
7S,15R-Dihydroxy-16S,17S-Epoxy-Docosapentaenoic Acid, a Novel DHA Epoxy Derivative, Inhibits Colorectal Cancer Stemness through Repolarization of Tumor-Associated Macrophage Functions and the ROS/STAT3 Signaling Pathway

 and
and
Abstract
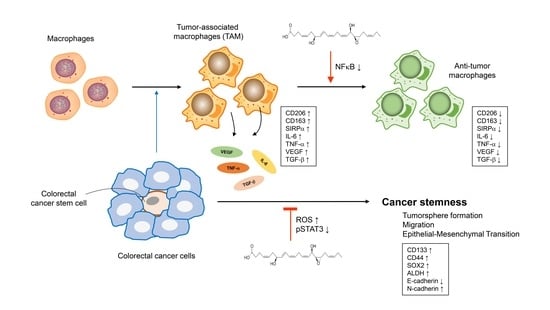
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Determination of the Cytokines IL-6, TNF-α, VEGF and TGF-β1
2.3. Cancer Cell Culture and Collection of Conditioned Medium
2.4. TAM Polarization
2.5. Quantitative Measurement of Human Inflammatory Cytokines
2.6. Cell Proliferation
2.7. Phagocytosis Assay
2.8. Tumorspheres Formation
2.9. Scratch, Migration, and Invasion Assays
2.10. Flow Cytometric Analysis of ALDH Activity and CD47 Expression
2.11. Measurement of ROS Activity
2.12. Gene Expression Analysis
2.13. Western Blot Analysis
2.14. Detection of NF-κB (p65) and STAT3 Transcription Factor Activity
2.15. Chemotherapy of Colorectal Cancer Cell-Bearing BALB/c Mice
2.16. Statistical Analysis
3. Results
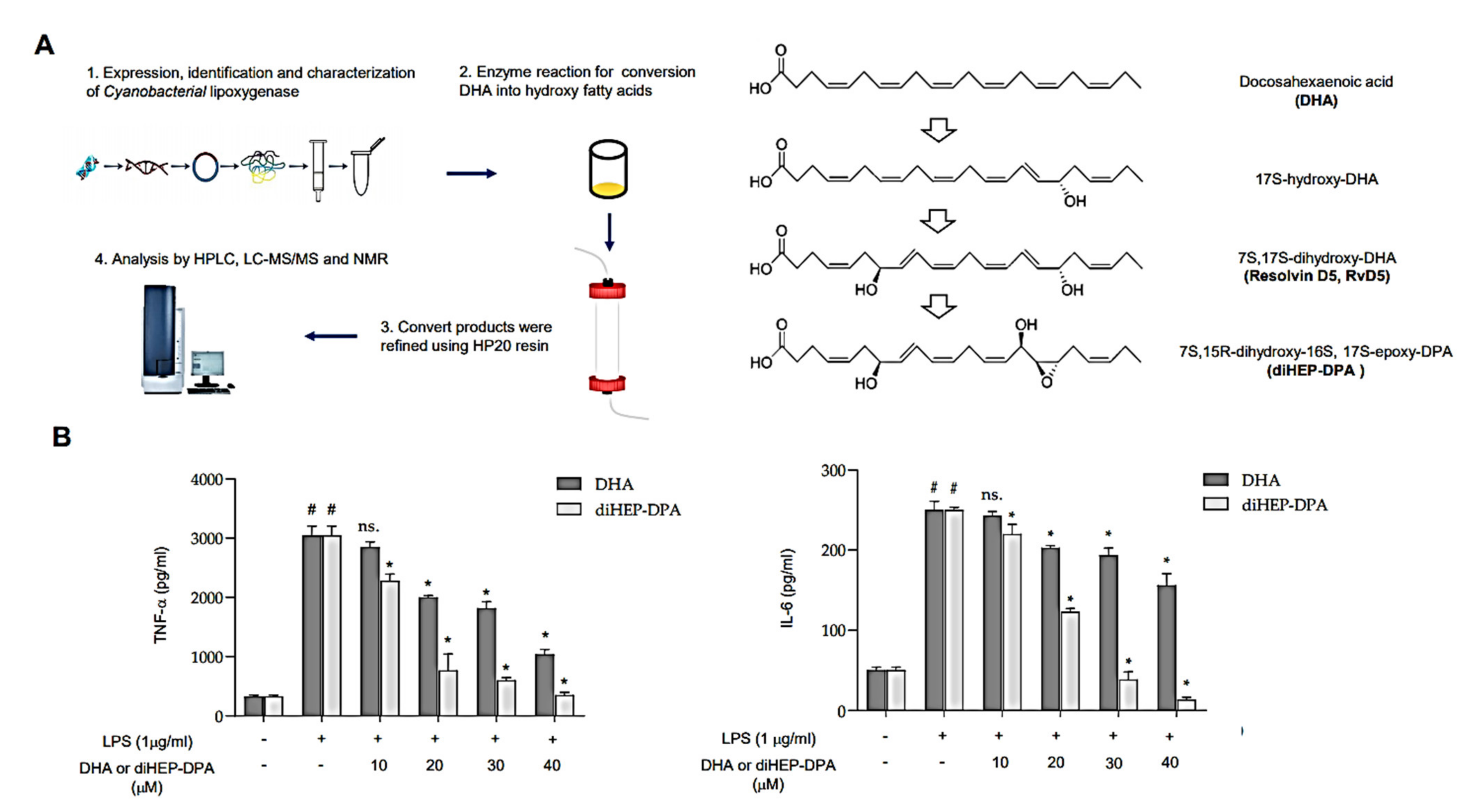
3.1. diHEP-DPA Suppresses Secretion of IL-6 and TNF-α by LPS-Stimulated Macrophage Cells
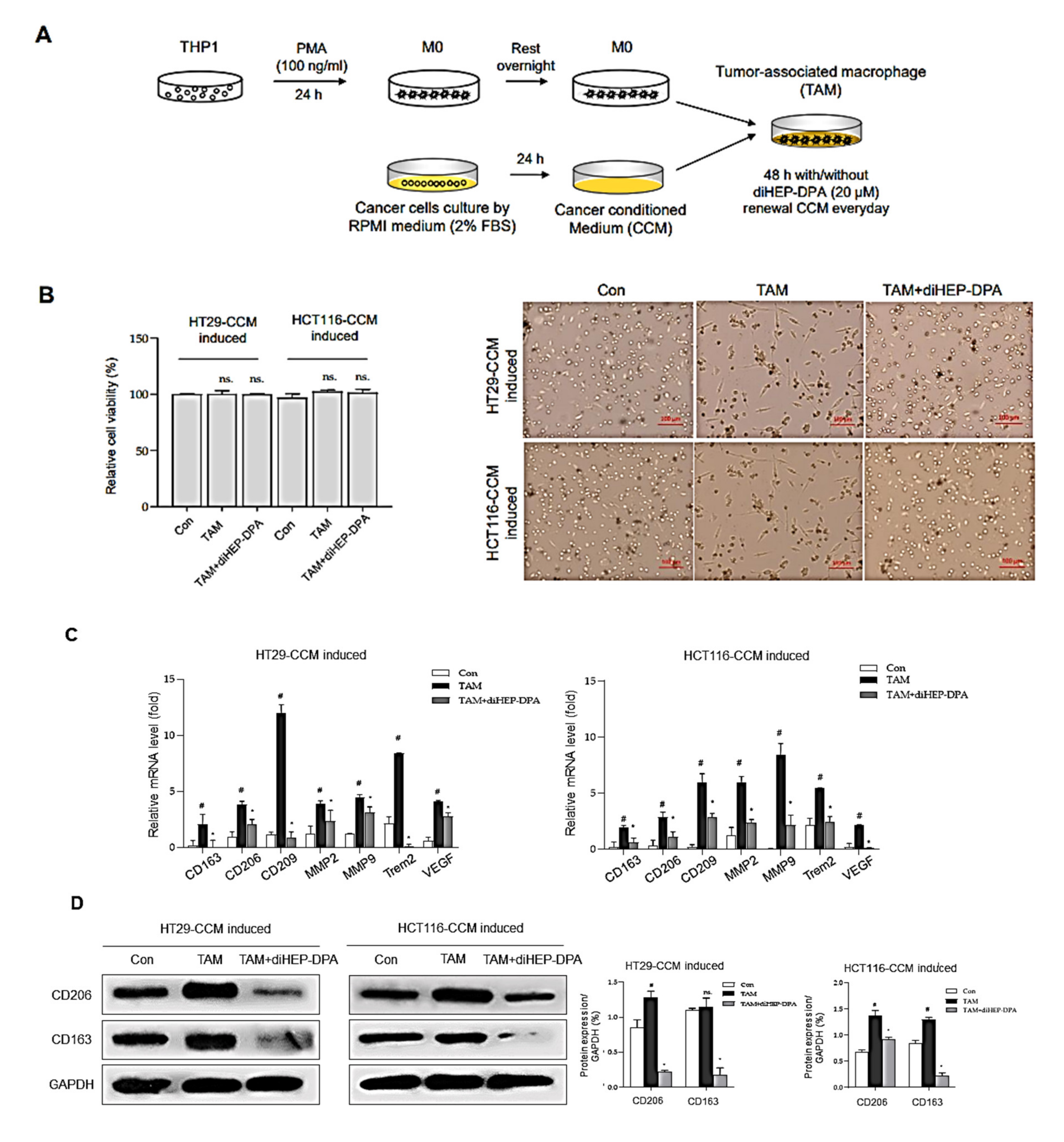
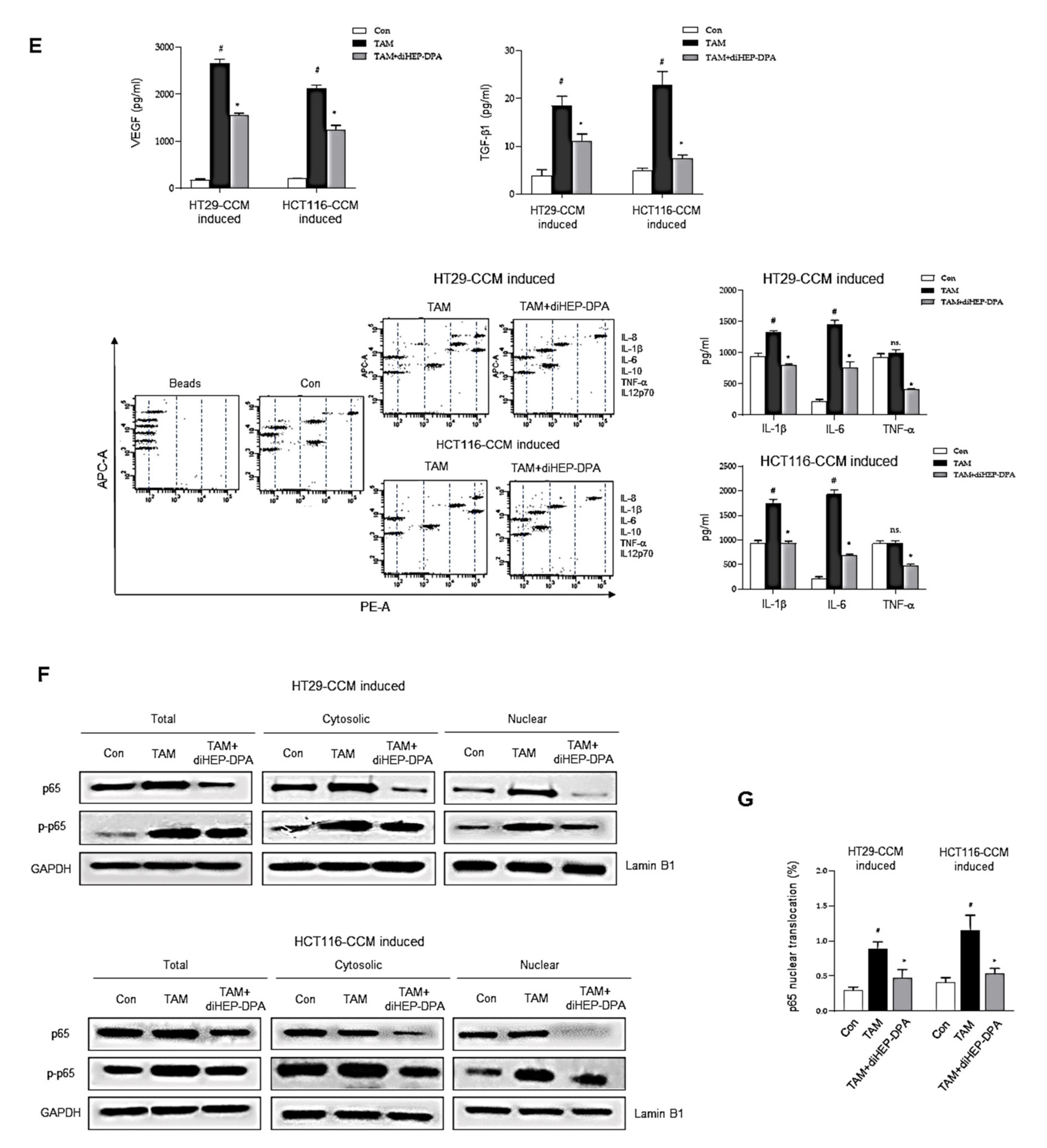
3.2. diHEP-DPA Modulates TAM Polarization via the NF-κB Signaling Pathway
3.3. DiHEP-DPA Enhances TAM Phagocytic Activity by Blocking the CD47/SIRPα Axis
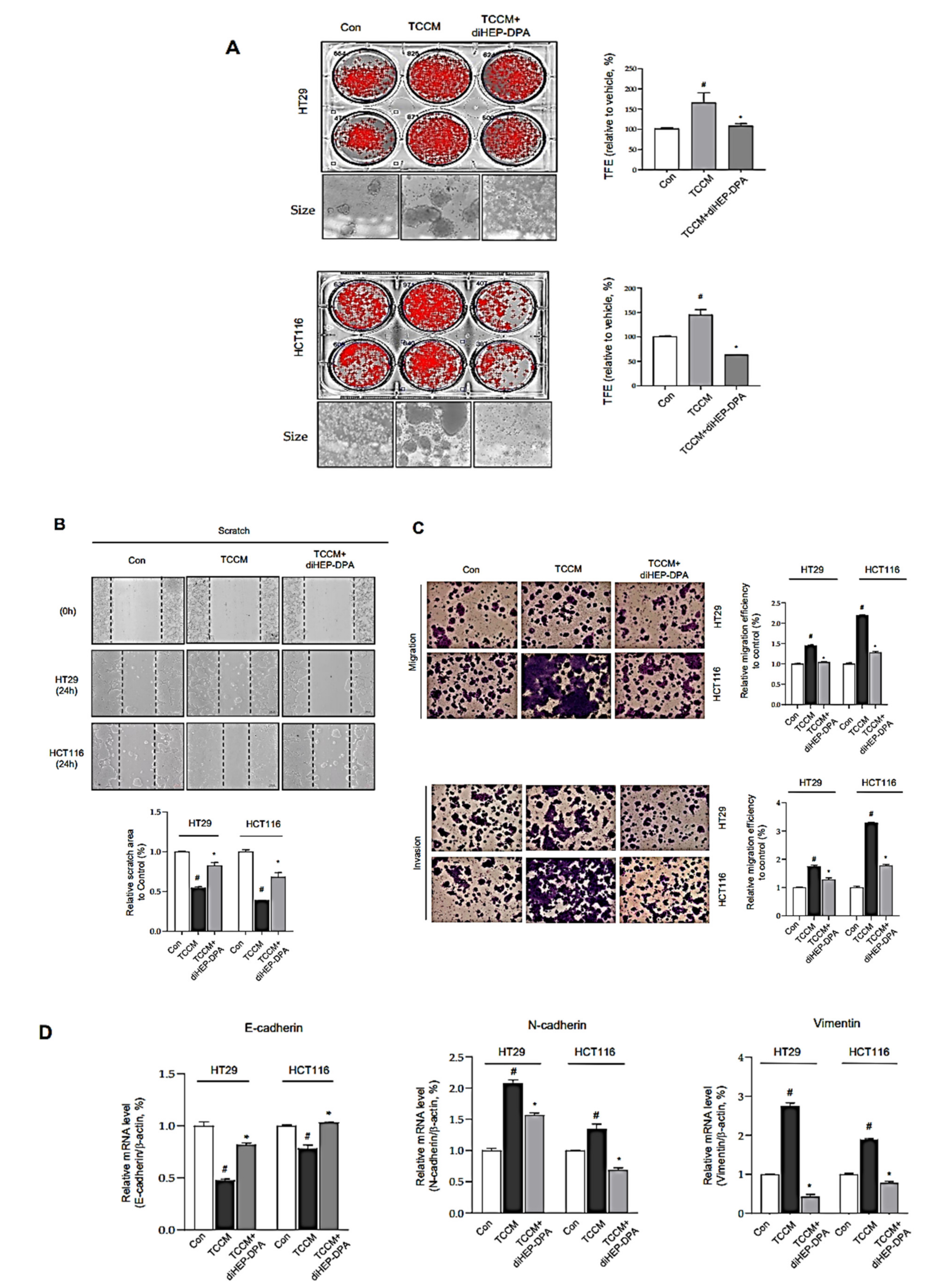
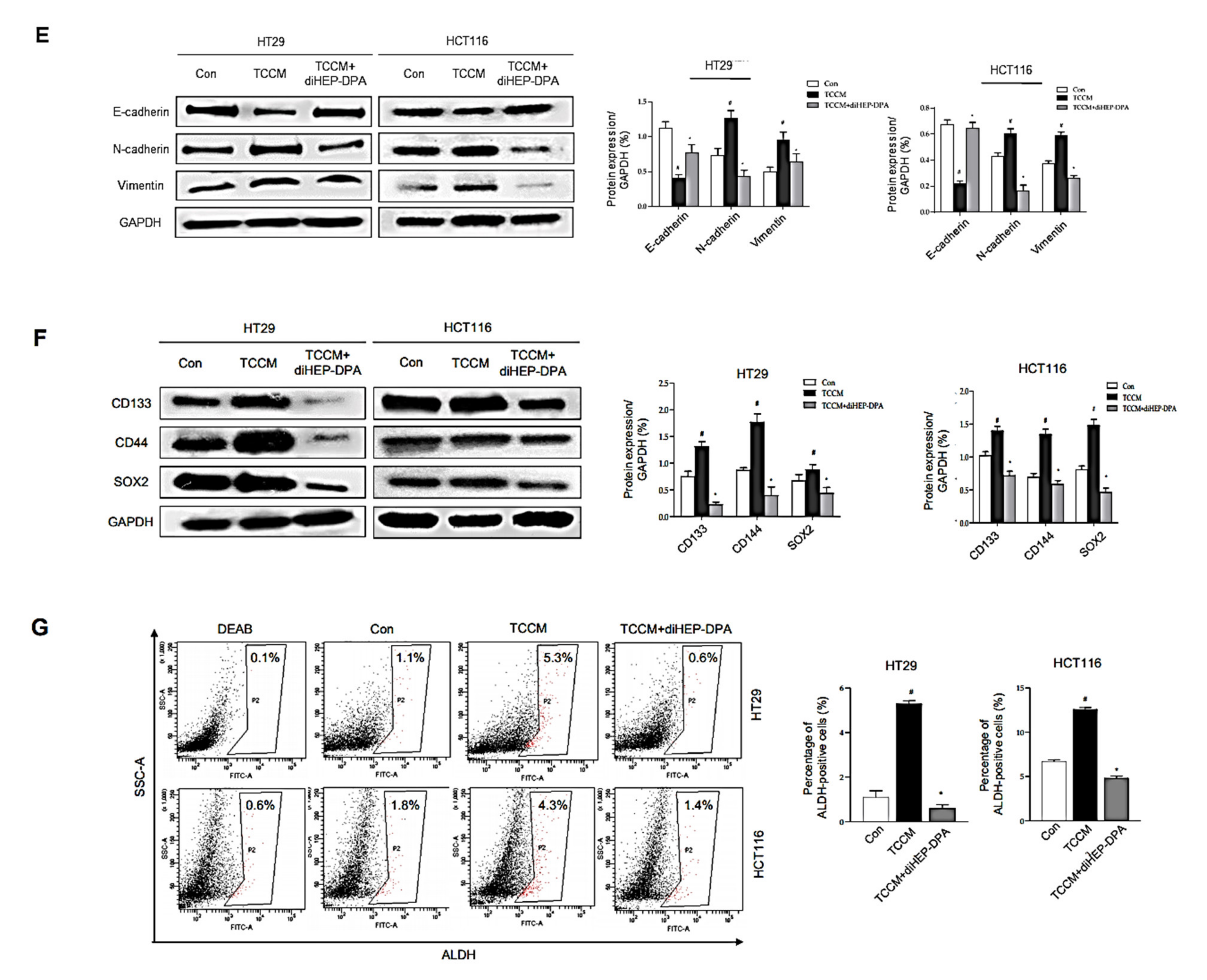
3.4. DiHEP-DPA Impedes TAM-Induced EMT and Acquisition of Stem-like Properties in CRCs
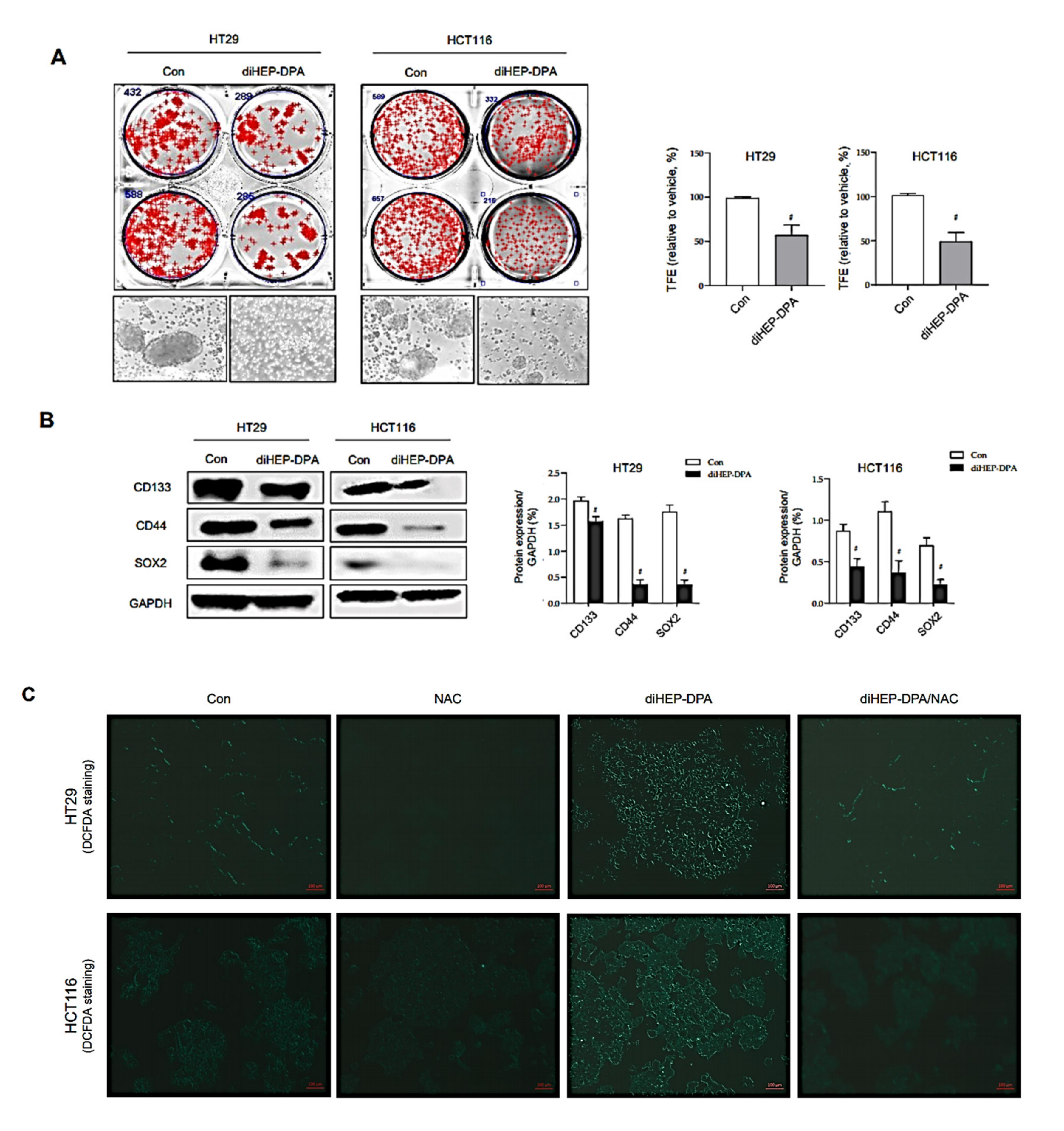
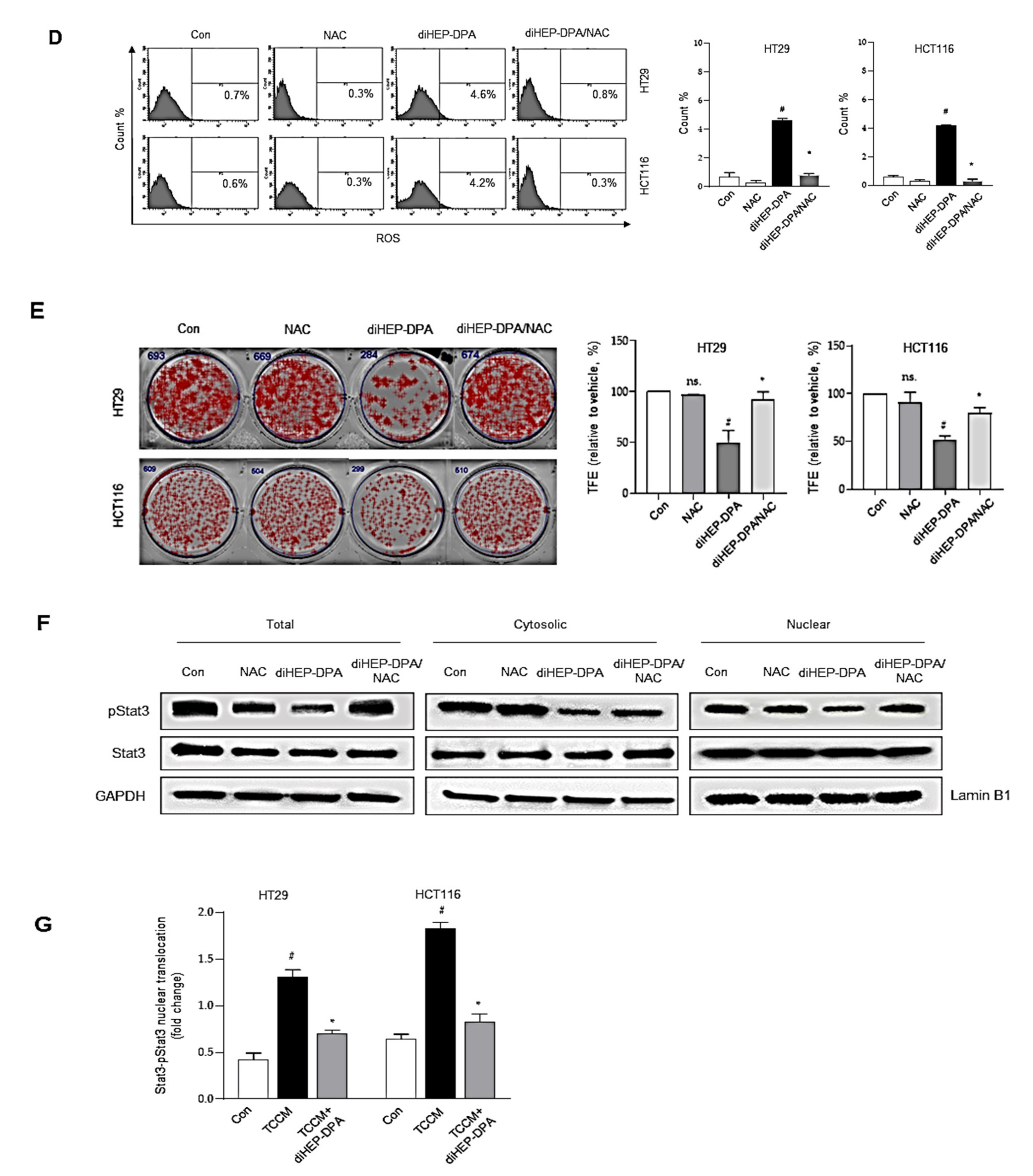
3.5. DiHEP-DPA Reduces Cancer Stemness by Increasing ROS Production
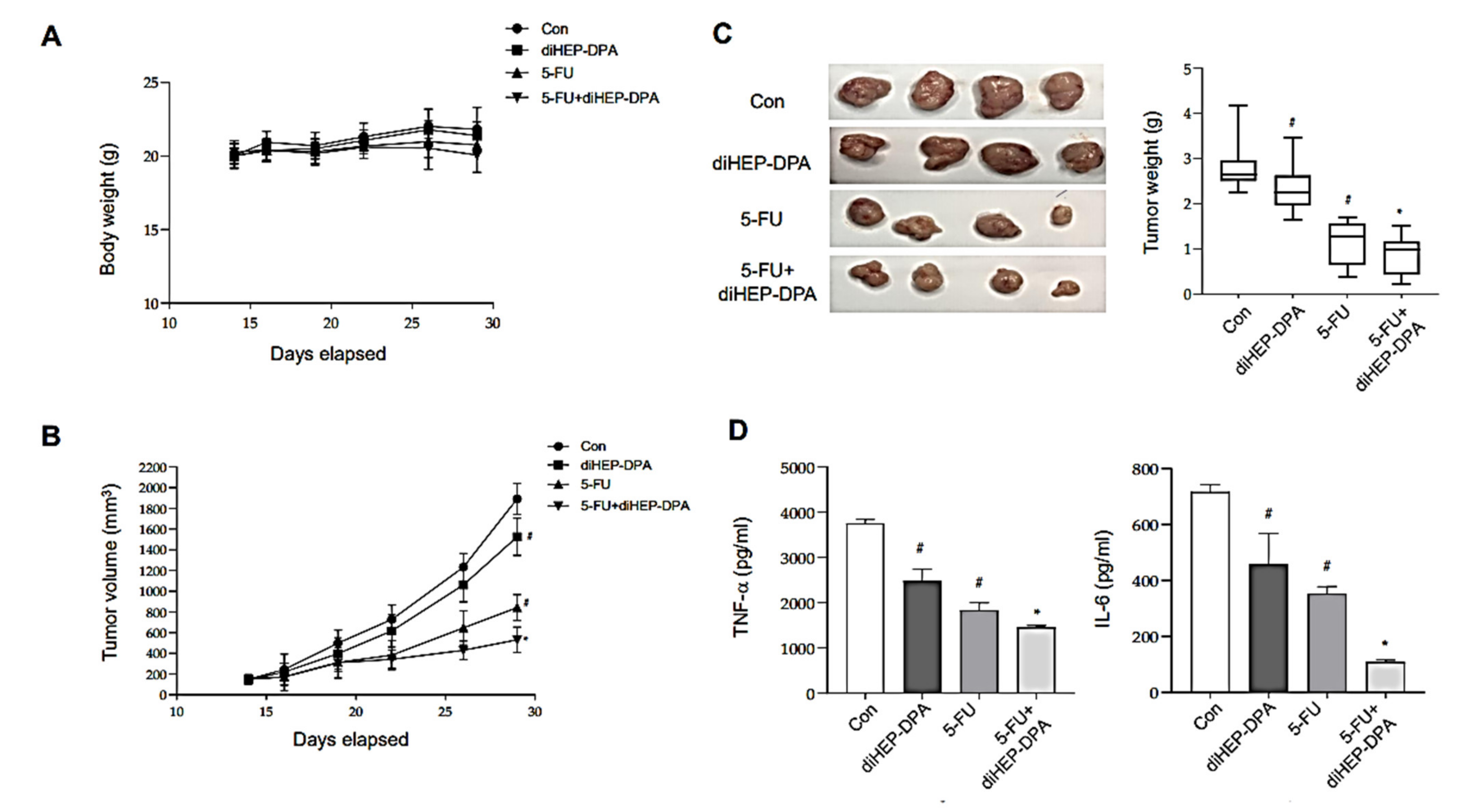
3.6. DiHEP-DPA Inhibits Tumor Growth in a Xenograft Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, J.J.; Lau, J.Y.; Goh, K.; Leung, W. For the Asia Pacific Working Group on Colorectal Cancer. Increasing incidence of colorectal cancer in Asia: Implications for screening. Lancet Oncol. 2005, 6, 871–876. [Google Scholar] [CrossRef]
- Meyer, J.E.; Narang, T.; Schnoll-Sussman, F.H.; Pochapin, M.B.; Christos, P.J.; Sherr, D.L. Increasing incidence of rectal cancer in patients aged younger than 40 years: An analysis of the surveillance, epidemiology, and end results database. Cancer 2010, 116, 4354–4359. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Hsiao, Y.-J.; Chen, H.-Y.; Chen, H.-W.; Ho, C.-C.; Chen, Y.-Y.; Liu, Y.-C.; Hong, T.-H.; Yu, S.-L.; Chen, J.J.W.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell. Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, E.; Lee, E.J.; Nam, G.-H.; Hong, Y.; Cho, E.; Yang, Y.; Kim, I.-S. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 2017, 121, 121–129. [Google Scholar] [CrossRef]
- Lian, S.; Xie, R.; Ye, Y.; Xie, X.; Li, S.; Lu, Y.; Li, B.; Cheng, Y.; Katanaev, V.L.; Jia, L. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine 2019, 42, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Dieter, S.M.; Glimm, H.; Ball, C.R. Colorectal cancer-initiating cells caught in the act. EMBO Mol. Med. 2017, 9, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Gerovska, D.; Garcia-Gallastegi, P.; Descarpentrie, J.; Crende, O.; Casado-Andres, M.; Martin, A.; Eguia, J.; Khatib, A.M.; Arauzo-Bravo, M.J.; Badiola, I. Proprotein convertases blockage up-regulates specifically metallothioneins coding genes in human colon cancer stem cells. Biochim. Et Biophys. Acta. Mol. Cell Res. 2020, 1868, 118912. [Google Scholar] [CrossRef] [PubMed]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Preciado, S.; Muntión, S.; Sánchez-Guijo, F. Improving hematopoietic engraftment: Potential role of mesenchymal stromal cell-derived extracellular vesicles. Stem Cells 2021, 39, 26–32. [Google Scholar]
- Adams, G.B.; Scadden, D.T. A niche opportunity for stem cell therapeutics. Gene Ther. 2008, 15, 96–99. [Google Scholar] [CrossRef] [Green Version]
- van der Zee, M.; Sacchetti, A.; Cansoy, M.; Joosten, R.; Teeuwssen, M.; Heijmans-Antonissen, C.; Ewing-Graham, P.C.; Burger, C.W.; Blok, L.J.; Fodde, R. IL6/JAK1/STAT3 Signaling Blockade in Endometrial Cancer Affects the ALDHhi/CD126+ Stem-like Component and Reduces Tumor Burden. Cancer Res. 2015, 75, 3608–3622. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Xie, J.; Zhao, S.; Du, R.; Luo, X.; He, H.; Jiang, S.; Hao, N.; Chen, C.; Guo, C.; et al. ICAM3 mediates inflammatory signaling to promote cancer cell stemness. Cancer Lett. 2018, 422, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Cheng, W.; Zhou, R.; Wu, X.; Feng, Y.; Wang, Y. The mechanisms for lung cancer risk of PM2. 5: Induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 2017, 32, 2341–2351. [Google Scholar] [CrossRef]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/β-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Xu, J.-Y.; Shi, X.-Y.; Huang, W.; Ruan, T.-Y.; Xie, P.; Ding, J.-L. M2-polarized tumor-associated macrophages promoted epithelial–mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: An update on production and actions. Essays Biochem. 2020, 64, 443–462. [Google Scholar]
- Serhan, C. Systems approach to inflammation resolution: Identification of novel anti-inflammatory and pro-resolving mediators. J. Thromb. Haemost. 2009, 7, 44–48. [Google Scholar] [CrossRef]
- Fiala, M.; Halder, R.; Almasi, A.; Sagong, B.; Leung, J.; Jewett, A. Curcuminoids and ω-3 fatty acids with anti-oxidants potentiate cytotoxicity of natural killer cells against pancreatic ductal adenocarcinoma cells and inhibit interferon γ production. Front. Physiol. 2015, 6, 129. [Google Scholar] [CrossRef]
- Huang, L.; Wang, C.-F.; Serhan, C.N.; Strichartz, G. Enduring prevention and transient reduction of postoperative pain by intrathecal resolvin D1. Pain 2011, 152, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Xu, Z.Z.; Strichartz, G.; Serhan, C.N. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011, 34, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Kuang, H.; Hua, X.; Zhou, J.; Yang, R. Resolvin D1 and E1 alleviate the progress of hepatitis toward liver cancer in long-term concanavalin A-induced mice through inhibition of NF-κB activity. Oncol. Rep. 2016, 35, 307–317. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, M.K.; Lee, E.J.; Lee, C.H. Resolvin D1 inhibits TGF-beta1-induced epithelial mesenchymal transition of A549 lung cancer cells via lipoxin A4 receptor/formyl peptide receptor 2 and GPR32. Int. J. Biochem. Cell Biol. 2013, 45, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Prevete, N.; Liotti, F.; Amoresano, A.; Pucci, P.; de Paulis, A.; Melillo, R.M. New perspectives in cancer: Modulation of lipid metabolism and inflammation resolution. Pharmacol. Res. 2018, 128, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.-F.; Strichartz, G.R. Prevention of Chronic Post-Thoracotomy Pain in Rats By Intrathecal Resolvin D1 and D2: Effectiveness of Perioperative and Delayed Drug Delivery. J. Pain 2017, 18, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Z.; Zhang, L.; Liu, T.; Park, J.Y.; Berta, T.; Yang, R.; Serhan, C.N.; Ji, R.R. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010, 16, 592–597, 591p following 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Lee, H.-N.; Surh, Y.-J. RvD1 inhibits TNFα-induced c-Myc expression in normal intestinal epithelial cells and destabilizes hyper-expressed c-Myc in colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 316–323. [Google Scholar] [CrossRef]
- Ye, Y.; Scheff, N.N.; Bernabe, D.; Salvo, E.; Ono, K.; Liu, C.; Veeramachaneni, R.; Viet, C.T.; Viet, D.T.; Dolan, J.C.; et al. Anti-cancer and analgesic effects of resolvin D2 in oral squamous cell carcinoma. Neuropharmacology 2018, 139, 182–193. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, X.; Li, W.; Cao, Q.; Shu, Y. Aspirin-triggered resolvin D1 inhibits TGF-beta1-induced EMT through the inhibition of the mTOR pathway by reducing the expression of PKM2 and is closely linked to oxidative stress. Int. J. Mol. Med. 2016, 38, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, Y.; Wang, L.; Yao, B.; Chen, T.; Li, Q.; Liu, Z.; Liu, R.; Niu, Y.; Song, T.; et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J. Exp. Clin. Cancer Res. CR 2019, 38, 170. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Feng, N.; Cui, J.; Wang, S.; Qu, H.; Fu, G.; Li, J.; Chen, H.; Wang, X.; Wang, R. Resolvin D1 and D2 inhibit tumour growth and inflammation via modulating macrophage polarization. J. Cell. Mol. Med. 2020, 24, 8045–8056. [Google Scholar] [CrossRef]
- Yi, J.J.; Heo, S.Y.; Ju, J.H.; Oh, B.R.; Son, W.S.; Seo, J.W. Synthesis of two new lipid mediators from docosahexaenoic acid by combinatorial catalysis involving enzymatic and chemical reaction. Sci. Rep. 2020, 10, 18849. [Google Scholar] [CrossRef]
- Wang, L.; Choi, H.S.; Lee, B.; Choi, J.H.; Jang, Y.S.; Seo, J.W. 13R, 20-Dihydroxydocosahexaenoic Acid, a Novel Dihydroxy-DHA Derivative, Inhibits Breast Cancer Stemness through Regulation of the Stat3/IL-6 Signaling Pathway by Inducing ROS Production. Antioxidants 2021, 10, 457. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Pisanti, S.; Napolitano, F.; Di Somma, S.; Martinelli, R.; Portella, G. Tumor-associated macrophage status in cancer treatment. Cancers 2020, 12, 1987. [Google Scholar] [CrossRef]
- Ko, J.L.; Liu, H.-C.; Loh, H.H. Role of an AP-2-like element in transcriptional regulation of mouse μ-opioid receptor gene. Mol. Brain Res. 2003, 112, 153–162. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, H.; Meng, X.; Wang, F.; Wang, P. Aloe-emodin from rhubarb (Rheum rhabarbarum) inhibits lipopolysaccharide-induced inflammatory responses in RAW264. 7 macrophages. J. Ethnopharmacol. 2014, 153, 846–853. [Google Scholar] [CrossRef]
- Chun, H.W.; Kim, S.J.; Pham, T.H.; Bak, Y.; Oh, J.; Ryu, H.W.; Oh, S.R.; Hong, J.T.; Yoon, D.Y. Epimagnolin A inhibits IL-6 production by inhibiting p38/NF-κB and AP-1 signaling pathways in PMA-stimulated THP-1 cells. Environ. Toxicol. 2019, 34, 796–803. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Jin, W.; Chen, C.; Shao, Z.; Wu, J. Tumor Associated Macrophage × Cancer Cell Hybrids May Acquire Cancer Stem Cell Properties in Breast Cancer. PLoS ONE 2012, 7, e41942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werfel, T.A.; Cook, R.S. Efferocytosis in the tumor microenvironment. Semin. Immunopathol. 2018, 40, 545–554. [Google Scholar] [CrossRef]
- Zhou, Y.; Yao, Y.; Deng, Y.; Shao, A. Regulation of efferocytosis as a novel cancer therapy. Cell Commun. Signal. 2020, 18, 1–11. [Google Scholar] [CrossRef]
- Cabrales, P. RRx-001 acts as a dual small molecule checkpoint inhibitor by downregulating CD47 on cancer cells and SIRP-α on monocytes/macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, D.-X.; Yu, X.-J.; Sun, H.-W.; Xu, Y.-T.; Zhang, Y.-J.; Xu, J. Macrophages induce CD47 upregulation via IL-6 and correlate with poor survival in hepatocellular carcinoma patients. Oncoimmunology 2019, 8, e1652540. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, Z.; Guo, S.; Zhang, L.; Sharma, A.; Robertson, G.P.; Huang, L. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 2013, 21, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [Green Version]
- Iribarren, K.; Buque, A.; Mondragon, L.; Xie, W.; Lévesque, S.; Pol, J.; Zitvogel, L.; Kepp, O.; Kroemer, G. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology 2019, 8, 1550619. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Tanaka, Y.; Piao, J.; Tanimine, N.; Oue, N.; Hinoi, T.; Garcia, N.V.; Miyasaka, M.; Matozaki, T.; Yasui, W. Signal regulatory protein alpha blockade potentiates tumoricidal effects of macrophages on gastroenterological neoplastic cells in syngeneic immunocompetent mice. Ann. Gastroenterol. Surg. 2018, 2, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.-Y.; Yuan, A.; Chen, J.J.-W.; Yang, P.-C. Tumor-associated macrophage: Its role in cancer invasion and metastasis. J. Cancer Mol. 2006, 2, 101–106. [Google Scholar]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. OncoTargets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, M.J.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Chen, J.; Hu, C.; Teng, M.; Jiao, K.; Shen, Z.; Zhu, D.; Yue, J.; Li, Z. The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells. Toxicol. Vitr. 2017, 45, 10–18. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Pope, N.H.; Salmon, M.; Davis, J.P.; Chatterjee, A.; Su, G.; Conte, M.S.; Ailawadi, G.; Upchurch Jr, G.R. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016, 30, 4192–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Lamont, G.J.; Lamont, R.J.; Uriarte, S.M.; Wang, H.; Scott, D.A. Resolvin D1, resolvin D2 and maresin 1 activate the GSK3β anti-inflammatory axis in TLR4-engaged human monocytes. Innate Immun. 2016, 22, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Famenini, S.; Rigali, E.A.; Olivera-Perez, H.M.; Dang, J.; Chang, M.T.; Halder, R.; Rao, R.V.; Pellegrini, M.; Porter, V.; Bredesen, D. Increased intermediate M1-M2 macrophage polarization and improved cognition in mild cognitive impairment patients on ω-3 supplementation. FASEB J. 2017, 31, 148–160. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [Green Version]
- Bingle, L.; Brown, N.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Zhang, R.; Sun, S.; Ji, F.; Liu, C.; Lin, H.; Xie, L.; Yang, H.; Tang, W.; Zhou, Y.; Xu, J. CNTN-1 enhances chemoresistance in human lung adenocarcinoma through induction of epithelial-mesenchymal transition by targeting the PI3K/Akt pathway. Cell. Physiol. Biochem. 2017, 43, 465–480. [Google Scholar] [CrossRef] [Green Version]
- da Silva, I.L.; Montero-Montero, L.; Martín-Villar, E.; Martin-Pérez, J.; Sainz, B.; Renart, J.; Simões, R.T.; Veloso, É.S.; Teixeira, C.S.; de Oliveira, M.C. Reduced expression of the murine HLA-G homolog Qa-2 is associated with malignancy, epithelial-mesenchymal transition and stemness in breast cancer cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Joseph, M.; Enting, D. Immune responses in bladder cancer-role of immune cell populations, prognostic factors and therapeutic implications. Front. Oncol. 2019, 9, 1270. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Gao, J.; Li, C.; Xu, X.; Hu, Y.; Huang, S. Reactive Oxygen Species Induce Endothelial Differentiation of Liver Cancer Stem-Like Sphere Cells through the Activation of Akt/IKK Signaling Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 1621687. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Choi, H.S.; Su, Y.; Lee, B.; Song, J.J.; Jang, Y.-S.; Seo, J.-W. 7S,15R-Dihydroxy-16S,17S-Epoxy-Docosapentaenoic Acid, a Novel DHA Epoxy Derivative, Inhibits Colorectal Cancer Stemness through Repolarization of Tumor-Associated Macrophage Functions and the ROS/STAT3 Signaling Pathway. Antioxidants 2021, 10, 1459. https://doi.org/10.3390/antiox10091459
Wang L, Choi HS, Su Y, Lee B, Song JJ, Jang Y-S, Seo J-W. 7S,15R-Dihydroxy-16S,17S-Epoxy-Docosapentaenoic Acid, a Novel DHA Epoxy Derivative, Inhibits Colorectal Cancer Stemness through Repolarization of Tumor-Associated Macrophage Functions and the ROS/STAT3 Signaling Pathway. Antioxidants. 2021; 10(9):1459. https://doi.org/10.3390/antiox10091459
Chicago/Turabian StyleWang, Lifang, Hack Sun Choi, Yan Su, Binna Lee, Jae Jun Song, Yong-Suk Jang, and Jeong-Woo Seo. 2021. "7S,15R-Dihydroxy-16S,17S-Epoxy-Docosapentaenoic Acid, a Novel DHA Epoxy Derivative, Inhibits Colorectal Cancer Stemness through Repolarization of Tumor-Associated Macrophage Functions and the ROS/STAT3 Signaling Pathway" Antioxidants 10, no. 9: 1459. https://doi.org/10.3390/antiox10091459
APA StyleWang, L., Choi, H. S., Su, Y., Lee, B., Song, J. J., Jang, Y.-S., & Seo, J.-W. (2021). 7S,15R-Dihydroxy-16S,17S-Epoxy-Docosapentaenoic Acid, a Novel DHA Epoxy Derivative, Inhibits Colorectal Cancer Stemness through Repolarization of Tumor-Associated Macrophage Functions and the ROS/STAT3 Signaling Pathway. Antioxidants, 10(9), 1459. https://doi.org/10.3390/antiox10091459