez, C.; Mazat, J.-P.; Elezgaray, J.; Marrink, S.-J.; Periole, X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. [Google Scholar] [CrossRef]
- Panov, A. Perhydroxyl Radical (HO2•) as Inducer of the Isoprostane Lipid Peroxidation in Mitochondria. Mol. Biol. 2018, 52, 295–305. [Google Scholar] [CrossRef]
- Miranda, É.G.A.; Araujo-Chaves, J.C.; Kawai, C.; Brito, A.M.M.; Dias, I.W.R.; Arantes, J.T.; Nantes-Cardoso, I.L. Cardiolipin Structure and Oxidation Are Affected by Ca2+ at the Interface of Lipid Bilayers. Front. Chem. 2019, 7, 930. [Google Scholar] [CrossRef] [PubMed]
- Cipolla-Neto, J.; Amaral, F.G.; Afeche, S.C.; Tan, D.X.; Reiter, R.J. Melatonin, Energy Metabolism, and Obesity: A Review. J. Pineal. Res. 2014, 56, 371–381. [Google Scholar] [CrossRef]
- Sustarsic, E.G.; Ma, T.; Lynes, M.D.; Larsen, M.; Karavaeva, I.; Havelund, J.F.; Nielsen, C.H.; Jedrychowski, M.P.; Moreno-Torres, M.; Lundh, M.; et al. Cardiolipin Synthesis in Brown and Beige Fat Mitochondria Is Essential for Systemic Energy Homeostasis. Cell Metab. 2018, 28, 159–174.e11. [Google Scholar] [CrossRef]
- Von Bank, H.; Hurtado-Thiele, M.; Oshimura, N.; Simcox, J. Mitochondrial Lipid Signaling and Adaptive Thermogenesis. Metabolites 2021, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Willers, C.; Kunji, E.R.S.; Crichton, P.G. Uncoupling Protein 1 Binds One Nucleotide per Monomer and Is Stabilized by Tightly Bound Cardiolipin. Proc. Natl. Acad. Sci. USA 2015, 112, 6973–6978. [Google Scholar] [CrossRef]
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin Increases Brown Adipose Tissue Mass and Function in Zücker Diabetic Fatty Rats: Implications for Obesity Control. J. Pineal. Res. 2018, 64, e12472. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin Increases the Activity of the Oxidative Phosphorylation Enzymes and the Production of ATP in Rat Brain and Liver Mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357. [Google Scholar] [CrossRef]
- Chen, X.; Hao, B.; Li, D.; Reiter, R.J.; Bai, Y.; Abay, B.; Chen, G.; Lin, S.; Zheng, T.; Ren, Y.; et al. Melatonin Inhibits Lung Cancer Development by Reversing the Warburg Effect via Stimulating the SIRT3/PDH Axis. J. Pineal. Res. 2021, e12755. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Rosales-Corral, S. Anti-Warburg Effect of Melatonin: A Proposed Mechanism to Explain Its Inhibition of Multiple Diseases. Int. J. Mol. Sci. 2021, 22, 764. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rorsales-Corral, S.; de Almeida Chuffa, L.G. Melatonin Inhibits Warburg-Dependent Cancer by Redirecting Glucose Oxidation to the Mitochondria: A Mechanistic Hypothesis. Cell. Mol. Life Sci. 2020, 77, 2527–2542. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Pires de Campos Zuccari, D.A.; de Almeida Chuffa, L.G.; Manucha, W.; Rodriguez, C. Melatonin Synthesis in and Uptake by Mitochondria: Implications for Diseased Cells with Dysfunctional Mitochondria. Future Med. Chem. 2021, 13, 335–339. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, S.; Zeng, S.; Zhao, Y.; Zhu, C.; Deng, B.; Zhu, G.; Yin, Y.; Wang, W.; Hardeland, R.; et al. Melatonin in Macrophage Biology: Current Understanding and Future Perspectives. J. Pineal. Res. 2019, 66, e12547. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q. Switching Diseased Cells from Cytosolic Aerobic Glycolysis to Mitochondrial Oxidative Phosphorylation: A Metabolic Rhythm Regulated by Melatonin? J. Pineal. Res. 2021, 70, e12677. [Google Scholar] [CrossRef]
- Fuller, G.G.; Han, T.; Freeberg, M.A.; Moresco, J.J.; Ghanbari Niaki, A.; Roach, N.P.; Yates, J.R.; Myong, S.; Kim, J.K. RNA Promotes Phase Separation of Glycolysis Enzymes into Yeast G Bodies in Hypoxia. Elife 2020, 9, e48480. [Google Scholar] [CrossRef]
- Jin, M.; Fuller, G.G.; Han, T.; Yao, Y.; Alessi, A.F.; Freeberg, M.A.; Roach, N.P.; Moresco, J.J.; Karnovsky, A.; Baba, M.; et al. Glycolytic Enzymes Coalesce in G Bodies under Hypoxic Stress. Cell Rep. 2017, 20, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mondal, J. Mechanistic Insights on ATP’s Role as a Hydrotrope. J. Phys. Chem. B 2021, 125, 7717–7731. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Lemasters, J.J. ATP/ADP Ratio, the Missed Connection between Mitochondria and the Warburg Effect. Mitochondrion 2014, 19 Pt A, 78–84. [Google Scholar] [CrossRef]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924. [Google Scholar] [CrossRef]
- Lu, J.; Qian, J.; Xu, Z.; Yin, S.; Zhou, L.; Zheng, S.; Zhang, W. Emerging Roles of Liquid-Liquid Phase Separation in Cancer: From Protein Aggregation to Immune-Associated Signaling. Front. Cell Dev. Biol. 2021, 9, 631486. [Google Scholar] [CrossRef] [PubMed]
- Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Passos, Y.M.; Mota, M.F.; Jakobus, B.; de Sousa, G.D.S.; Pereira da Costa, F.; Felix, A.L.; Ferretti, G.D.S.; et al. Phase Separation of p53 Precedes Aggregation and Is Affected by Oncogenic Mutations and Ligands. Chem. Sci. 2021, 12, 7334–7349. [Google Scholar] [CrossRef] [PubMed]
- Gargini, R.; Segura-Collar, B.; Sánchez-Gómez, P. Novel Functions of the Neurodegenerative-Related Gene Tau in Cancer. Front. Aging Neurosci. 2019, 11, 231. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The Involvement of Tau in Nucleolar Transcription and the Stress Response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Reddi, P.P. Transcription and Splicing Factor TDP-43: Role in Regulation of Gene Expression in Testis. Semin. Reprod. Med. 2017, 35, 167–172. [Google Scholar] [CrossRef]
- Morera, A.A.; Ahmed, N.S.; Schwartz, J.C. TDP-43 Regulates Transcription at Protein-Coding Genes and Alu Retrotransposons. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194434. [Google Scholar] [CrossRef]
- Yang, L.; Gal, J.; Chen, J.; Zhu, H. Self-Assembled FUS Binds Active Chromatin and Regulates Gene Transcription. Proc. Natl. Acad. Sci. USA 2014, 111, 17809–17814. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Organization and Regulation of Gene Transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Pappu, R.V. Conformations of Intrinsically Disordered Proteins Are Influenced by Linear Sequence Distributions of Oppositely Charged Residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shi, X.; Wang, X. RNA and Liquid-Liquid Phase Separation. Noncoding RNA Res. 2021, 6, 92–99. [Google Scholar] [CrossRef]
- Fay, M.M.; Anderson, P.J. The Role of RNA in Biological Phase Separations. J. Mol. Biol. 2018, 430, 4685–4701. [Google Scholar] [CrossRef] [PubMed]
- Roden, C.; Gladfelter, A.S. RNA Contributions to the Form and Function of Biomolecular Condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Conn, G.L.; Gittis, A.G.; Lattman, E.E.; Misra, V.K.; Draper, D.E. A Compact RNA Tertiary Structure Contains a Buried Backbone-K+ Complex. J. Mol. Biol. 2002, 318, 963–973. [Google Scholar] [CrossRef]
- Drobot, B.; Iglesias-Artola, J.M.; Le Vay, K.; Mayr, V.; Kar, M.; Kreysing, M.; Mutschler, H.; Tang, T.-Y.D. Compartmentalised RNA Catalysis in Membrane-Free Coacervate Protocells. Nat. Commun. 2018, 9, 3643. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.A.; Bevilacqua, P.C.; Keating, C.D. Polyamine/Nucleotide Coacervates Provide Strong Compartmentalization of Mg2+, Nucleotides, and RNA. Langmuir 2016, 32, 2041–2049. [Google Scholar] [CrossRef]
- Kirschbaum, J.; Zwicker, D. Controlling Biomolecular Condensates via Chemical Reactions. arXiv 2021, arXiv:2103.02921. [Google Scholar]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Zwicker, D.; Sohrabi-Jahromi, S.; Boehning, M.; Kirschbaum, J. Mechanisms for Active Regulation of Biomolecular Condensates. Trends Cell Biol. 2020, 30, 4–14. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer Physics of Intracellular Phase Transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Schisa, J.A.; Elaswad, M.T. An Emerging Role for Post-Translational Modifications in Regulating RNP Condensates in the Germ Line. Frontiers in Molecular Biosciences 2021, 8, 230. [Google Scholar] [CrossRef]
- Hofweber, M.; Dormann, D. Friend or Foe-Post-Translational Modifications as Regulators of Phase Separation and RNP Granule Dynamics. J. Biol. Chem. 2019, 294, 7137–7150. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.; Fukuda, H.; Imajoh-Ohmi, S.; Saito, H.; Takekawa, M. Formation of Stress Granules Inhibits Apoptosis by Suppressing Stress-Responsive MAPK Pathways. Nat. Cell Biol. 2008, 10, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Advani, V.M.; Ivanov, P. Stress Granule Subtypes: An Emerging Link to Neurodegeneration. Cell. Mol. Life Sci. 2020, 77, 4827–4845. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA Self-Assembly Contributes to Stress Granule Formation and Defining the Stress Granule Transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef]
- Gaete-Argel, A.; Velásquez, F.; Márquez, C.L.; Rojas-Araya, B.; Bueno-Nieto, C.; Marín-Rojas, J.; Cuevas-Zúñiga, M.; Soto-Rifo, R.; Valiente-Echeverría, F. Tellurite Promotes Stress Granules and Nuclear SG-Like Assembly in Response to Oxidative Stress and DNA Damage. Front. Cell Dev. Biol. 2021, 9, 622057. [Google Scholar] [CrossRef]
- Lian, X.J.; Gallouzi, I.-E. Oxidative Stress Increases the Number of Stress Granules in Senescent Cells and Triggers a Rapid Decrease in p21waf1/cip1 Translation. J. Biol. Chem. 2009, 284, 8877–8887. [Google Scholar] [CrossRef]
- Curdy, N.; Lanvin, O.; Cadot, S.; Laurent, C.; Fournié, J.-J.; Franchini, D.-M. Stress Granules in the Post-Transcriptional Regulation of Immune Cells. Front. Cell Dev. Biol. 2020, 8, 611185. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins in Overcrowded Milieu: Membrane-Less Organelles, Phase Separation, and Intrinsic Disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of Tumor Suppressor p53 and Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress Granule Assembly Is Mediated by Prion-like Aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.J.; Wolozin, B. Pathological Stress Granules in Alzheimer’s Disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S. Matter over Mind: Liquid Phase Separation and Neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef]
- Santofimia-Castaño, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velázquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting Intrinsically Disordered Proteins Involved in Cancer. Cell. Mol. Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef]
- Wolozin, B. Regulated Protein Aggregation: Stress Granules and Neurodegeneration. Mol. Neurodegener. 2012, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Brown, C.J.; Dunker, A.K.; Uversky, V.N. Intrinsically Disordered Regions of p53 Family Are Highly Diversified in Evolution. Biochim. Biophys. Acta 2013, 1834, 725–738. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Lei, S.; Shattuck, J.; Boudeau, S.; Carlomagno, Y.; Medalla, M.; Mashimo, B.L.; Socorro, G.; Al-Mohanna, L.F.A.; Jiang, L.; et al. TIA1 Potentiates Tau Phase Separation and Promotes Generation of Toxic Oligomeric Tau. Proc. Natl. Acad. Sci. USA 2021, 118, e2014188118. [Google Scholar] [CrossRef]
- Asakawa, K.; Handa, H.; Kawakami, K. Optogenetic Modulation of TDP-43 Oligomerization Accelerates ALS-Related Pathologies in the Spinal Motor Neurons. Nat. Commun. 2020, 11, 1004. [Google Scholar] [CrossRef]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically Disordered Sequences Enable Modulation of Protein Phase Separation through Distributed Tyrosine Motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef]
- Jeon, P.; Lee, J.A. Dr. Jekyll and Mr. Hyde? Physiology and Pathology of Neuronal Stress Granules. Front Cell Dev. Biol. 2021, 9, 609698. [Google Scholar] [CrossRef] [PubMed]
- Farny, N.G.; Kedersha, N.L.; Silver, P.A. Metazoan Stress Granule Assembly Is Mediated by P-eIF2alpha-Dependent and -Independent Mechanisms. RNA 2009, 15, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Yu, H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting Pathology of the Stress Granule Proteins TIA-1 and G3BP in Tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef]
- Jenkins, S.M.; Zinnerman, M.; Garner, C.; Johnson, G.V. Modulation of Tau Phosphorylation and Intracellular Localization by Cellular Stress. Biochem. J. 2000, 345 Pt 2, 263–270. [Google Scholar] [CrossRef]
- Cruz, A.; Verma, M.; Wolozin, B. The Pathophysiology of Tau and Stress Granules in Disease. In Tau Biology; Takashima, A., Wolozin, B., Buee, L., Eds.; Springer: Singapore, 2019; pp. 359–372. [Google Scholar] [CrossRef]
- Dobra, I.; Pankivskyi, S.; Samsonova, A.; Pastre, D.; Hamon, L. Relation Between Stress Granules and Cytoplasmic Protein Aggregates Linked to Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2018, 18, 107. [Google Scholar] [CrossRef]
- Yoshida, Y.; Izumi, H.; Torigoe, T.; Ishiguchi, H.; Yoshida, T.; Itoh, H.; Kohno, K. Binding of RNA to p53 Regulates Its Oligomerization and DNA-Binding Activity. Oncogene 2004, 23, 4371–4379. [Google Scholar] [CrossRef]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA Buffers the Phase Separation Behavior of Prion-like RNA Binding Proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules That Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef]
- Kovachev, P.S.; Banerjee, D.; Rangel, L.P.; Eriksson, J.; Pedrote, M.M.; Martins-Dinis, M.M.D.C.; Edwards, K.; Cordeiro, Y.; Silva, J.L.; Sanyal, S. Distinct Modulatory Role of RNA in the Aggregation of the Tumor Suppressor Protein p53 Core Domain. J. Biol. Chem. 2017, 292, 9345–9357. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.S.; Wang, Z.; Tailor, K.; Kolomeisky, A.B.; Conrad, J.C.; Vekilov, P.G. Anomalous Dense Liquid Condensates Host the Nucleation of Tumor Suppressor p53 Fibrils. Science 2019, 12, 342–355. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-Mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef]
- Ding, Q.; Chaplin, J.; Morris, M.J.; Hilliard, M.A.; Wolvetang, E.; Ng, D.C.H.; Noakes, P.G. TDP-43 Mutation Affects Stress Granule Dynamics in Differentiated NSC-34 Motoneuron-Like Cells. Front. Cell Dev. Biol. 2021, 9, 611601. [Google Scholar] [CrossRef]
- Aulas, A.; Vande Velde, C. Alterations in Stress Granule Dynamics Driven by TDP-43 and FUS: A Link to Pathological Inclusions in ALS? Front. Cell Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.K.; Chian, R.-J.; Boggio, K.J.; Quaresma, A.J.C.; Nickerson, J.A.; Bosco, D.A. Amyotrophic Lateral Sclerosis-Linked FUS/TLS Alters Stress Granule Assembly and Dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, J.; De Santis, R.; de Turris, V.; Morlando, M.; Laneve, P.; Calvo, A.; Caliendo, V.; Chiò, A.; Rosa, A.; Bozzoni, I. ALS Mutant FUS Proteins Are Recruited into Stress Granules in Induced Pluripotent Stem Cell-Derived Motoneurons. Dis. Model. Mech. 2015, 8, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Vande Velde, C. TDP-43 Regulation of Stress Granule Dynamics in Neurodegenerative Disease-Relevant Cell Types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA Binding Protein-43 (TDP-43) Associates with Stress Granules: Analysis of Cultured Cells and Pathological Brain Tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular Mechanisms of Disease-Causing Missense Mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [PubMed]
- Kolonko-Adamska, M.; Uversky, V.N.; Greb-Markiewicz, B. The Participation of the Intrinsically Disordered Regions of the bHLH-PAS Transcription Factors in Disease Development. Int. J. Mol. Sci. 2021, 22, 2868. [Google Scholar] [CrossRef] [PubMed]
- Portz, B.; Lee, B.L.; Shorter, J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem. Sci. 2021, 46, 550–563. [Google Scholar] [CrossRef]
- Tsang, B.; Pritišanac, I.; Scherer, S.W.; Moses, A.M.; Forman-Kay, J.D. Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 2020, 183, 1742–1756. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS Low-Complexity Domain Disrupts Phase Separation, Aggregation, and Toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L.F. Amyotrophic Lateral Sclerosis-Linked Mutations Increase the Viscosity of Liquid-like TDP-43 RNP Granules in Neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–E2475. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.-D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- King, O.D.; Gitler, A.D.; Shorter, J. The Tip of the Iceberg: RNA-Binding Proteins with Prion-like Domains in Neurodegenerative Disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef]
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016, 14, e1002338. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 α-Helical Structure Tunes Liquid-Liquid Phase Separation and Function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-L.; Zhao, J.; Yin, X.-F.; He, W.-T.; Yang, H.; Che, M.-X.; Hu, H.-Y. Two Mutations G335D and Q343R within the Amyloidogenic Core Region of TDP-43 Influence Its Aggregation and Inclusion Formation. Sci. Rep. 2016, 6, 23928. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Lee, V.M.-Y.; Trojanowski, J.Q. Mutations in TDP-43 Link Glycine-Rich Domain Functions to Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2009, 18, R156–R162. [Google Scholar] [CrossRef]
- Genc, S.; Kurnaz, I.A.; Ozilgen, M. Astrocyte-Neuron Lactate Shuttle May Boost More ATP Supply to the Neuron under Hypoxic Conditions--in Silico Study Supported by in Vitro Expression Data. BMC Syst. Biol. 2011, 5, 162. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct Responses of Neurons and Astrocytes to TDP-43 Proteinopathy in Amyotrophic Lateral Sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS Disease Protein TDP-43 Is Actively Transported in Motor Neuron Axons and Regulates Axon Outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef]
- Dang, M.; Kang, J.; Lim, L.; Li, Y.; Wang, L.; Song, J. ATP Is a Cryptic Binder of TDP-43 RRM Domains to Enhance Stability and Inhibit ALS/AD-Associated Fibrillation. Biochem. Biophys. Res. Commun. 2020, 522, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Sorarù, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; et al. Mutations of FUS Gene in Sporadic Amyotrophic Lateral Sclerosis. J. Med. Genet. 2010, 47, 190–194. [Google Scholar] [CrossRef]
- Ticozzi, N.; Silani, V.; LeClerc, A.L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T.J.; McKenna-Yasek, D.M.; Sapp, P.C.; et al. Analysis of FUS Gene Mutation in Familial Amyotrophic Lateral Sclerosis within an Italian Cohort. Neurology 2009, 73, 1180–1185. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS from DNA Repair to Stress Response: Implications for ALS. ASN Neuro 2014, 6, 1759091414544472. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, T.; Wang, H.; Wang, T.; Qin, M.; Bao, P.; Wang, R.; Liu, Y.; Chang, H.-C.; Yan, J.; et al. Neurodegeneration-Associated FUS Is a Novel Regulator of Circadian Gene Expression. Transl. Neurodegener. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS Mutations of FUS Suppress Protein Translation and Disrupt the Regulation of Nonsense-Mediated Decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Tran, S.E.F.; Eriksson, J.E.; Sistonen, L. Multisite Phosphorylation Provides Sophisticated Regulation of Transcription Factors. Trends Biochem. Sci. 2002, 27, 619–627. [Google Scholar] [CrossRef]
- Filtz, T.M.; Vogel, W.K.; Leid, M. Regulation of Transcription Factor Activity by Interconnected Post-Translational Modifications. Trends Pharmacol. Sci. 2014, 35, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J.; Davis, R.J. Regulation of Transcription Factor Function by Phosphorylation. Cell. Mol. Life Sci. 2000, 57, 1172–1183. [Google Scholar] [CrossRef]
- Sotthibundhu, A.; Ekthuwapranee, K.; Govitrapong, P. Comparison of Melatonin with Growth Factors in Promoting Precursor Cells Proliferation in Adult Mouse Subventricular Zone. EXCLI J. 2016, 15, 829–841. [Google Scholar] [CrossRef]
- Onaolapo, O.J.; Onaolapo, A.Y.; Olowe, O.A.; Udoh, M.O.; Udoh, D.O.; Nathaniel, T.I. Melatonin and Melatonergic Influence on Neuronal Transcription Factors: Implications for the Development of Novel Therapies for Neurodegenerative Disorders. Curr. Neuropharmacol. 2020, 18, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, W.M.; Keating, C.D. Phosphorylation-Mediated RNA/peptide Complex Coacervation as a Model for Intracellular Liquid Organelles. Nat. Chem. 2016, 8, 129–137. [Google Scholar] [CrossRef]
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like Droplet Formation by Tumor Suppressor p53 Induced by Multivalent Electrostatic Interactions between Two Disordered Domains. Sci. Rep. 2020, 10, 580. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Zhou, H.-X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Milovanovic, D.; Wu, Y.; Bian, X.; De Camilli, P. A Liquid Phase of Synapsin and Lipid Vesicles. Science 2018, 361, 604–607. [Google Scholar] [CrossRef]
- Beutel, O.; Maraspini, R.; Pombo-García, K.; Martin-Lemaitre, C.; Honigmann, A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936.e11. [Google Scholar] [CrossRef]
- Wang, J.T.; Smith, J.; Chen, B.-C.; Schmidt, H.; Rasoloson, D.; Paix, A.; Lambrus, B.G.; Calidas, D.; Betzig, E.; Seydoux, G. Regulation of RNA Granule Dynamics by Phosphorylation of Serine-Rich, Intrinsically Disordered Proteins in C. Elegans. Elife 2014, 3, e04591. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.A.; Wessel, G.M. DEAD-Box Helicases: Posttranslational Regulation and Function. Biochem. Biophys. Res. Commun. 2010, 395, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-Box Helicase DDX3X Is a Critical Component of the TANK-Binding Kinase 1-Dependent Innate Immune Response. EMBO J. 2008, 27, 2135–2146. [Google Scholar] [CrossRef]
- Ron, D. Translational Control in the Endoplasmic Reticulum Stress Response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with Stress: eIF2 Kinases and Translational Control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α Kinases: Their Structures and Functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The Small Molecule ISRIB Reverses the Effects of eIF2α Phosphorylation on Translation and Stress Granule Assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct Stages in Stress Granule Assembly and Disassembly. Elife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Visibly Stressed: The Role of eIF2, TIA-1, and Stress Granules in Protein Translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Mateju, D.; Eichenberger, B.; Voigt, F.; Eglinger, J.; Roth, G.; Chao, J.A. Single-Molecule Imaging Reveals Translation of mRNAs Localized to Stress Granules. Cell 2020, 183, 1801–1812.e13. [Google Scholar] [CrossRef]
- Baumann, K. mRNA Translation in Stress Granules Is Not Uncommon. Nat. Rev. Mol. Cell Biol. 2021, 22, 164. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032813. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback Inhibition of the Unfolded Protein Response by GADD34-Mediated Dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.-E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of Ribosome Recruitment Induces Stress Granule Formation Independently of Eukaryotic Initiation Factor 2alpha Phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation Reinitiation at Alternative Open Reading Frames Regulates Gene Expression in an Integrated Stress Response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Chen, X.; Chen, M.; Schafer, N.P.; Wolynes, P.G. Exploring the Interplay between Fibrillization and Amorphous Aggregation Channels on the Energy Landscapes of Tau Repeat Isoforms. Proc. Natl. Acad. Sci. USA 2020, 117, 4125–4130. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging. Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau Stabilizes Microtubules by Binding at the Interface between Tubulin Heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.V.; Hartigan, J.A. Tau Protein in Normal and Alzheimer’s Disease Brain: An Update. J. Alzheimers. Dis. 1999, 1, 329–351. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-Liquid Phase Separation of the Microtubule-Binding Repeats of the Alzheimer-Related Protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-Liquid Phase Separation Induces Pathogenic Tau Conformations in Vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The Physiological Roles of Tau and Aβ: Implications for Alzheimer’s Disease Pathology and Therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Ferrer, I.; Andrés-Benito, P.; Zelaya, M.V.; Aguirre, M.E.E.; Carmona, M.; Ausín, K.; Lachén-Montes, M.; Fernández-Irigoyen, J.; Santamaría, E.; Del Rio, J.A. Familial Globular Glial Tauopathy Linked to MAPT Mutations: Molecular Neuropathology and Seeding Capacity of a Prototypical Mixed Neuronal and Glial Tauopathy. Acta Neuropathol. 2020, 139, 735–771. [Google Scholar] [CrossRef]
- McAleese, K.E.; Firbank, M.; Dey, M.; Colloby, S.J.; Walker, L.; Johnson, M.; Beverley, J.R.; Taylor, J.P.; Thomas, A.J.; O’Brien, J.T.; et al. Cortical Tau Load Is Associated with White Matter Hyperintensities. Acta Neuropathol. Commun. 2015, 3, 60. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Moon, S.; Zhang, Q.; Volitakis, I.; Finkelstein, D.I.; Bush, A.I. Motor and Cognitive Deficits in Aged Tau Knockout Mice in Two Background Strains. Mol. Neurodegener. 2014, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ferreira, E.; Tran, A.; Turner, E.C.; Belfiore, R.; Branca, C.; Oddo, S. Acute Tau Knockdown in the Hippocampus of Adult Mice Causes Learning and Memory Deficits. Aging Cell 2018, 17, e12775. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced Tau Protein Translation by Hyper-Excitation. Front. Aging. Neurosci. 2019, 11, 322. [Google Scholar] [CrossRef]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau Deletion Promotes Brain Insulin Resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef]
- Wijesekara, N.; Gonçalves, R.A.; Ahrens, R.; De Felice, F.G.; Fraser, P.E. Tau Ablation in Mice Leads to Pancreatic β Cell Dysfunction and Glucose Intolerance. FASEB J. 2018, 32, 3166–3173. [Google Scholar] [CrossRef]
- Adams, J.N.; Lockhart, S.N.; Li, L.; Jagust, W.J. Relationships Between Tau and Glucose Metabolism Reflect Alzheimer’s Disease Pathology in Cognitively Normal Older Adults. Cereb. Cortex 2019, 29, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a Key Player in Neuronal DNA Protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Savastano, A.; Singh, P.; Mukhopadhyay, S.; Zweckstetter, M. Liquid-Liquid Phase Separation of Tau: From Molecular Biophysics to Physiology and Disease. Protein Sci. 2021, 30, 1294–1314. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of Tau, a Microtubule-Associated Protein That Induces Assembly of Microtubules from Purified Tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic Instability of Microtubule Growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Panda, D.; Daijo, J.E.; Jordan, M.A.; Wilson, L. Kinetic Stabilization of Microtubule Dynamics at Steady State in Vitro by Substoichiometric Concentrations of Tubulin-Colchicine Complex. Biochemistry 1995, 34, 9921–9929. [Google Scholar] [CrossRef]
- Qiang, L.; Yu, W.; Andreadis, A.; Luo, M.; Baas, P.W. Tau Protects Microtubules in the Axon from Severing by Katanin. J. Neurosci. 2006, 26, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Boyko, S.; Surewicz, K.; Surewicz, W.K. Regulatory Mechanisms of Tau Protein Fibrillation under the Conditions of Liquid-Liquid Phase Separation. Proc. Natl. Acad. Sci. USA 2020, 117, 31882–31890. [Google Scholar] [CrossRef]
- Liu, M.; Dexheimer, T.; Sui, D.; Hovde, S.; Deng, X.; Kwok, R.; Bochar, D.A.; Kuo, M.-H. Hyperphosphorylated Tau Aggregation and Cytotoxicity Modulators Screen Identified Prescription Drugs Linked to Alzheimer’s Disease and Cognitive Functions. Sci. Rep. 2020, 10, 16551. [Google Scholar] [CrossRef]
- Brunello, C.A.; Merezhko, M.; Uronen, R.-L.; Huttunen, H.J. Mechanisms of Secretion and Spreading of Pathological Tau Protein. Cell Mol. Life Sci. 2020, 77, 1721–1744. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fichou, Y.; Zeng, Z.; Hu, N.Y.; Han, S. Electrostatically Driven Complex Coacervation and Amyloid Aggregation of Tau Are Independent Processes with Overlapping Conditions. ACS Chem. Neurosci. 2020, 11, 615–627. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. RNA Stores Tau Reversibly in Complex Coacervates. PLoS Biol. 2017, 15, e2002183. [Google Scholar] [CrossRef]
- Boyko, S.; Qi, X.; Chen, T.-H.; Surewicz, K.; Surewicz, W.K. Liquid-Liquid Phase Separation of Tau Protein: The Crucial Role of Electrostatic Interactions. J. Biol. Chem. 2019, 294, 11054–11059. [Google Scholar] [CrossRef]
- Lin, Y.; McCarty, J.; Rauch, J.N.; Delaney, K.T.; Kosik, K.S.; Fredrickson, G.H.; Shea, J.-E.; Han, S. Narrow Equilibrium Window for Complex Coacervation of Tau and RNA under Cellular Conditions. Elife 2019, 8, e42571. [Google Scholar] [CrossRef]
- Lin, Y.; Fichou, Y.; Longhini, A.P.; Llanes, L.C.; Yin, P.; Bazan, G.C.; Kosik, K.S.; Han, S. Liquid-Liquid Phase Separation of Tau Driven by Hydrophobic Interaction Facilitates Fibrillization of Tau. J. Mol. Biol. 2021, 433, 166731. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Ruben, G.C.; Ciardelli, T.L.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer Disease Hyperphosphorylated Tau Aggregates Hydrophobically. Synapse 1997, 27, 208–229. [Google Scholar] [CrossRef]
- Takashima, A. Hyperphosphorylated Tau Is a Cause of Neuronal Dysfunction in Tauopathy. J. Alzheimers. Dis. 2008, 14, 371–375. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef]
- Wang, J.Z.; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer Paired Helical Filaments by Protein Phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Restoration of Biological Activity of Alzheimer Abnormally Phosphorylated Tau by Dephosphorylation with Protein Phosphatase-2A, -2B and -1. Brain Res. Mol. Brain Res. 1996, 38, 200–208. [Google Scholar] [CrossRef]
- Poppek, D.; Keck, S.; Ermak, G.; Jung, T.; Stolzing, A.; Ullrich, O.; Davies, K.J.A.; Grune, T. Phosphorylation Inhibits Turnover of the Tau Protein by the Proteasome: Influence of RCAN1 and Oxidative Stress. Biochem. J. 2006, 400, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-Associated Protein Tau. Abnormal Phosphorylation of a Non-Paired Helical Filament Pool in Alzheimer Disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Khatoon, S.; Grundke-Iqbal, I.; Iqbal, K. Brain Levels of Microtubule-Associated Protein Tau Are Elevated in Alzheimer’s Disease: A Radioimmuno-Slot-Blot Assay for Nanograms of the Protein. J. Neurochem. 1992, 59, 750–753. [Google Scholar] [CrossRef]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Ait-Bouziad, N.; Chiki, A.; Limorenko, G.; Xiao, S.; Eliezer, D.; Lashuel, H.A. Phosphorylation of the Overlooked Tyrosine 310 Regulates the Structure, Aggregation, and Microtubule- and Lipid-Binding Properties of Tau. J. Biol. Chem. 2020, 295, 7905–7922. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of Abnormally Phosphorylated Tau Precedes the Formation of Neurofibrillary Tangles in Alzheimer’s Disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef]
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear Magnetic Resonance Spectroscopy Characterization of Interaction of Tau with DNA and Its Regulation by Phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R.-Q. Tau Could Protect DNA Double Helix Structure. Biochim. Biophys. Acta 2003, 1645, 205–211. [Google Scholar] [CrossRef]
- Castellani, R.J.; Nunomura, A.; Lee, H.-G.; Perry, G.; Smith, M.A. Phosphorylated Tau: Toxic, Protective, or None of the above. J. Alzheimers. Dis. 2008, 14, 377–383. [Google Scholar] [CrossRef]
- Luna-Muñoz, J.; Harrington, C.R.; Wischik, C.M.; Flores-Rodríguez, P.; Avila, J.; Zamudio, S.R.; la Cruz, F.D.; Mena, R.; Meraz-Ríos, M.A.; Floran-Garduño, B. Phosphorylation of Tau Protein Associated as a Protective Mechanism in the Presence of Toxic, C-Terminally Truncated Tau in Alzheimer’s Disease. In Understanding Alzheimer’s Disease; Zerr, I., Ed.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Castellani, R.J.; Honda, K.; Zhu, X.; Cash, A.D.; Nunomura, A.; Perry, G.; Smith, M.A. Contribution of Redox-Active Iron and Copper to Oxidative Damage in Alzheimer Disease. Ageing. Res. Rev. 2004, 3, 319–326. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron Accumulation in Alzheimer Disease Is a Source of Redox-Generated Free Radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Sayre, L.M.; Perry, G.; Harris, P.L.; Liu, Y.; Schubert, K.A.; Smith, M.A. In Situ Oxidative Catalysis by Neurofibrillary Tangles and Senile Plaques in Alzheimer’s Disease: A Central Role for Bound Transition Metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef]
- Smith, M.A.; Casadesus, G.; Joseph, J.A.; Perry, G. Amyloid-β and τ Serve Antioxidant Functions in the Aging and Alzheimer Brain. Free. Radic. Biol. Med. 2002, 33, 1194–1199. [Google Scholar] [CrossRef]
- Li, H.-L.; Wang, H.-H.; Liu, S.-J.; Deng, Y.-Q.; Zhang, Y.-J.; Tian, Q.; Wang, X.-C.; Chen, X.-Q.; Yang, Y.; Zhang, J.-Y.; et al. Phosphorylation of Tau Antagonizes Apoptosis by Stabilizing Beta-Catenin, a Mechanism Involved in Alzheimer’s Neurodegeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 3591–3596. [Google Scholar] [CrossRef]
- Wang, H.-H.; Li, H.-L.; Liu, R.; Zhang, Y.; Liao, K.; Wang, Q.; Wang, J.-Z.; Liu, S.-J. Tau Overexpression Inhibits Cell Apoptosis with the Mechanisms Involving Multiple Viability-Related Factors. J. Alzheimers. Dis. 2010, 21, 167–179. [Google Scholar] [CrossRef]
- Haj-Yahya, M.; Gopinath, P.; Rajasekhar, K.; Mirbaha, H.; Diamond, M.I.; Lashuel, H.A. Site-Specific Hyperphosphorylation Inhibits, Rather than Promotes, Tau Fibrillization, Seeding Capacity, and Its Microtubule Binding. Angew. Chem. Int. Ed Engl. 2020, 59, 4059–4067. [Google Scholar] [CrossRef]
- Amir Mishan, M.; Rezaei Kanavi, M.; Shahpasand, K.; Ahmadieh, H. Pathogenic Tau Protein Species: Promising Therapeutic Targets for Ocular Neurodegenerative Diseases. J. Ophthalmic Vis. Res. 2019, 14, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Shi, R.; Li, L.; Chen, F.; Zhou, Y.; Tung, Y.C.; Hu, W.; Gong, C.-X.; Iqbal, K.; Liu, F. Pathological Tau from Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in Vivo. Front. Aging Neurosci. 2019, 11, 34. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in Physiology and Pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Fath, T.; Eidenmüller, J.; Brandt, R. Tau-Mediated Cytotoxicity in a Pseudohyperphosphorylation Model of Alzheimer’s Disease. J. Neurosci. 2002, 22, 9733–9741. [Google Scholar] [CrossRef]
- Di, J.; Cohen, L.S.; Corbo, C.P.; Phillips, G.R.; El Idrissi, A.; Alonso, A.D. Abnormal Tau Induces Cognitive Impairment through Two Different Mechanisms: Synaptic Dysfunction and Neuronal Loss. Sci. Rep. 2016, 6, 20833. [Google Scholar] [CrossRef]
- Tai, X.Y.; Koepp, M.; Duncan, J.S.; Fox, N.; Thompson, P.; Baxendale, S.; Liu, J.Y.W.; Reeves, C.; Michalak, Z.; Thom, M. Hyperphosphorylated Tau in Patients with Refractory Epilepsy Correlates with Cognitive Decline: A Study of Temporal Lobe Resections. Brain 2016, 139, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical Perspectives in Neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Srinivasan, V.; Pandi-Perumal, S.R.; Maestroni, G.J.; Esquifino, A.I.; Hardeland, R.; Cardinali, D.P. Role of Melatonin in Neurodegenerative Diseases. Neurotox. Res. 2005, 7, 293–318. [Google Scholar] [CrossRef]
- Kong, P.-J.; Byun, J.-S.; Lim, S.-Y.; Lee, J.-J.; Hong, S.-J.; Kwon, K.-J.; Kim, S.-S. Melatonin Induces Akt Phosphorylation through Melatonin Receptor- and PI3K-Dependent Pathways in Primary Astrocytes. Korean J. Physiol. Pharmacol. 2008, 12, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kilic, U.; Caglayan, A.B.; Beker, M.C.; Gunal, M.Y.; Caglayan, B.; Yalcin, E.; Kelestemur, T.; Gundogdu, R.Z.; Yulug, B.; Yılmaz, B.; et al. Particular Phosphorylation of PI3K/Akt on Thr308 via PDK-1 and PTEN Mediates Melatonin’s Neuroprotective Activity after Focal Cerebral Ischemia in Mice. Redox Biol. 2017, 12, 657–665. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and Tau: Two Convergence Points in Alzheimer’s Disease. J. Alzheimers. Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin Ameliorates Amyloid Beta-Induced Memory Deficits, Tau Hyperphosphorylation and Neurodegeneration via PI3/Akt/GSk3β Pathway in the Mouse Hippocampus. J. Pineal. Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Luengo, E.; Buendia, I.; Fernández-Mendívil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernández-García, B.; Sánchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological Doses of Melatonin Impede Cognitive Decline in Tau-Related Alzheimer Models, Once Tauopathy Is Initiated, by Restoring the Autophagic Flux. J. Pineal. Res. 2019, 67, e12578. [Google Scholar] [CrossRef]
- Shi, C.; Zeng, J.; Li, Z.; Chen, Q.; Hang, W.; Xia, L.; Wu, Y.; Chen, J.; Shi, A. Melatonin Mitigates Kainic Acid-Induced Neuronal Tau Hyperphosphorylation and Memory Deficits through Alleviating ER Stress. Front. Mol. Neurosci. 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Zhao, Y.-F.; Zu, H.-B. Melatonin Receptor Stimulation by Agomelatine Prevents Aβ-Induced Tau Phosphorylation and Oxidative Damage in PC12 Cells. Drug Des. Devel. Ther. 2019, 13, 387–396. [Google Scholar] [CrossRef]
- Heberle, A.M.; Razquin Navas, P.; Langelaar-Makkinje, M.; Kasack, K.; Sadik, A.; Faessler, E.; Hahn, U.; Marx-Stoelting, P.; Opitz, C.A.; Sers, C.; et al. The PI3K and MAPK/p38 Pathways Control Stress Granule Assembly in a Hierarchical Manner. Life Sci. Alliance 2019, 2, e201800257. [Google Scholar] [CrossRef]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Melatonin Reduces GSK3β-Mediated Tau Phosphorylation, Enhances Nrf2 Nuclear Translocation and Anti-Inflammation. ASN Neuro. 2020, 12, 1759091420981204. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Effect of Melatonin on Tau Aggregation and Tau-Mediated Cell Surface Morphology. Int. J. Biol. Macromol. 2020, 152, 30–39. [Google Scholar] [CrossRef]
- Schwalbe, M.; Kadavath, H.; Biernat, J.; Ozenne, V.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Structural Impact of Tau Phosphorylation at Threonine 231. Structure 2015, 23, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Raz, Y.; Miller, Y. Interactions between Aβ and Mutated Tau Lead to Polymorphism and Induce Aggregation of Aβ-Mutated Tau Oligomeric Complexes. PLoS ONE 2013, 8, e73303. [Google Scholar] [CrossRef]
- Skribanek, Z.; Baláspiri, L.; Mák, M. Interaction between Synthetic Amyloid-Beta-Peptide (1-40) and Its Aggregation Inhibitors Studied by Electrospray Ionization Mass Spectrometry. J. Mass Spectrom. 2001, 36, 1226–1229. [Google Scholar] [CrossRef]
- He, H.; Dong, W.; Huang, F. Anti-Amyloidogenic and Anti-Apoptotic Role of Melatonin in Alzheimer Disease. Curr. Neuropharmacol. 2010, 8, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, W.M.; Hansmann, U.H.E. Structure and Dynamics of Amyloid-β Segmental Polymorphisms. PLoS ONE 2012, 7, e41479. [Google Scholar] [CrossRef]
- Fraser, P.E.; Nguyen, J.T.; Surewicz, W.K.; Kirschner, D.A. pH-Dependent Structural Transitions of Alzheimer Amyloid Peptides. Biophys. J. 1991, 60, 1190–1201. [Google Scholar] [CrossRef]
- Huang, T.H.; Fraser, P.E.; Chakrabartty, A. Fibrillogenesis of Alzheimer Abeta Peptides Studied by Fluorescence Energy Transfer. J. Mol. Biol. 1997, 269, 214–224. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric Amyloid Beta Protein Rapidly Accumulates in Lipid Rafts Followed by Apolipoprotein E and Phosphorylated Tau Accumulation in the Tg2576 Mouse Model of Alzheimer’s Disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Whitehead, S.N.; Gangaraju, S.; Aylsworth, A.; Hou, S.T. Membrane Raft Disruption Results in Neuritic Retraction prior to Neuronal Death in Cortical Neurons. Biosci. Trends 2012, 6, 183–191. [Google Scholar] [CrossRef]
- Pooler, A.M.; Hanger, D.P. Functional Implications of the Association of Tau with the Plasma Membrane. Biochem. Soc. Trans. 2010, 38, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic Association of Tau with Neuronal Membranes Is Regulated by Phosphorylation. Neurobiol. Aging. 2012, 33, 431.e27–e38. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Pérez, M.; Avila, J. Tau Dephosphorylation at Tau-1 Site Correlates with Its Association to Cell Membrane. Neurochem. Res. 2000, 25, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, F.J.; Shea, T.B. Phosphorylation of Tau Alters Its Association with the Plasma Membrane. Cell. Mol. Neurobiol. 2000, 20, 497–508. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Reid, P.C.; Sperry, J.B.; Furukawa, K.; Takeya, M.; Chang, C.C.Y.; Chang, T.-Y. Plasma Membrane Rafts Complete Cholesterol Synthesis by Participating in Retrograde Movement of Precursor Sterols. J. Biol. Chem. 2007, 282, 34994–35004. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, N.; Gong, J.-S.; Chang, T.-Y.; Yanagisawa, K.; Michikawa, M. Promotion of Tau Phosphorylation by MAP Kinase Erk1/2 Is Accompanied by Reduced Cholesterol Level in Detergent-Insoluble Membrane Fraction in Niemann-Pick C1-Deficient Cells. J. Neurochem. 2003, 84, 1086–1096. [Google Scholar] [CrossRef]
- Glöckner, F.; Ohm, T.G. Tau Pathology Induces Intraneuronal Cholesterol Accumulation. J. Neuropathol. Exp. Neurol. 2014, 73, 846–854. [Google Scholar] [CrossRef]
- Jones, E.M.; Dubey, M.; Camp, P.J.; Vernon, B.C.; Biernat, J.; Mandelkow, E.; Majewski, J.; Chi, E.Y. Interaction of Tau Protein with Model Lipid Membranes Induces Tau Structural Compaction and Membrane Disruption. Biochemistry 2012, 51, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.K.; Greiss, F.; Khmelinskaia, A.; Schwille, P. Control of Lipid Domain Organization by a Biomimetic Contractile Actomyosin Cortex. Elife 2017, 6, e24350. [Google Scholar] [CrossRef] [PubMed]
- Connell, S.D.; Smith, D.A. The Atomic Force Microscope as a Tool for Studying Phase Separation in Lipid Membranes. Mol. Membr. Biol. 2006, 23, 17–28. [Google Scholar] [CrossRef]
- Lee, C.-W.; Jang, L.-L.; Pan, H.-J.; Chen, Y.-R.; Chen, C.-C.; Lee, C.-H. Membrane Roughness as a Sensitive Parameter Reflecting the Status of Neuronal Cells in Response to Chemical and Nanoparticle Treatments. J. Nanobiotechnology 2016, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 Aggregation, Interactions with Tau, and Impaired DNA Damage Response in Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Slade, N.; Hof, P.R.; Šimić, G. The Interactions of p53 with Tau and Aß as Potential Therapeutic Targets for Alzheimer’s Disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef]
- Hooper, C.; Meimaridou, E.; Tavassoli, M.; Melino, G.; Lovestone, S.; Killick, R. p53 Is Upregulated in Alzheimer’s Disease and Induces Tau Phosphorylation in HEK293a Cells. Neurosci. Lett. 2007, 418, 34–37. [Google Scholar] [CrossRef]
- Rueda-Rincon, N.; Bloch, K.; Derua, R.; Vyas, R.; Harms, A.; Hankemeier, T.; Khan, N.A.; Dehairs, J.; Bagadi, M.; Binda, M.M.; et al. p53 Attenuates AKT Signaling by Modulating Membrane Phospholipid Composition. Oncotarget 2015, 6, 21240–21254. [Google Scholar] [CrossRef]
- Pascual, A.S.; Espejo, L.S.; Moore, J.L.; Leyva, K.J.; Hull, E.E. Properties of Lipid Rafts in Two Epigenetically Distinct Subtypes of the Oncogenic Cell-line SW13. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in Response to DNA Damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef]
- Reisman, D.; Takahashi, P.; Polson, A.; Boggs, K. Transcriptional Regulation of the p53 Tumor Suppressor Gene in S-Phase of the Cell-Cycle and the Cellular Response to DNA Damage. Biochem. Res. Int. 2012, 2012, 808934. [Google Scholar] [CrossRef]
- Beckerman, R.; Prives, C. Transcriptional Regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A Complex Barcode Underlies the Heterogeneous Response of p53 to Stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Crawford, L.V. T Antigen Is Bound to a Host Protein in SV40-Transformed Cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A Model for p53-Induced Apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 Family and Programmed Cell Death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does p53 Induce Apoptosis and How Does This Relate to p53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Lane, D.P. p53 and Human Cancers. Br. Med. Bull. 1994, 50, 582–599. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. p53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the Cellular Gatekeeper for Growth and Division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a Novel p53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 Suppresses the Nrf2-Dependent Transcription of Antioxidant Response Genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase Separation of 53BP1 Determines Liquid-like Behavior of DNA Repair Compartments. EMBO J. 2019, 38, e101379. [Google Scholar] [CrossRef]
- Xu, Y. Regulation of p53 Responses by Post-Translational Modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational Modification of p53: Cooperative Integrators of Function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. p53 Post-Translational Modification: Deregulated in Tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef]
- Asher, G.; Shaul, Y. p53 Proteasomal Degradation: Poly-Ubiquitination Is Not the Whole Story. Cell Cycle 2005, 4, 1015–1018. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. Dynamics in the p53-Mdm2 Ubiquitination Pathway. Cell Cycle 2004, 3, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Reuven, N.; Shaul, Y. Ubiquitin-Independent p53 Proteasomal Degradation. Cell Death Differ. 2010, 17, 103–108. [Google Scholar] [CrossRef]
- Pant, V.; Lozano, G. Limiting the Power of p53 through the Ubiquitin Proteasome Pathway. Genes Dev. 2014, 28, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. The Role of Ubiquitin Modification in the Regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Perri, F.; Pisconti, S.; Della Vittoria Scarpati, G. P53 Mutations and Cancer: A Tight Linkage. Ann. Transl. Med. 2016, 4, 522. [Google Scholar] [CrossRef] [PubMed]
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190.e8. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Miñana, B.; Valcárcel, J.; Gabaldón, T.; Lehner, B. Synonymous Mutations Frequently Act as Driver Mutations in Human Cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef]
- Royds, J.A.; Iacopetta, B. p53 and Disease: When the Guardian Angel Fails. Cell Death Differ. 2006, 13, 1017–1026. [Google Scholar] [CrossRef]
- Hainaut, P.; Butcher, S.; Milner, J. Temperature Sensitivity for Conformation Is an Intrinsic Property of Wild-Type p53. Br. J. Cancer 1995, 71, 227–231. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening Guardian Angels: Drugging the p53 Pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Krois, A.S.; Dyson, H.J.; Wright, P.E. Long-Range Regulation of p53 DNA Binding by Its Intrinsically Disordered N-Terminal Transactivation Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E11302–E11310. [Google Scholar] [CrossRef]
- Laptenko, O.; Shiff, I.; Freed-Pastor, W.; Zupnick, A.; Mattia, M.; Freulich, E.; Shamir, I.; Kadouri, N.; Kahan, T.; Manfredi, J.; et al. The p53 C Terminus Controls Site-Specific DNA Binding and Promotes Structural Changes within the Central DNA Binding Domain. Mol. Cell 2015, 57, 1034–1046. [Google Scholar] [CrossRef]
- Kim, H.; Kim, K.; Choi, J.; Heo, K.; Baek, H.J.; Roeder, R.G.; An, W. p53 Requires an Intact C-Terminal Domain for DNA Binding and Transactivation. J. Mol. Biol. 2012, 415, 843–854. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Saiz Rubio, M.; Hompes, D.; Naus, E.; De Baets, G.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear Inclusion Bodies of Mutant and Wild-Type p53 in Cancer: A Hallmark of p53 Inactivation and Proteostasis Remodelling by p53 Aggregation. J. Pathol. 2017, 242, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Ano Bom, A.P.D.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.I.; Cepeda, A.O.T.; Stumbo, A.C.; et al. Mutant p53 Aggregates into Prion-like Amyloid Oligomers and Fibrils: Implications for Cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef]
- Rangel, L.P.; Costa, D.C.F.; Vieira, T.C.R.G.; Silva, J.L. The Aggregation of Mutant p53 Produces Prion-like Properties in Cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef]
- Silva, J.L.; De Moura Gallo, C.V.; Costa, D.C.F.; Rangel, L.P. Prion-like Aggregation of Mutant p53 in Cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 Amyloid Formation Leading to Its Loss of Function: Implications in Cancer Pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef]
- Park, S.-K.; Park, S.; Pentek, C.; Liebman, S.W. Tumor Suppressor Protein p53 Expressed in Yeast Can Remain Diffuse, Form a Prion, or Form Unstable Liquid-like Droplets. iScience 2021, 24, 102000. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Yang, X.; Zhao, Y.; Petersen, R.B.; Liu, X.; Liu, Y.; Huang, K. Amyloidogenicity of p53: A Hidden Link between Protein Misfolding and Cancer. Curr. Protein Pept. Sci. 2015, 16, 135–146. [Google Scholar] [CrossRef]
- Ishimaru, D.; Andrade, L.R.; Teixeira, L.S.P.; Quesado, P.A.; Maiolino, L.M.; Lopez, P.M.; Cordeiro, Y.; Costa, L.T.; Heckl, W.M.; Weissmüller, G.; et al. Fibrillar Aggregates of the Tumor Suppressor p53 Core Domain. Biochemistry 2003, 42, 9022–9027. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of Function of Mutant p53 by Coaggregation with Multiple Tumor Suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.S.; Norberto, D.R.; Spohr, T.C.L.S.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A.P. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 Amyloids in Cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Buizza, L.; Cenini, G.; Lanni, C.; Ferrari-Toninelli, G.; Prandelli, C.; Govoni, S.; Buoso, E.; Racchi, M.; Barcikowska, M.; Styczynska, M.; et al. Conformational Altered p53 as an Early Marker of Oxidative Stress in Alzheimer’s Disease. PLoS ONE 2012, 7, e29789. [Google Scholar] [CrossRef]
- Amor-Gutiérrez, O.; Costa-Rama, E.; Arce-Varas, N.; Martínez-Rodríguez, C.; Novelli, A.; Fernández-Sánchez, M.T.; Costa-García, A. Competitive Electrochemical Immunosensor for the Detection of Unfolded p53 Protein in Blood as Biomarker for Alzheimer’s Disease. Anal. Chim. Acta 2020, 1093, 28–34. [Google Scholar] [CrossRef]
- Abate, G.; Frisoni, G.B.; Bourdon, J.-C.; Piccirella, S.; Memo, M.; Uberti, D. The Pleiotropic Role of p53 in Functional/dysfunctional Neurons: Focus on Pathogenesis and Diagnosis of Alzheimer’s Disease. Alzheimers. Res. Ther. 2020, 12, 1–10. [Google Scholar] [CrossRef]
- Fusée, L.T.S.; Marín, M.; Fåhraeus, R.; López, I. Alternative Mechanisms of p53 Action During the Unfolded Protein Response. Cancers 2020, 12, 401. [Google Scholar] [CrossRef]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered Longevity-Assurance Activity of p53:p44 in the Mouse Causes Memory Loss, Neurodegeneration and Premature Death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the “Longevity-Assurance” Isoform of P53, Regulates Tau Phosphorylation and Is Activated in an Age-Dependent Fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 Stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 Is a Ubiquitin Ligase E3 for Tumor Suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Sola, M.; Magrin, C.; Pedrioli, G.; Pinton, S.; Salvadè, A.; Papin, S.; Paganetti, P. Tau Affects P53 Function and Cell Fate during the DNA Damage Response. Commun. Biol. 2020, 3, 245. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Petrenko, O. The MDM2-p53 Interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar] [PubMed]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Alves-Rodrigues, A.; Gregori, L.; Figueiredo-Pereira, M.E. Ubiquitin, Cellular Inclusions and Their Role in Neurodegeneration. Trends Neurosci. 1998, 21, 516–520. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef]
- Markmiller, S.; Fulzele, A.; Higgins, R.; Leonard, M.; Yeo, G.W.; Bennett, E.J. Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 2019, 27, 1356–1363.e3. [Google Scholar] [CrossRef]
- Dao, T.P.; Kolaitis, R.-M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978.e6. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like Protein Conjugation and the Ubiquitin-Proteasome System as Drug Targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Gallego, L.D.; Schneider, M.; Mittal, C.; Romanauska, A.; Gudino Carrillo, R.M.; Schubert, T.; Pugh, B.F.; Köhler, A. Phase Separation Directs Ubiquitination of Gene-Body Nucleosomes. Nature 2020, 579, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.-C.; Schuman, E.M. Ubiquitin, the Proteasome and Protein Degradation in Neuronal Function and Dysfunction. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Taguchi, K.; Tanaka, M. Ubiquitin, Autophagy and Neurodegenerative Diseases. Cells 2020, 9, 2022. [Google Scholar] [CrossRef]
- Park, C.-W.; Jung, B.-K.; Ryu, K.-Y. Reduced Free Ubiquitin Levels and Proteasome Activity in Cultured Neurons and Brain Tissues Treated with Amyloid Beta Aggregates. Mol. Brain 2020, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Bellia, F.; Lanza, V.; García-Viñuales, S.; Ahmed, I.M.M.; Pietropaolo, A.; Iacobucci, C.; Malgieri, G.; D’Abrosca, G.; Fattorusso, R.; Nicoletti, V.G.; et al. Ubiquitin Binds the Amyloid β Peptide and Interferes with Its Clearance Pathways. Chem. Sci. 2019, 10, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Benanti, J.A. Coordination of Cell Growth and Division by the Ubiquitin-Proteasome System. Semin. Cell Dev. Biol. 2012, 23, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z. The Role of the Ubiquitin-Proteasome Pathway in Apoptosis. Cell Death Differ. 1999, 6, 303–313. [Google Scholar] [CrossRef]
- Wójcik, C. Regulation of Apoptosis by the Ubiquitin and Proteasome Pathway. J. Cell Mol. Med. 2002, 6, 25–48. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Trivedi, A.K. Regulation of Apoptosis by E3 Ubiquitin Ligases in Ubiquitin Proteasome System. Cell Biol. Int. 2020, 44, 721–734. [Google Scholar] [CrossRef]
- Marfella, R.; D’Amico, M.; Esposito, K.; Baldi, A.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Portoghese, M.; Cirillo, F.; Cacciapuoti, F.; et al. The Ubiquitin-Proteasome System and Inflammatory Activity in Diabetic Atherosclerotic Plaques: Effects of Rosiglitazone Treatment. Diabetes 2006, 55, 622–632. [Google Scholar] [CrossRef]
- Goetzke, C.C.; Ebstein, F.; Kallinich, T. Role of Proteasomes in Inflammation. J. Clin. Med. Res. 2021, 10, 1783. [Google Scholar] [CrossRef]
- Muratani, M.; Tansey, W.P. How the Ubiquitin-Proteasome System Controls Transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Wenzel, S.; Tansey, W.P. Ubiquitin and Proteasomes in Transcription. Annu. Rev. Biochem. 2012, 81, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. The Ubiquitin-Proteasome System and Signal Transduction Pathways Regulating Epithelial Mesenchymal Transition of Cancer. J. Biomed. Sci. 2012, 19, 67. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Maldonado, M.A. The Ubiquitin-Proteasome System and Its Role in Inflammatory and Autoimmune Diseases. Cell Mol. Immunol. 2006, 3, 255–261. [Google Scholar] [PubMed]
- Hershko, A. The Ubiquitin System for Protein Degradation and Some of Its Roles in the Control of the Cell Division Cycle. Cell Death Differ. 2005, 12, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. ATP-Dependent Proteases in Prokaryotic and Eukaryotic Cells. Semin. Cell Biol. 1990, 1, 423–432. [Google Scholar]
- Baker, T.A.; Sauer, R.T. ATP-Dependent Proteases of Bacteria: Recognition Logic and Operating Principles. Trends Biochem. Sci. 2006, 31, 647–653. [Google Scholar] [CrossRef]
- Tomko Jr, R.J.; Hochstrasser, M. Molecular Architecture and Assembly of the Eukaryotic Proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin Chain-Induced p62 Phase Separation Drives Autophagic Cargo Segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and Ubiquitylation-Dependent Phase Separation of the Proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, S.; Wu, Z.; Li, X.; Wang, W.L.; Zhu, Y.; Stoilova-McPhie, S.; Lu, Y.; Finley, D.; Mao, Y. Cryo-EM Structures and Dynamics of Substrate-Engaged Human 26S Proteasome. Nature 2019, 565, 49–55. [Google Scholar] [CrossRef]
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP Costs and Time Required to Degrade Ubiquitinated Proteins by the 26 S Proteasome. J. Biol. Chem. 2013, 288, 29215–29222. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying Intracellular Rates of Glycolytic and Oxidative ATP Production and Consumption Using Extracellular Flux Measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Schurr, A. Lactate, Not Pyruvate, Is the End Product of Glucose Metabolism via Glycolysis. In Carbohydrate; Caliskan, M., Kavakli, I.H., Oz, G.C., Eds.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of Pyruvate Metabolism and Human Disease. Cell Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef]
- Atas, E.; Oberhuber, M.; Kenner, L. The Implications of PDK1-4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 2020, 10, 583217. [Google Scholar] [CrossRef]
- Hall, C.N.; Klein-Flügge, M.C.; Howarth, C.; Attwell, D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jeon, S.; Suk, K. Pyruvate Dehydrogenase Kinases in the Nervous System: Their Principal Functions in Neuronal-Glial Metabolic Interaction and Neuro-Metabolic Disorders. Curr. Neuropharmacol. 2012, 10, 393–403. [Google Scholar] [CrossRef]
- Blass, J.P.; Sheu, R.K.; Gibson, G.E. Inherent Abnormalities in Energy Metabolism in Alzheimer Disease. Interaction with Cerebrovascular Compromise. Ann. N. Y. Acad. Sci. 2000, 903, 204–221. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Marin, R.; Ramírez, C.M.; González, M.; González-Muñoz, E.; Zorzano, A.; Camps, M.; Alonso, R.; Díaz, M. Voltage-Dependent Anion Channel (VDAC) Participates in Amyloid Beta-Induced Toxicity and Interacts with Plasma Membrane Estrogen Receptor Alpha in Septal and Hippocampal Neurons. Mol. Membr. Biol. 2007, 24, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Paschon, V.; Morena, B.C.; Correia, F.F.; Beltrame, G.R.; Dos Santos, G.B.; Cristante, A.F.; Kihara, A.H. VDAC1 Is Essential for Neurite Maintenance and the Inhibition of Its Oligomerization Protects Spinal Cord from Demyelination and Facilitates Locomotor Function Recovery after Spinal Cord Injury. Sci. Rep. 2019, 9, 14063. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ Promotes VDAC1 Channel Dephosphorylation in Neuronal Lipid Rafts. Relevance to the Mechanisms of Neurotoxicity in Alzheimer’s Disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Cesarini, E.; Cerioni, L.; Canonico, B.; Di Sario, G.; Guidarelli, A.; Lattanzi, D.; Savelli, D.; Guescini, M.; Nasoni, M.G.; Bigini, N.; et al. Melatonin Protects Hippocampal HT22 Cells from the Effects of Serum Deprivation Specifically Targeting Mitochondria. PLoS ONE 2018, 13, e0203001. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 Pathway Revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Song, J.; Ma, S.-J.; Luo, J.-H.; Zhang, H.; Wang, R.-X.; Liu, H.; Li, L.; Zhang, Z.-G.; Zhou, R.-X. Melatonin Induces the Apoptosis and Inhibits the Proliferation of Human Gastric Cancer Cells via Blockade of the AKT/MDM2 Pathway. Oncol. Rep. 2018, 39, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fåhraeus, R. The p53 mRNA: An Integral Part of the Cellular Stress Response. Nucleic Acids Res. 2019, 47, 3257–3271. [Google Scholar] [CrossRef]
- Kasteri, J.; Das, D.; Zhong, X.; Persaud, L.; Francis, A.; Muharam, H.; Sauane, M. Translation Control by p53. Cancers 2018, 10, 133. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Qin, Z.-H.; Wang, J. The Role of p53 in Cell Metabolism. Acta Pharmacol. Sin. 2010, 31, 1208–1212. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-Associated Mutant p53 Drives the Warburg Effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Roe, J.-S.; Lee, J.-E.; Cho, E.-J.; Youn, H.-D. p53 Regulates Glucose Metabolism by miR-34a. Biochem. Biophys. Res. Commun. 2013, 437, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 Regulates Glucose Metabolism through an IKK-NF-kappaB Pathway and Inhibits Cell Transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Sung, H.J.; Park, J.Y.; Matoba, S.; Hwang, P.M. A Pivotal Role for p53: Balancing Aerobic Respiration and Glycolysis. J. Bioenerg. Biomembr. 2007, 39, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Contractor, T.; Harris, C.R. p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res. 2012, 72, 560–567. [Google Scholar] [CrossRef]
- Mediavilla, M.D.; Cos, S.; Sánchez-Barceló, E.J. Melatonin Increases p53 and p21WAF1 Expression in MCF-7 Human Breast Cancer Cells in Vitro. Life Sci. 1999, 65, 415–420. [Google Scholar] [CrossRef]
- Kim, C.H.; Yoo, Y.-M. Melatonin Induces Apoptotic Cell Death via p53 in LNCaP Cells. Korean J. Physiol. Pharmacol. 2010, 14, 365–369. [Google Scholar] [CrossRef]
- Ma, L.; Liu, Q.; Tian, M.; Tian, X.; Gao, L. Mechanisms of Melatonin in Anti-Aging and Its Regulation Effects in Radiation-Induced Premature Senescence. Radiat. Med. Prot. 2021, 2, 33–37. [Google Scholar] [CrossRef]
- Santoro, R.; Marani, M.; Blandino, G.; Muti, P.; Strano, S. Melatonin Triggers p53Ser Phosphorylation and Prevents DNA Damage Accumulation. Oncogene 2012, 31, 2931–2942. [Google Scholar] [CrossRef]
- Santoro, R.; Mori, F.; Marani, M.; Grasso, G.; Cambria, M.A.; Blandino, G.; Muti, P.; Strano, S. Blockage of Melatonin Receptors Impairs p53-Mediated Prevention of DNA Damage Accumulation. Carcinogenesis 2013, 34, 1051–1061. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; Dobrowolny, G.; D’Anselmi, F.; Dinicola, S.; Masiello, M.G.; Pasqualato, A.; Palombo, A.; Morini, V.; Reiter, R.J.; et al. Melatonin down-Regulates MDM2 Gene Expression and Enhances p53 Acetylation in MCF-7 Cells. J. Pineal. Res. 2014, 57, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.I.; Roderick, J.E.; Gannon, H.S.; Kelliher, M.A.; Jones, S.N. Mdm2 Phosphorylation Regulates Its Stability and Has Contrasting Effects on Oncogene and Radiation-Induced Tumorigenesis. Cell Rep. 2016, 16, 2618–2629. [Google Scholar] [CrossRef] [PubMed]
- Facchin, S.; Ruzzene, M.; Peggion, C.; Sartori, G.; Carignani, G.; Marin, O.; Brustolon, F.; Lopreiato, R.; Pinna, L.A. Phosphorylation and Activation of the Atypical Kinase p53-Related Protein Kinase (PRPK) by Akt/PKB. Cell. Mol. Life Sci. 2007, 64, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical Role for p53-Serine 15 Phosphorylation in Stimulating Transactivation at p53-Responsive Promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Matani, A.S.; Braun, S.M.G.; Milanesi, F.; Rodewald, L.W.; Wahl, G.M. Functional Analysis and Consequences of Mdm2 E3 Ligase Inhibition in Human Tumor Cells. Oncogene 2012, 31, 4789–4797. [Google Scholar] [CrossRef]
- Hou, H.; Sun, D.; Zhang, X. The Role of MDM2 Amplification and Overexpression in Therapeutic Resistance of Malignant Tumors. Cancer Cell Int. 2019, 19, 216. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Leal, J.F.M.; Seger, R.; Taya, Y.; Oren, M. Cross-Talk between Akt, p53 and Mdm2: Possible Implications for the Regulation of Apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-Mediated p53 Acetylation Is Commonly Induced by p53-Activating Agents and Inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Fu, M.; Sauve, A.A.; Wang, F.; Quong, A.A.; Perkins, N.D.; Hay, R.T.; Gu, W.; Pestell, R.G. SIRT1 Deacetylation and Repression of p300 Involves Lysine Residues 1020/1024 within the Cell Cycle Regulatory Domain 1. J. Biol. Chem. 2005, 280, 10264–10276. [Google Scholar] [CrossRef]
- Lohrum, M.A.E.; Ludwig, R.L.; Kubbutat, M.H.G.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 Activity by the Ribosomal Protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef]
- Bursać, S.; Brdovčak, M.C.; Pfannkuchen, M.; Orsolić, I.; Golomb, L.; Zhu, Y.; Katz, C.; Daftuar, L.; Grabušić, K.; Vukelić, I.; et al. Mutual Protection of Ribosomal Proteins L5 and L11 from Degradation Is Essential for p53 Activation upon Ribosomal Biogenesis Stress. Proc. Natl. Acad. Sci. USA 2012, 109, 20467–20472. [Google Scholar] [CrossRef]
- Zhu, Y.; Poyurovsky, M.V.; Li, Y.; Biderman, L.; Stahl, J.; Jacq, X.; Prives, C. Ribosomal Protein S7 Is Both a Regulator and a Substrate of MDM2. Mol. Cell 2009, 35, 316–326. [Google Scholar] [CrossRef]
- Dai, M.-S.; Zeng, S.X.; Jin, Y.; Sun, X.-X.; David, L.; Lu, H. Ribosomal Protein L23 Activates p53 by Inhibiting MDM2 Function in Response to Ribosomal Perturbation but Not to Translation Inhibition. Mol. Cell. Biol. 2004, 24, 7654–7668. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and Activation of p53 by Ribosomal Protein L23. Mol. Cell. Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef]
- Dai, M.-S.; Lu, H. Inhibition of MDM2-Mediated p53 Ubiquitination and Degradation by Ribosomal Protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer Res. 2016, 2, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Blyszcz, T.; González-Prieto, R.; Vertegaal, A.C.O.; Goldberg, A.L. Inhibiting Ubiquitination Causes an Accumulation of SUMOylated Newly Synthesized Nuclear Proteins at PML Bodies. J. Biol. Chem. 2019, 294, 15218–15234. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Jentsch, S. Principles of Ubiquitin and SUMO Modifications in DNA Repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Moldovan, G.-L.; Vinciguerra, P.; Murai, J.; Takeda, S.; D’Andrea, A.D. Regulation of the Fanconi Anemia Pathway by a SUMO-like Delivery Network. Genes Dev. 2011, 25, 1847–1858. [Google Scholar] [CrossRef]
- Jackson, S.P.; Durocher, D. Regulation of DNA Damage Responses by Ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef]
- Pellegrino, S.; Altmeyer, M. Interplay between Ubiquitin, SUMO, and Poly(ADP-Ribose) in the Cellular Response to Genotoxic Stress. Front. Genet. 2016, 7, 63. [Google Scholar] [CrossRef]
- Lee, C.C.; Li, B.; Yu, H.; Matunis, M.J. A Method for SUMO Modification of Proteins in Vitro. Bio Protoc 2018, 8, e3033. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO Pathway: Emerging Mechanisms That Shape Specificity, Conjugation and Recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef]
- Van Wijk, S.J.L.; Müller, S.; Dikic, I. Shared and Unique Properties of Ubiquitin and SUMO Interaction Networks in DNA Repair. Genes Dev. 2011, 25, 1763–1769. [Google Scholar] [CrossRef][Green Version]
- Lamoliatte, F.; McManus, F.P.; Maarifi, G.; Chelbi-Alix, M.K.; Thibault, P. Uncovering the SUMOylation and Ubiquitylation Crosstalk in Human Cells Using Sequential Peptide Immunopurification. Nat. Commun. 2017, 8, 14109. [Google Scholar] [CrossRef]
- Gill, G. SUMO and Ubiquitin in the Nucleus: Different Functions, Similar Mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef]
- Ulrich, H.D. Mutual Interactions between the SUMO and Ubiquitin Systems: A Plea of No Contest. Trends Cell Biol. 2005, 15, 525–532. [Google Scholar] [CrossRef]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-Targeted Ubiquitin Ligases. Biochim. Biophys. Acta 2014, 1843, 75–85. [Google Scholar] [CrossRef]
- Cubeñas-Potts, C.; Matunis, M.J. SUMO: A Multifaceted Modifier of Chromatin Structure and Function. Dev. Cell 2013, 24, 1–12. [Google Scholar] [CrossRef]
- Cardone, L.; Hirayama, J.; Giordano, F.; Tamaru, T.; Palvimo, J.J.; Sassone-Corsi, P. Circadian Clock Control by SUMOylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Berndt, A.; Wilkinson, K.A.; Henley, J.M. Regulation of Neuronal Protein Trafficking and Translocation by SUMOylation. Biomolecules 2012, 2, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Nakamura, Y.; Henley, J.M. Targets and Consequences of Protein SUMOylation in Neurons. Brain Res. Rev. 2010, 64, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ashikaga, E.; Rubin, P.P.; Heimann, M.J.; Hildick, K.L.; Bishop, P.; Girach, F.; Josa-Prado, F.; Tang, L.T.H.; Carmichael, R.E.; et al. Receptor Trafficking and the Regulation of Synaptic Plasticity by SUMO. Neuromolecular Med. 2013, 15, 692–706. [Google Scholar] [CrossRef]
- Meinecke, I.; Cinski, A.; Baier, A.; Peters, M.A.; Dankbar, B.; Wille, A.; Drynda, A.; Mendoza, H.; Gay, R.E.; Hay, R.T.; et al. Modification of Nuclear PML Protein by SUMO-1 Regulates Fas-Induced Apoptosis in Rheumatoid Arthritis Synovial Fibroblasts. Proc. Natl. Acad. Sci. USA 2007, 104, 5073–5078. [Google Scholar] [CrossRef]
- Liebelt, F.; Vertegaal, A.C.O. Ubiquitin-Dependent and Independent Roles of SUMO in Proteostasis. Am. J. Physiol. Cell Physiol. 2016, 311, C284–C296. [Google Scholar] [CrossRef]
- Song, J.; Durrin, L.K.; Wilkinson, T.A.; Krontiris, T.G.; Chen, Y. Identification of a SUMO-Binding Motif That Recognizes SUMO-Modified Proteins. Proc. Natl. Acad. Sci. USA. 2004, 101, 14373–14378. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O. SUMO Junction-What’s Your Function? New Insights through SUMO-Interacting Motifs. EMBO Rep. 2007, 8, 550–555. [Google Scholar] [CrossRef] [PubMed]
- González-Prieto, R.; Eifler-Olivi, K.; Claessens, L.A.; Willemstein, E.; Xiao, Z.; Talavera Ormeno, C.M.P.; Ovaa, H.; Ulrich, H.D.; Vertegaal, A.C.O. Global Non-Covalent SUMO Interaction Networks Reveal SUMO-Dependent Stabilization of the Non-Homologous End Joining Complex. Cell Rep. 2021, 34, 108691. [Google Scholar] [CrossRef] [PubMed]
- Nacerddine, K.; Lehembre, F.; Bhaumik, M.; Artus, J.; Cohen-Tannoudji, M.; Babinet, C.; Pandolfi, P.P.; Dejean, A. The SUMO Pathway Is Essential for Nuclear Integrity and Chromosome Segregation in Mice. Dev. Cell 2005, 9, 769–779. [Google Scholar] [CrossRef]
- Jin, J. Interplay between Ubiquitylation and SUMOylation: Empowered by Phase Separation. J. Biol. Chem. 2019, 294, 15235–15236. [Google Scholar] [CrossRef]
- Keiten-Schmitz, J.; Röder, L.; Hornstein, E.; Müller-McNicoll, M.; Müller, S. SUMO: Glue or Solvent for Phase-Separated Ribonucleoprotein Complexes and Molecular Condensates? Front. Mol. Biosci. 2021, 8, 673038. [Google Scholar] [CrossRef]
- Mattsson, K.; Pokrovskaja, K.; Kiss, C.; Klein, G.; Szekely, L. Proteins Associated with the Promyelocytic Leukemia Gene Product (PML)-Containing Nuclear Body Move to the Nucleolus upon Inhibition of Proteasome-Dependent Protein Degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Mediani, L.; Guillén-Boixet, J.; Alberti, S.; Carra, S. Nucleoli and Promyelocytic Leukemia Protein (PML) Bodies Are Phase Separated Nuclear Protein Quality Control Compartments for Misfolded Proteins. Mol. Cell Oncol. 2019, 6, e1415624. [Google Scholar] [CrossRef]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid Degradation of a Large Fraction of Newly Synthesized Proteins by Proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.M.; Bowman, M.A.; Soto Santarriaga, I.F.; Rodriguez, A.; Clark, P.L. Synonymous Codon Substitutions Perturb Cotranslational Protein Folding in Vivo and Impair Cell Fitness. Proc. Natl. Acad. Sci. USA 2020, 117, 3528–3534. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Wilke, C.O. The Evolutionary Consequences of Erroneous Protein Synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Adaptation of mRNA Structure to Control Protein Folding. RNA Biol. 2017, 14, 1649–1654. [Google Scholar] [CrossRef]
- Gout, J.-F.; Li, W.; Fritsch, C.; Li, A.; Haroon, S.; Singh, L.; Hua, D.; Fazelinia, H.; Smith, Z.; Seeholzer, S.; et al. The Landscape of Transcription Errors in Eukaryotic Cells. Sci. Adv. 2017, 3, e1701484. [Google Scholar] [CrossRef]
- Jamar, N.H.; Kritsiligkou, P.; Grant, C.M. Loss of mRNA Surveillance Pathways Results in Widespread Protein Aggregation. Sci. Rep. 2018, 8, 3894. [Google Scholar] [CrossRef]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis Impairment in Protein-Misfolding and -Aggregation Diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- Wu, H. Higher-Order Assemblies in a New Paradigm of Signal Transduction. Cell 2013, 153, 287–292. [Google Scholar] [CrossRef]
- Kuroda, Y. Biophysical Studies of Protein Solubility and Amorphous Aggregation by Systematic Mutational Analysis and a Helical Polymerization Model. Biophys. Rev. 2018, 10, 473–480. [Google Scholar] [CrossRef]
- Koo, E.H.; Lansbury, P.T.; Kelly, J.W. Amyloid Diseases: Abnormal Protein Aggregation in Neurodegeneration. Proc. Natl. Acad. Sci. USA 1999, 96, 9989–9990. [Google Scholar] [CrossRef]
- Dorval, V.; Fraser, P.E. SUMO on the Road to Neurodegeneration. Biochim. Biophys. Acta 2007, 1773, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.-Y.; Molina, O.; Courey, A.J. SUMOylation in Development and Neurodegeneration. Development 2020, 147, dev175703. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Min, D.S.; Rhim, H.; Kim, J.; Paik, S.R.; Chung, K.C. Parkin Ubiquitinates and Promotes the Degradation of RanBP2*. J. Biol. Chem. 2006, 281, 3595–3603. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein Folding in the Cytoplasm and the Heat Shock Response. Cold Spring Harb. Perspect. Biol. 2010, 2, a004390. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The Stress of Protein Misfolding: From Single Cells to Multicellular Organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704. [Google Scholar] [CrossRef]
- Ruddock, L.W.; Klappa, P. Oxidative Stress: Protein Folding with a Novel Redox Switch. Curr. Biol. 1999, 9, R400–R402. [Google Scholar] [CrossRef][Green Version]
- Stadmiller, S.S.; Gorensek-Benitez, A.H.; Guseman, A.J.; Pielak, G.J. Osmotic Shock Induced Protein Destabilization in Living Cells and Its Reversal by Glycine Betaine. J. Mol. Biol. 2017, 429, 1155–1161. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Parker, R. Multiple Modes of Protein-Protein Interactions Promote RNP Granule Assembly. J. Mol. Biol. 2018, 430, 4636–4649. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Tolay, N.; Buchberger, A. Comparative Profiling of Stress Granule Clearance Reveals Differential Contributions of the Ubiquitin System. Life Sci Alliance 2021, 4. [Google Scholar] [CrossRef] [PubMed]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Müller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef] [PubMed]
- Jongjitwimol, J.; Baldock, R.A.; Morley, S.J.; Watts, F.Z. Sumoylation of eIF4A2 Affects Stress Granule Formation. J. Cell Sci. 2016, 129, 2407–2415. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Hsu, W.-L.; Ma, Y.-L.; Lee, E.H.Y. Melatonin Induction of APP Intracellular Domain 50 SUMOylation Alleviates AD through Enhanced Transcriptional Activation and Aβ Degradation. Mol. Ther. 2021, 29, 376–395. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Sarge, K.D. Sumoylation of Amyloid Precursor Protein Negatively Regulates Abeta Aggregate Levels. Biochem. Biophys. Res. Commun. 2008, 374, 673–678. [Google Scholar] [CrossRef]
- Agbor, T.A.; Taylor, C.T. SUMO, Hypoxia and the Regulation of Metabolism. Biochem. Soc. Trans. 2008, 36 Pt 3, 445–448. [Google Scholar] [CrossRef]
- Lee, Y.J.; Castri, P.; Bembry, J.; Maric, D.; Auh, S.; Hallenbeck, J.M. SUMOylation Participates in Induction of Ischemic Tolerance. J. Neurochem. 2009, 109, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Bossis, G.; Melchior, F. Regulation of SUMOylation by Reversible Oxidation of SUMO Conjugating Enzymes. Mol. Cell 2006, 21, 349–357. [Google Scholar] [CrossRef]
- Silveirinha, V.; Stephens, G.J.; Cimarosti, H. Molecular Targets Underlying SUMO-Mediated Neuroprotection in Brain Ischemia. J. Neurochem. 2013, 127, 580–591. [Google Scholar] [CrossRef]
- Lee, L.; Sakurai, M.; Matsuzaki, S.; Arancio, O.; Fraser, P. SUMO and Alzheimer’s Disease. Neuromolecular Med. 2013, 15, 720–736. [Google Scholar] [CrossRef]
- Boulanger, M.; Chakraborty, M.; Tempé, D.; Piechaczyk, M.; Bossis, G. SUMO and Transcriptional Regulation: The Lessons of Large-Scale Proteomic, Modifomic and Genomic Studies. Molecules 2021, 26, 828. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. RNA Evolution and the Origins of Life. Nature 1989, 338, 217–224. [Google Scholar] [CrossRef]
- Joyce, G.F. The Antiquity of RNA-Based Evolution. Nature 2002, 418, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.P.; Joyce, G.F. The Origins of the RNA World. Cold Spring Harb. Perspect. Biol. 2012, 4, a003608. [Google Scholar] [CrossRef]
- Hawkins, P.G.; Morris, K.V. RNA and Transcriptional Modulation of Gene Expression. Cell Cycle 2008, 7, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V. The Emerging Role of RNA in the Regulation of Gene Transcription in Human Cells. Semin. Cell Dev. Biol. 2011, 22, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.L.; Khalid, F.; Wagner, A. RNA-Mediated Gene Regulation Is Less Evolvable than Transcriptional Regulation. Proc. Natl. Acad. Sci. USA 2018, 115, E3481–E3490. [Google Scholar] [CrossRef]
- Gott, J.M.; Emeson, R.B. Functions and Mechanisms of RNA Editing. Annu. Rev. Genet. 2000, 34, 499–531. [Google Scholar] [CrossRef]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA Modifications: Form, Distribution, and Function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef]
- Szymański, M.; Barciszewska, M.Z.; Zywicki, M.; Barciszewski, J. Noncoding RNA Transcripts. J. Appl. Genet. 2003, 44, 1–19. [Google Scholar] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene Regulation by Non-Coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Potashkin, J.A.; Bottero, V.; Santiago, J.A.; Quinn, J.P. Computational Identification of Key Genes That May Regulate Gene Expression Reprogramming in Alzheimer’s Patients. PLoS ONE 2019, 14, e0222921. [Google Scholar] [CrossRef]
- He, C. Grand Challenge Commentary: RNA Epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef]
- Seo, K.W.; Kleiner, R.E. Mechanisms of Epitranscriptomic Gene Regulation. Biopolymers 2021, 112, e23403. [Google Scholar] [CrossRef]
- Aumiller, W.M.; Pir Cakmak, F.; Davis, B.W.; Keating, C.D. RNA-Based Coacervates as a Model for Membraneless Organelles: Formation, Properties, and Interfacial Liposome Assembly. Langmuir 2016, 32, 10042–10053. [Google Scholar] [CrossRef]
- Peng, L.; Li, E.-M.; Xu, L.-Y. From Start to End: Phase Separation and Transcriptional Regulation. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194641. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Hawley, Z.C.E.; Droppelmann, C.A.; Strong, M.J. The Integral Role of RNA in Stress Granule Formation and Function. Front. Cell Dev. Biol. 2021, 9, 621779. [Google Scholar] [CrossRef] [PubMed]
- Sanchez de Groot, N.; Armaos, A.; Graña-Montes, R.; Alriquet, M.; Calloni, G.; Vabulas, R.M.; Tartaglia, G.G. RNA Structure Drives Interaction with Proteins. Nat. Commun. 2019, 10, 3246. [Google Scholar] [CrossRef] [PubMed]
- Colvin, V.L.; Kulinowski, K.M. Nanoparticles as Catalysts for Protein Fibrillation. Proc. Natl. Acad. Sci. USA 2007, 104, 8679–8680. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.; Le Droumaguet, B.; Nicolas, J.; Hashemi, S.H.; Wu, L.-P.; Moghimi, S.M.; Couvreur, P.; Andrieux, K. Nanotechnologies for Alzheimer’s Disease: Diagnosis, Therapy, and Safety Issues. Nanomedicine 2011, 7, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Conlon, E.G.; Manley, J.L. RNA-Binding Proteins in Neurodegeneration: Mechanisms in Aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef]
- Kinoshita, C.; Kubota, N.; Aoyama, K. Interplay of RNA-Binding Proteins and microRNAs in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 5292. [Google Scholar] [CrossRef]
- Anders, M.; Chelysheva, I.; Goebel, I.; Trenkner, T.; Zhou, J.; Mao, Y.; Verzini, S.; Qian, S.-B.; Ignatova, Z. Dynamic m6A Methylation Facilitates mRNA Triaging to Stress Granules. Life Sci. Alliance 2018, 1, e201800113. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.-S.; Ping, X.-L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.-Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A Reader YTHDF3 Promotes mRNA Translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef]
- Rajecka, V.; Skalicky, T.; Vanacova, S. The Role of RNA Adenosine Demethylases in the Control of Gene Expression. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 343–355. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-Methyladenosine and Its Role in Cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-Methyladenosine Links RNA Metabolism to Cancer Progression. Cell Death Dis. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-Methyladenosine-Dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The Emerging Role of RNA Modifications in the Regulation of mRNA Stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Wu, J.; Liu, J.; Qin, Z.; Fan, H. N6-Methyladenosine: A Potential Breakthrough for Human Cancer. Mol. Ther. Nucleic Acids 2020, 19, 804–813. [Google Scholar] [CrossRef]
- Zhu, Z.-M.; Huo, F.-C.; Pei, D.-S. Function and Evolution of RNA N6-Methyladenosine Modification. Int. J. Biol. Sci. 2020, 16, 1929–1940. [Google Scholar] [CrossRef]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m6A Modification Controls the Innate Immune Response to Infection by Targeting Type I Interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Gao, Y.; Vasic, R.; Song, Y.; Teng, R.; Liu, C.; Gbyli, R.; Biancon, G.; Nelakanti, R.; Lobben, K.; Kudo, E.; et al. m6A Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity 2020, 52, 1007–1021.e8. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Z.; Li, R.; Wu, Y.; Chi, M.; Gao, S.; Sun, X.; Meng, X.; Wang, B. Potential Roles of N6-Methyladenosine (m6A) in Immune Cells. J. Transl. Med. 2021, 19, 251. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yu, G.-L.; Zhu, X.; Peng, T.-H.; Lv, Y.-C. Critical Roles of FTO-Mediated mRNA m6A Demethylation in Regulating Adipogenesis and Lipid Metabolism: Implications in Lipid Metabolic Disorders. Genes Dis. 2021. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m6A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e4. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, W.; Huang, J.-T.; Shen, F.; Xiong, J.; Yuan, E.-F.; Qin, S.-S.; Zhang, M.; Feng, Y.-Q.; Yuan, B.-F.; et al. Increased N6-Methyladenosine in Human Sperm RNA as a Risk Factor for Asthenozoospermia. Sci. Rep. 2016, 6, 24345. [Google Scholar] [CrossRef]
- Zhao, B.S.; He, C. “Gamete On” for m6A: YTHDF2 Exerts Essential Functions in Female Fertility. Mol. Cell 2017, 67, 903–905. [Google Scholar] [CrossRef]
- Tang, C.; Klukovich, R.; Peng, H.; Wang, Z.; Yu, T.; Zhang, Y.; Zheng, H.; Klungland, A.; Yan, W. ALKBH5-Dependent m6A Demethylation Controls Splicing and Stability of Long 3’-UTR mRNAs in Male Germ Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E325–E333. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lao, Y.; Wang, Y.; Li, R.; Fang, X.; Wang, Y.; Gao, X.; Dong, Z. Role of RNA N6-Methyladenosine Modification in Male Infertility and Genital System Tumors. Front. Cell Dev. Biol. 2021, 9, 676364. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G.; Dominissini, D. The m6A Epitranscriptome: Transcriptome Plasticity in Brain Development and Function. Nat. Rev. Neurosci. 2020, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef]
- Zou, S.; Toh, J.D.W.; Wong, K.H.Q.; Gao, Y.-G.; Hong, W.; Woon, E.C.Y. N(6)-Methyladenosine: A Conformational Marker That Regulates the Substrate Specificity of Human Demethylases FTO and ALKBH5. Sci. Rep. 2016, 6, 25677. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Gu, Q.; Ma, Y.; Yang, Y.; Zhu, J.; Zhang, Q. The Biological Function of m6A Demethylase ALKBH5 and Its Role in Human Disease. Cancer Cell Int. 2020, 20, 347. [Google Scholar] [CrossRef]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N6-Methyladenosine (m6A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Church, C.; Moir, L.; McMurray, F.; Girard, C.; Banks, G.T.; Teboul, L.; Wells, S.; Brüning, J.C.; Nolan, P.M.; Ashcroft, F.M.; et al. Overexpression of Fto Leads to Increased Food Intake and Results in Obesity. Nat. Genet. 2010, 42, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Church, C.; Lee, S.; Bagg, E.A.L.; McTaggart, J.S.; Deacon, R.; Gerken, T.; Lee, A.; Moir, L.; Mecinović, J.; Quwailid, M.M.; et al. A Mouse Model for the Metabolic Effects of the Human Fat Mass and Obesity Associated FTO Gene. PLoS Genet. 2009, 5, e1000599. [Google Scholar] [CrossRef]
- Barber, T.M.; Bennett, A.J.; Groves, C.J.; Sovio, U.; Ruokonen, A.; Martikainen, H.; Pouta, A.; Hartikainen, A.-L.; Elliott, P.; Lindgren, C.M.; et al. Association of Variants in the Fat Mass and Obesity Associated (FTO) Gene with Polycystic Ovary Syndrome. Diabetologia 2008, 51, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.-F.; Wei, B.; et al. RNA N6-Methyladenosine Demethylase FTO Promotes Breast Tumor Progression through Inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Shi, Y.; Liu, J.; Lin, L.; Chen, X. m6A Demethylase FTO Promotes Hepatocellular Carcinoma Tumorigenesis via Mediating PKM2 Demethylation. Am. J. Transl. Res. 2019, 11, 6084–6092. [Google Scholar]
- Yang, S.; Wei, J.; Cui, Y.-H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m6A mRNA Demethylase FTO Regulates Melanoma Tumorigenicity and Response to Anti-PD-1 Blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Jeschke, J.; Collignon, E.; Al Wardi, C.; Krayem, M.; Bizet, M.; Jia, Y.; Garaud, S.; Wimana, Z.; Calonne, E.; Hassabi, B.; et al. Downregulation of the FTO m6A RNA Demethylase Promotes EMT-Mediated Progression of Epithelial Tumors and Sensitivity to Wnt Inhibitors. Nature Cancer 2021, 2, 611–628. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by m6A-Dependent Translation Control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-Tumour Immunity Controlled through mRNA m6A Methylation and YTHDF1 in Dendritic Cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Hüttelmaier, S.; Liu, T. The Critical Role of RNA m6A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wu, R.; Ming, L. The Role of m6A RNA Methylation in Cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef]
- Huang, H.; Camats-Perna, J.; Medeiros, R.; Anggono, V.; Widagdo, J. Altered Expression of the m6A Methyltransferase METTL3 in Alzheimer’s Disease. Eneuro 2020, 7, 5. [Google Scholar] [CrossRef]
- Han, M.; Liu, Z.; Xu, Y.; Liu, X.; Wang, D.; Li, F.; Wang, Y.; Bi, J. Abnormality of m6A mRNA Methylation Is Involved in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 98. [Google Scholar] [CrossRef]
- Shafik, A.M.; Zhang, F.; Guo, Z.; Dai, Q.; Pajdzik, K.; Li, Y.; Kang, Y.; Yao, B.; Wu, H.; He, C.; et al. N6-Methyladenosine Dynamics in Neurodevelopment and Aging, and Its Potential Role in Alzheimer’s Disease. Genome Biol. 2021, 22, 17. [Google Scholar] [CrossRef]
- Yen, Y.-P.; Chen, J.-A. The m6A Epitranscriptome on Neural Development and Degeneration. J. Biomed. Sci. 2021, 28, 40. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Yue, M.; Wang, J.; Kumar, S.; Wechsler-Reya, R.J.; Zhang, Z.; Ogawa, Y.; Kellis, M.; Duester, G.; et al. N6-Methyladenosine RNA Modification Regulates Embryonic Neural Stem Cell Self-Renewal through Histone Modifications. Nat. Neurosci. 2018, 21, 195–206. [Google Scholar] [CrossRef]
- Wang, C.-X.; Cui, G.-S.; Liu, X.; Xu, K.; Wang, M.; Zhang, X.-X.; Jiang, L.-Y.; Li, A.; Yang, Y.; Lai, W.-Y.; et al. METTL3-Mediated m6A Modification Is Required for Cerebellar Development. PLoS Biol. 2018, 16, e2004880. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.-C.; Huang, C.; Shen, H.; Sun, B.; Cheng, X.; Zhang, Y.-J.; Yang, Y.-G.; Shu, Q.; Yang, Y.; et al. m6A Regulates Neurogenesis and Neuronal Development by Modulating Histone Methyltransferase Ezh2. Genom. Proteom. Bioinform. 2019, 17, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Edens, B.M.; Vissers, C.; Su, J.; Arumugam, S.; Xu, Z.; Shi, H.; Miller, N.; Rojas Ringeling, F.; Ming, G.-L.; He, C.; et al. FMRP Modulates Neural Differentiation through m6A-Dependent mRNA Nuclear Export. Cell Rep. 2019, 28, 845–854.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhai, Y.; Zhang, S.; Dai, X.; Li, Z. Roles of N6-Methyladenosine (m6A) in Stem Cell Fate Decisions and Early Embryonic Development in Mammals. Front. Cell Dev. Biol. 2020, 8, 782. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zang, L.; Zhang, F.; Chen, J.; Shen, H.; Shu, L.; Liang, F.; Feng, C.; Chen, D.; Tao, H.; et al. Fat Mass and Obesity-Associated (FTO) Protein Regulates Adult Neurogenesis. Hum. Mol. Genet. 2017, 26, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Yi, C.; Peng, J. Epitranscriptomics: Toward A Better Understanding of RNA Modifications. Genom. Proteom. Bioinform. 2017, 15, 147–153. [Google Scholar] [CrossRef]
- Yoon, K.-J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.-S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 2017, 171, 877–889.e17. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and Gene Expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef]
- Deng, X.; Su, R.; Weng, H.; Huang, H.; Li, Z.; Chen, J. RNA N6-Methyladenosine Modification in Cancers: Current Status and Perspectives. Cell Res. 2018, 28, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, C.M.; Batista, P.J. It’s Complicate m6A-Dependent Regulation of Gene Expression in Cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 382–393. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Guo, M.; Zheng, X.; Ali, S.; Huang, H.; Zhang, L.; Wang, S.; Huang, Y.; Qie, S.; et al. Down-Regulation of m6A mRNA Methylation Is Involved in Dopaminergic Neuronal Death. ACS Chem. Neurosci. 2019, 10, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Z.; Zhou, J.; Liu, Y.; Fan, R.; Sun, T. Transcriptome and N6-Methyladenosine RNA Methylome Analyses in Aortic Dissection and Normal Human Aorta. Front. Cardiovasc. Med. 2021, 8, 627380. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.-B.; Lei, S.-F.; Zhang, Y.-H.; Zhang, H. Examination of the Associations between m6A-Associated Single-Nucleotide Polymorphisms and Blood Pressure. Hypertens. Res. 2019, 42, 1582–1589. [Google Scholar] [CrossRef]
- Kmietczyk, V.; Riechert, E.; Kalinski, L.; Boileau, E.; Malovrh, E.; Malone, B.; Gorska, A.; Hofmann, C.; Varma, E.; Jürgensen, L.; et al. m6A-mRNA Methylation Regulates Cardiac Gene Expression and Cellular Growth. Life Sci. Alliance 2019, 2, e201800233. [Google Scholar] [CrossRef]
- Kwok, C.-T.; Marshall, A.D.; Rasko, J.E.J.; Wong, J.J.L. Genetic Alterations of m6A Regulators Predict Poorer Survival in Acute Myeloid Leukemia. J. Hematol. Oncol. 2017, 10, 39. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The Rebel Angel: Mutant p53 as the Driving Oncogene in Breast Cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why Are There Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Kang, N.; Wang, Y.; Guo, S.; Ou, Y.; Wang, G.; Chen, J.; Li, D.; Zhan, Q. Mutant TP53 G245C and R273H Promote Cellular Malignancy in Esophageal Squamous Cell Carcinoma. BMC Cell Biol. 2018, 19, 16. [Google Scholar] [CrossRef]
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.-X.J. Hotspot Mutant p53-R273H Inhibits KLF6 Expression to Promote Cell Migration and Tumor Metastasis. Cell Death Dis. 2020, 11, 595. [Google Scholar] [CrossRef]
- Uddin, M.B.; Roy, K.R.; Hosain, S.B.; Khiste, S.K.; Hill, R.A.; Jois, S.D.; Zhao, Y.; Tackett, A.J.; Liu, Y.-Y. An N6-Methyladenosine at the Transited Codon 273 of p53 Pre-mRNA Promotes the Expression of R273H Mutant Protein and Drug Resistance of Cancer Cells. Biochem. Pharmacol. 2019, 160, 134–145. [Google Scholar] [CrossRef]
- Fu, Y.; Zhuang, X. m6A-Binding YTHDF Proteins Promote Stress Granule Formation. Nat. Chem. Biol. 2020, 16, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A Enhances the Phase Separation Potential of mRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef]
- Gao, Y.; Pei, G.; Li, D.; Li, R.; Shao, Y.; Zhang, Q.C.; Li, P. Multivalent m6A Motifs Promote Phase Separation of YTHDF Proteins. Cell Res. 2019, 29, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Feng, Y.; Wu, J.-J.; Zou, M.-L.; Sun, Z.-L.; Li, X.; Yuan, F.-L. m6 A Facilitates YTHDF-Independent Phase Separation. J. Cell. Mol. Med. 2020, 24, 2070–2072. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.B.; Salbego, C.G.; Cimarosti, H. SUMOylation: Novel Neuroprotective Approach for Alzheimer’s Disease? Aging. Dis. 2015, 6, 322–330. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on Its 25th Anniversary: Mechanisms, Pathology, and Emerging Concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Eckermann, K. SUMO and Parkinson’s Disease. Neuromolecular Med. 2013, 15, 737–759. [Google Scholar] [CrossRef]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A Cellular System That Degrades Misfolded Proteins and Protects against Neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.-H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation Inhibits Alpha-Synuclein Aggregation and Toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Kim, Y.M.; Mouradian, M.M.; Chung, K.C. Human Polycomb Protein 2 Promotes α-Synuclein Aggregate Formation through Covalent SUMOylation. Brain Res. 2011, 1381, 78–89. [Google Scholar] [CrossRef]
- Guo, C.; Henley, J.M. Wrestling with Stress: Roles of Protein SUMOylation and deSUMOylation in Cell Stress Response. IUBMB Life 2014, 66, 71–77. [Google Scholar] [CrossRef]
- Enserink, J.M. Sumo and the Cellular Stress Response. Cell Div. 2015, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Karhausen, J.; Ulloa, L.; Yang, W. SUMOylation Connects Cell Stress Responses and Inflammatory Control: Lessons from the Gut as a Model Organ. Front. Immunol. 2021, 12, 646633. [Google Scholar] [CrossRef]
- He, J.; Cheng, J.; Wang, T. SUMOylation-Mediated Response to Mitochondrial Stress. Int. J. Mol. Sci. 2020, 21, 5657. [Google Scholar] [CrossRef]
- Gill, G. Something about SUMO Inhibits Transcription. Curr. Opin. Genet. Dev. 2005, 15, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.D.; Lee, Y.-J.; Liu, K.; Li, G.; Lin, F.-T.; Lin, W.-C. E2F1 Sumoylation as a Protective Cellular Mechanism in Oxidative Stress Response. Proc. Natl. Acad. Sci. USA. 2020, 117, 14958–14969. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide Dismutase 1 Acts as a Nuclear Transcription Factor to Regulate Oxidative Stress Resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef]
- Fei, E.; Jia, N.; Yan, M.; Ying, Z.; Sun, Q.; Wang, H.; Zhang, T.; Ma, X.; Ding, H.; Yao, X.; et al. SUMO-1 Modification Increases Human SOD1 Stability and Aggregation. Biochem. Biophys. Res. Commun. 2006, 347, 406–412. [Google Scholar] [CrossRef]
- Zhang, H.; Kuai, X.; Ji, Z.; Li, Z.; Shi, R. Over-Expression of Small Ubiquitin-Related Modifier-1 and Sumoylated p53 in Colon Cancer. Cell Biochem. Biophys. 2013, 67, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.-B.; Xia, Y.-Y.; Shu, X.-J.; Liu, Z.-C.; Feng, Y.; Liu, X.-H.; Yu, G.; Yin, G.; Xiong, Y.-S.; Zeng, K.; et al. SUMOylation at K340 Inhibits Tau Degradation through Deregulating Its Phosphorylation and Ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, N.T.; Gozal, Y.M.; Dammer, E.B.; Xia, Q.; Duong, D.M.; Cheng, D.; Lah, J.J.; Levey, A.I.; Peng, J. Multiplex SILAC Analysis of a Cellular TDP-43 Proteinopathy Model Reveals Protein Inclusions Associated with SUMOylation and Diverse Polyubiquitin Chains. Mol. Cell Proteom. 2010, 9, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Dangoumau, A.; Veyrat-Durebex, C.; Blasco, H.; Praline, J.; Corcia, P.; Andres, C.R.; Vourc’h, P. Protein SUMOylation, an Emerging Pathway in Amyotrophic Lateral Sclerosis. Int. J. Neurosci. 2013, 123, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Foran, E.; Rosenblum, L.; Bogush, A.I.; Trotti, D. Sumoylation of Critical Proteins in Amyotrophic Lateral Sclerosis: Emerging Pathways of Pathogenesis. Neuromolecular Med. 2013, 15, 760–770. [Google Scholar] [CrossRef]
- Du, Y.; Hou, G.; Zhang, H.; Dou, J.; He, J.; Guo, Y.; Li, L.; Chen, R.; Wang, Y.; Deng, R.; et al. SUMOylation of the m6A-RNA Methyltransferase METTL3 Modulates Its Function. Nucleic Acids Res. 2018, 46, 5195–5208. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Abe, Y.; Niikura, T. SENP1 and SENP2 Regulate SUMOylation of Amyloid Precursor Protein. Heliyon 2018, 4, e00601. [Google Scholar] [CrossRef]
- Xu, H.; Wang, H.; Zhao, W.; Fu, S.; Li, Y.; Ni, W.; Xin, Y.; Li, W.; Yang, C.; Bai, Y.; et al. SUMO1 Modification of Methyltransferase-like 3 Promotes Tumor Progression via Regulating Snail mRNA Homeostasis in Hepatocellular Carcinoma. Theranostics 2020, 10, 5671–5686. [Google Scholar] [CrossRef]
- Hou, G.; Zhao, X.; Li, L.; Yang, Q.; Liu, X.; Huang, C.; Lu, R.; Chen, R.; Wang, Y.; Jiang, B.; et al. SUMOylation of YTHDF2 Promotes mRNA Degradation and Cancer Progression by Increasing Its Binding Affinity with m6A-Modified mRNAs. Nucleic Acids Res. 2021, 49, 2859–2877. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-Methylcytosine Promotes mRNA Export—NSUN2 as the Methyltransferase and ALYREF as an m5C Reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Dai, X.; Gonzalez, G.; Li, L.; Li, J.; You, C.; Miao, W.; Hu, J.; Fu, L.; Zhao, Y.; Li, R.; et al. YTHDF2 Binds to 5-Methylcytosine in RNA and Modulates the Maturation of Ribosomal RNA. Anal. Chem. 2020, 92, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Ji, X.; Wu, X.; Chen, J.; Li, X.; Jiang, H.; Fu, H.; Wang, H.; Lin, Z.; Tang, X.; et al. Melatonin Antagonizes Ovarian Aging via YTHDF2-MAPK-NF-κB Pathway. Genes Dis. 2020. [Google Scholar] [CrossRef]
- Lv, Y.; Li, T.; Yang, M.; Su, L.; Zhu, Z.; Zhao, S.; Zeng, W.; Zheng, Y. Melatonin Attenuates Chromium (VI)-Induced Spermatogonial Stem Cell/Progenitor Mitophagy by Restoration of METTL3-Mediated RNA N6-Methyladenosine Modification. Front. Cell Dev. Biol. 2021, 9, 684398. [Google Scholar] [CrossRef]
- Li, H.; Chen, Q.; Li, S.; Yao, W.; Li, L.; Shi, X.; Wang, L.; Castranova, V.; Vallyathan, V.; Ernst, E.; et al. Effect of Cr(VI) Exposure on Sperm Quality: Human and Animal Studies. Ann. Occup. Hyg. 2001, 45, 505–511. [Google Scholar] [CrossRef]
- Das, J.; Kang, M.-H.; Kim, E.; Kwon, D.-N.; Choi, Y.-J.; Kim, J.-H. Hexavalent Chromium Induces Apoptosis in Male Somatic and Spermatogonial Stem Cells via Redox Imbalance. Sci. Rep. 2015, 5, 13921. [Google Scholar] [CrossRef]
- Zhao, B.S.; He, C. Fate by RNA Methylation: m6A Steers Stem Cell Pluripotency. Genome Biol. 2015, 16, 43. [Google Scholar] [CrossRef]
- Yang, L.; Liu, X.; Song, L.; Su, G.; Di, A.; Bai, C.; Wei, Z.; Li, G. Melatonin Restores the Pluripotency of Long-Term-Cultured Embryonic Stem Cells through Melatonin Receptor-Dependent m6A RNA Regulation. J. Pineal. Res. 2020, 69, e12669. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-Methyladenosine Modification Destabilizes Developmental Regulators in Embryonic Stem Cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Aguilo, F.; Zhang, F.; Sancho, A.; Fidalgo, M.; Di Cecilia, S.; Vashisht, A.; Lee, D.-F.; Chen, C.-H.; Rengasamy, M.; Andino, B.; et al. Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell 2015, 17, 689–704. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, C.; Qi, S.; Guo, S.; Chen, Y.; Du, E.; Zhang, H.; Wang, X.; Liu, R.; Qiao, B.; et al. Elevated Expression of ZNF217 Promotes Prostate Cancer Growth by Restraining Ferroportin-Conducted Iron Egress. Oncotarget 2016, 7, 84893–84906. [Google Scholar] [CrossRef]
- Matsumoto, T.; Uchiumi, T.; Monji, K.; Yagi, M.; Setoyama, D.; Amamoto, R.; Matsushima, Y.; Shiota, M.; Eto, M.; Kang, D. Doxycycline Induces Apoptosis via ER Stress Selectively to Cells with a Cancer Stem Cell-like Properties: Importance of Stem Cell Plasticity. Oncogenesis 2017, 6, 397. [Google Scholar] [CrossRef] [PubMed]
- Gerken, T.; Girard, C.A.; Tung, Y.-C.L.; Webby, C.J.; Saudek, V.; Hewitson, K.S.; Yeo, G.S.H.; McDonough, M.A.; Cunliffe, S.; McNeill, L.A.; et al. The Obesity-Associated FTO Gene Encodes a 2-Oxoglutarate-Dependent Nucleic Acid Demethylase. Science 2007, 318, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64–80.e9. [Google Scholar] [CrossRef]
- Shah, A.; Rashid, F.; Awan, H.M.; Hu, S.; Wang, X.; Chen, L.; Shan, G. The DEAD-Box RNA Helicase DDX3 Interacts with m6A RNA Demethylase ALKBH5. Stem Cells Int. 2017, 2017, 8596135. [Google Scholar] [CrossRef]
- Kerr, C.L.; Bol, G.M.; Vesuna, F.; Raman, V. Targeting RNA Helicase DDX3 in Stem Cell Maintenance and Teratoma Formation. Genes Cancer 2019, 10, 11–20. [Google Scholar] [CrossRef][Green Version]
- Hilbert, M.; Karow, A.R.; Klostermeier, D. The Mechanism of ATP-Dependent RNA Unwinding by DEAD Box Proteins. Biol. Chem. 2009, 390, 1237–1250. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; Russell, R. DEAD-Box Proteins as RNA Helicases and Chaperones. Wiley Interdiscip. Rev. RNA 2011, 2, 135–152. [Google Scholar] [CrossRef]
- Ledoux, S.; Guthrie, C. Regulation of the Dbp5 ATPase Cycle in mRNP Remodeling at the Nuclear Pore: A Lively New Paradigm for DEAD-Box Proteins. Genes Dev. 2011, 25, 1109–1114. [Google Scholar] [CrossRef]
- Young, C.L.; Khoshnevis, S.; Karbstein, K. Cofactor-Dependent Specificity of a DEAD-Box Protein. Proc. Natl. Acad. Sci. USA 2013, 110, E2668–E2676. [Google Scholar] [CrossRef] [PubMed]
- Conaway, R.C.; Conaway, J.W. ATP Activates Transcription Initiation from Promoters by RNA Polymerase II in a Reversible Step prior to RNA Synthesis. J. Biol. Chem. 1988, 263, 2962–2968. [Google Scholar] [CrossRef]
- Sassanfar, M.; Szostak, J.W. An RNA Motif That Binds ATP. Nature 1993, 364, 550–553. [Google Scholar] [CrossRef]
- White, H.B. Coenzymes as Fossils of an Earlier Metabolic State. J. Mol. Evol. 1976, 7, 101–104. [Google Scholar] [CrossRef]
- Goldman, A.D.; Kacar, B. Cofactors Are Remnants of Life’s Origin and Early Evolution. J. Mol. Evol. 2021, 89, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Lymn, R.W.; Taylor, E.W. Mechanism of Adenosine Triphosphate Hydrolysis by Actomyosin. Biochemistry 1971, 10, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Theissen, B.; Karow, A.R.; Köhler, J.; Gubaev, A.; Klostermeier, D. Cooperative Binding of ATP and RNA Induces a Closed Conformation in a DEAD Box RNA Helicase. Proc. Natl. Acad. Sci. USA 2008, 105, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Henn, A.; Bradley, M.J.; De La Cruz, E.M. ATP Utilization and RNA Conformational Rearrangement by DEAD-Box Proteins. Annu. Rev. Biophys. 2012, 41, 247–267. [Google Scholar] [CrossRef]
- Song, H.; Ji, X. The Mechanism of RNA Duplex Recognition and Unwinding by DEAD-Box Helicase DDX3X. Nat. Commun. 2019, 10, 3085. [Google Scholar] [CrossRef]
- Liu, F.; Putnam, A.A.; Jankowsky, E. DEAD-Box Helicases Form Nucleotide-Dependent, Long-Lived Complexes with RNA. Biochemistry 2014, 53, 423–433. [Google Scholar] [CrossRef]
- Hooper, C.; Hilliker, A. Packing Them up and Dusting Them off: RNA Helicases and mRNA Storage. Biochim. Biophys. Acta 2013, 1829, 824–834. [Google Scholar] [CrossRef]
- Pimentel, J.; Boccaccio, G.L. Translation and Silencing in RNA Granules: A Tale of Sand Grains. Front. Mol. Neurosci. 2014, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.F.; Mortreux, F.; Auboeuf, D. The Multiple Functions of RNA Helicases as Drivers and Regulators of Gene Expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Beißel, C.; Grosse, S.; Krebber, H. Dbp5/DDX19 between Translational Readthrough and Nonsense Mediated Decay. Int. J. Mol. Sci. 2020, 21, 1085. [Google Scholar] [CrossRef] [PubMed]
- Alluri, R.K.; Li, Z.; McCrae, K.R. Stress Granule-Mediated Oxidized RNA Decay in P-Body: Hypothetical Role of ADAR1, Tudor-SN, and STAU1. Front. Mol. Biosci. 2021, 8, 672988. [Google Scholar] [CrossRef]
- Tseng, S.S.; Weaver, P.L.; Liu, Y.; Hitomi, M.; Tartakoff, A.M.; Chang, T.H. Dbp5p, a Cytosolic RNA Helicase, Is Required for poly(A)+ RNA Export. EMBO J. 1998, 17, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Hodge, C.A.; Colot, H.V.; Stafford, P.; Cole, C.N. Rat8p/Dbp5p Is a Shuttling Transport Factor That Interacts with Rat7p/Nup159p and Gle1p and Suppresses the mRNA Export Defect of xpo1-1 Cells. EMBO J. 1999, 18, 5778–5788. [Google Scholar] [CrossRef]
- Hodge, C.A.; Tran, E.J.; Noble, K.N.; Alcazar-Roman, A.R.; Ben-Yishay, R.; Scarcelli, J.J.; Folkmann, A.W.; Shav-Tal, Y.; Wente, S.R.; Cole, C.N. The Dbp5 Cycle at the Nuclear Pore Complex during mRNA Export I: dbp5 Mutants with Defects in RNA Binding and ATP Hydrolysis Define Key Steps for Nup159 and Gle1. Genes Dev. 2011, 25, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Noble, K.N.; Tran, E.J.; Alcázar-Román, A.R.; Hodge, C.A.; Cole, C.N.; Wente, S.R. The Dbp5 Cycle at the Nuclear Pore Complex during mRNA Export II: Nucleotide Cycling and mRNP Remodeling by Dbp5 Are Controlled by Nup159 and Gle1. Genes Dev. 2011, 25, 1065–1077. [Google Scholar] [CrossRef]
- Okamura, M.; Yamanaka, Y.; Shigemoto, M.; Kitadani, Y.; Kobayashi, Y.; Kambe, T.; Nagao, M.; Kobayashi, I.; Okumura, K.; Masuda, S. Depletion of mRNA Export Regulator DBP5/DDX19, GLE1 or IPPK That Is a Key Enzyme for the Production of IP6, Resulting in Differentially Altered Cytoplasmic mRNA Expression and Specific Cell Defect. PLoS ONE 2018, 13, e0197165. [Google Scholar] [CrossRef]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martinez, S.; Tsanov, N.; Abou Merhi, R.; Maiorano, D. An ATR-Dependent Function for the Ddx19 RNA Helicase in Nuclear R-Loop Metabolism. EMBO J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef]
- Hodroj, D.; Serhal, K.; Maiorano, D. Ddx19 Links mRNA Nuclear Export with Progression of Transcription and Replication and Suppresses Genomic Instability upon DNA Damage in Proliferating Cells. Nucleus 2017, 8, 489–495. [Google Scholar] [CrossRef]
- Lin, D.H.; Correia, A.R.; Cai, S.W.; Huber, F.M.; Jette, C.A.; Hoelz, A. Structural and Functional Analysis of mRNA Export Regulation by the Nuclear Pore Complex. Nat. Commun. 2018, 9, 2319. [Google Scholar] [CrossRef] [PubMed]
- Sumner, M.C.; Brickner, J. The Nuclear Pore Complex as a Transcription Regulator. Cold Spring Harb. Perspect. Biol. 2021, a039438. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.Z.; Klostermeier, D. The DEAD-Box Helicase eIF4A: Paradigm or the Odd One Out? RNA Biol. 2013, 10, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Tauber, D.; Tauber, G.; Khong, A.; Van Treeck, B.; Pelletier, J.; Parker, R. Modulation of RNA Condensation by the DEAD-Box Protein eIF4A. Cell 2020, 180, 411–426.e16. [Google Scholar] [CrossRef]
- Torbati, M.; Lele, T.P.; Agrawal, A. Ultradonut Topology of the Nuclear Envelope. Proc. Natl. Acad. Sci. USA 2016, 113, 11094–11099. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Lele, T.P. Mechanics of Nuclear Membranes. J. Cell Sci. 2019, 132, jcs229245. [Google Scholar] [CrossRef] [PubMed]
- Fiserova, J.; Kiseleva, E.; Goldberg, M.W. Nuclear Envelope and Nuclear Pore Complex Structure and Organization in Tobacco BY-2 Cells. Plant J. 2009, 59, 243–255. [Google Scholar] [CrossRef]
- Mattaj, I.W. Sorting out the Nuclear Envelope from the Endoplasmic Reticulum. Nat. Rev. Mol. Cell Biol. 2004, 5, 65–69. [Google Scholar] [CrossRef]
- De Magistris, P.; Antonin, W. The Dynamic Nature of the Nuclear Envelope. Curr. Biol. 2018, 28, R487–R497. [Google Scholar] [CrossRef]
- Anderson, D.J.; Hetzer, M.W. Shaping the Endoplasmic Reticulum into the Nuclear Envelope. J. Cell Sci. 2008, 121 Pt 2, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Dazzoni, R.; Grélard, A.; Morvan, E.; Bouter, A.; Applebee, C.J.; Loquet, A.; Larijani, B.; Dufourc, E.J. The Unprecedented Membrane Deformation of the Human Nuclear Envelope, in a Magnetic Field, Indicates Formation of Nuclear Membrane Invaginations. Sci. Rep. 2020, 10, 5147. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.H.; Bardarov, S.; Jiang, Z. Molecular Aspects of Diagnostic Nucleolar and Nuclear Envelope Changes in Prostate Cancer. J. Cell. Biochem. 2004, 91, 170–184. [Google Scholar] [CrossRef]
- Herlan, G.; Giese, G.; Wunderlich, F. Influence of Nuclear Membrane Lipid Fluidity on Nuclear RNA Release. Exp. Cell Res. 1979, 118, 305–309. [Google Scholar] [CrossRef]
- Hodge, C.A.; Choudhary, V.; Wolyniak, M.J.; Scarcelli, J.J.; Schneiter, R.; Cole, C.N. Integral Membrane Proteins Brr6 and Apq12 Link Assembly of the Nuclear Pore Complex to Lipid Homeostasis in the Endoplasmic Reticulum. J. Cell Sci. 2010, 123 Pt 1, 141–151. [Google Scholar] [CrossRef]
- Scarcelli, J.J.; Hodge, C.A.; Cole, C.N. The Yeast Integral Membrane Protein Apq12 Potentially Links Membrane Dynamics to Assembly of Nuclear Pore Complexes. J. Cell Biol. 2007, 178, 799–812. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Kennedy, K.A.; Trush, M.A.; Sinha, B.K. Adriamycin-Enhanced Membrane Lipid Peroxidation in Isolated Rat Nuclei. Cancer Res. 1985, 45, 3296–3304. [Google Scholar]
- Vaca, C.E.; Harms-Ringdahl, M. Nuclear Membrane Lipid Peroxidation Products Bind to Nuclear Macromolecules. Arch. Biochem. Biophys. 1989, 269, 548–554. [Google Scholar] [CrossRef]
- Jarsch, I.K.; Daste, F.; Gallop, J.L. Membrane Curvature in Cell Biology: An Integration of Molecular Mechanisms. J. Cell Biol. 2016, 214, 375–387. [Google Scholar] [CrossRef]
- Antonin, W.; Ellenberg, J.; Dultz, E. Nuclear Pore Complex Assembly through the Cell Cycle: Regulation and Membrane Organization. FEBS Lett. 2008, 582, 2004–2016. [Google Scholar] [CrossRef]
- Antonin, W.; Mattaj, I.W. Nuclear Pore Complexes: Round the Bend? Nat. Cell Biol. 2005, 7, 10–12. [Google Scholar] [CrossRef]
- Mészáros, N.; Cibulka, J.; Mendiburo, M.J.; Romanauska, A.; Schneider, M.; Köhler, A. Nuclear Pore Basket Proteins Are Tethered to the Nuclear Envelope and Can Regulate Membrane Curvature. Dev. Cell 2015, 33, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Schooley, A.; Sachdev, R.; Eisenhardt, N.; Schneider, A.M.; Sieverding, C.; Madlung, J.; Gerken, U.; Macek, B.; Antonin, W. Dimerization and Direct Membrane Interaction of Nup53 Contribute to Nuclear Pore Complex Assembly. EMBO J. 2012, 31, 4072–4084. [Google Scholar] [CrossRef] [PubMed]
- Bahmanyar, S.; Schlieker, C. Lipid and Protein Dynamics That Shape Nuclear Envelope Identity. Mol. Biol. Cell 2020, 31, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Rangamani, P.; Mandadap, K.K.; Oster, G. Protein-Induced Membrane Curvature Alters Local Membrane Tension. Biophys. J. 2014, 107, 751–762. [Google Scholar] [CrossRef]
- Hinderliter, A.; May, S. Cooperative Adsorption of Proteins onto Lipid Membranes. J. Phys. Condens. Matter 2006, 18, S1257–S1270. [Google Scholar] [CrossRef] [PubMed]
- Cascianelli, G.; Villani, M.; Tosti, M.; Marini, F.; Bartoccini, E.; Magni, M.V.; Albi, E. Lipid Microdomains in Cell Nucleus. Mol. Biol. Cell 2008, 19, 5289–5295. [Google Scholar] [CrossRef]
- Vlassov, A.; Khvorova, A.; Yarus, M. Binding and Disruption of Phospholipid Bilayers by Supramolecular RNA Complexes. Proc. Natl. Acad. Sci. USA 2001, 98, 7706–7711. [Google Scholar] [CrossRef]
- Khvorova, A.; Kwak, Y.G.; Tamkun, M.; Majerfeld, I.; Yarus, M. RNAs That Bind and Change the Permeability of Phospholipid Membranes. Proc. Natl. Acad. Sci. USA 1999, 96, 10649–10654. [Google Scholar] [CrossRef] [PubMed]
- Janas, T.; Janas, T.; Yarus, M. Human tRNA(Sec) Associates with HeLa Membranes, Cell Lipid Liposomes, and Synthetic Lipid Bilayers. RNA 2012, 18, 2260–2268. [Google Scholar] [CrossRef]
- Janas, T.; Janas, T.; Yarus, M. Specific RNA Binding to Ordered Phospholipid Bilayers. Nucleic Acids Res. 2006, 34, 2128–2136. [Google Scholar] [CrossRef]
- Lehman, N. The RNA World: 4,000,000,050 Years Old. Life 2015, 5, 1583–1586. [Google Scholar] [CrossRef]
- Horning, D.P.; Joyce, G.F. Amplification of RNA by an RNA Polymerase Ribozyme. Proc. Natl. Acad. Sci. USA 2016, 113, 9786–9791. [Google Scholar] [CrossRef]
- Xu, J.; Green, N.J.; Gibard, C.; Krishnamurthy, R.; Sutherland, J.D. Prebiotic Phosphorylation of 2-Thiouridine Provides Either Nucleotides or DNA Building Blocks via Photoreduction. Nat. Chem. 2019, 11, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Watkins, K.P.; Williams-Carrier, R.; Chotewutmontri, P.; Friso, G.; Teubner, M.; Belcher, S.; Ruwe, H.; Schmitz-Linneweber, C.; van Wijk, K.J.; Barkan, A. Exploring the Proteome Associated with the mRNA Encoding the D1 Reaction Center Protein of Photosystem II in Plant Chloroplasts. Plant J. 2020, 102, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Bock, R. Chloroplast Translation: Structural and Functional Organization, Operational Control, and Regulation. Plant Cell 2018, 30, 745–770. [Google Scholar] [CrossRef] [PubMed]
- Gawroński, P.; Enroth, C.; Kindgren, P.; Marquardt, S.; Karpiński, S.; Leister, D.; Jensen, P.E.; Vinther, J.; Scharff, L.B. Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region. Cells 2021, 10, 322. [Google Scholar] [CrossRef]
- Preiss, S.; Schrader, S.; Johanningmeier, U. Rapid, ATP-Dependent Degradation of a Truncated D1 Protein in the Chloroplast. Eur. J. Biochem. 2001, 268, 4562–4569. [Google Scholar] [CrossRef]
- Pévet, P. Melatonin in Animal Models. Dialogues Clin. Neurosci. 2003, 5, 343–352. [Google Scholar]
- Zheng, X.; Tan, D.X.; Allan, A.C.; Zuo, B.; Zhao, Y.; Reiter, R.J.; Wang, L.; Wang, Z.; Guo, Y.; Zhou, J.; et al. Chloroplastic Biosynthesis of Melatonin and Its Involvement in Protection of Plants from Salt Stress. Sci. Rep. 2017, 7, 41236. [Google Scholar] [CrossRef]
- Hwang, O.J.; Kang, K.; Back, K. Effects of Light Quality and Phytochrome Form on Melatonin Biosynthesis in Rice. Biomolecules 2020, 10, 523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, H.; Cao, K.; Hu, L.; Du, T.; Baluška, F.; Zou, Z. Beneficial Roles of Melatonin on Redox Regulation of Photosynthetic Electron Transport and Synthesis of D1 Protein in Tomato Seedlings under Salt Stress. Front. Plant Sci. 2016, 7, 1823. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, D.; Reiter, R.J. Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants 2021, 10, 1483. https://doi.org/10.3390/antiox10091483
Loh D, Reiter RJ. Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants. 2021; 10(9):1483. https://doi.org/10.3390/antiox10091483
Chicago/Turabian StyleLoh, Doris, and Russel J. Reiter. 2021. "Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders" Antioxidants 10, no. 9: 1483. https://doi.org/10.3390/antiox10091483
APA StyleLoh, D., & Reiter, R. J. (2021). Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants, 10(9), 1483. https://doi.org/10.3390/antiox10091483