
Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression

 ,
,  , ,
, ,  ,
,  ,
, 
Abstract
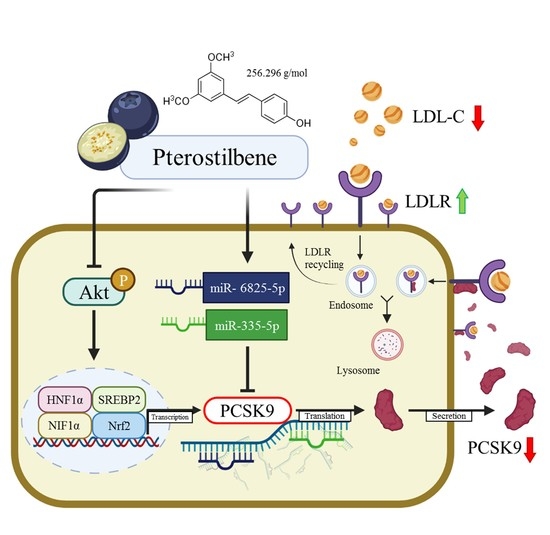
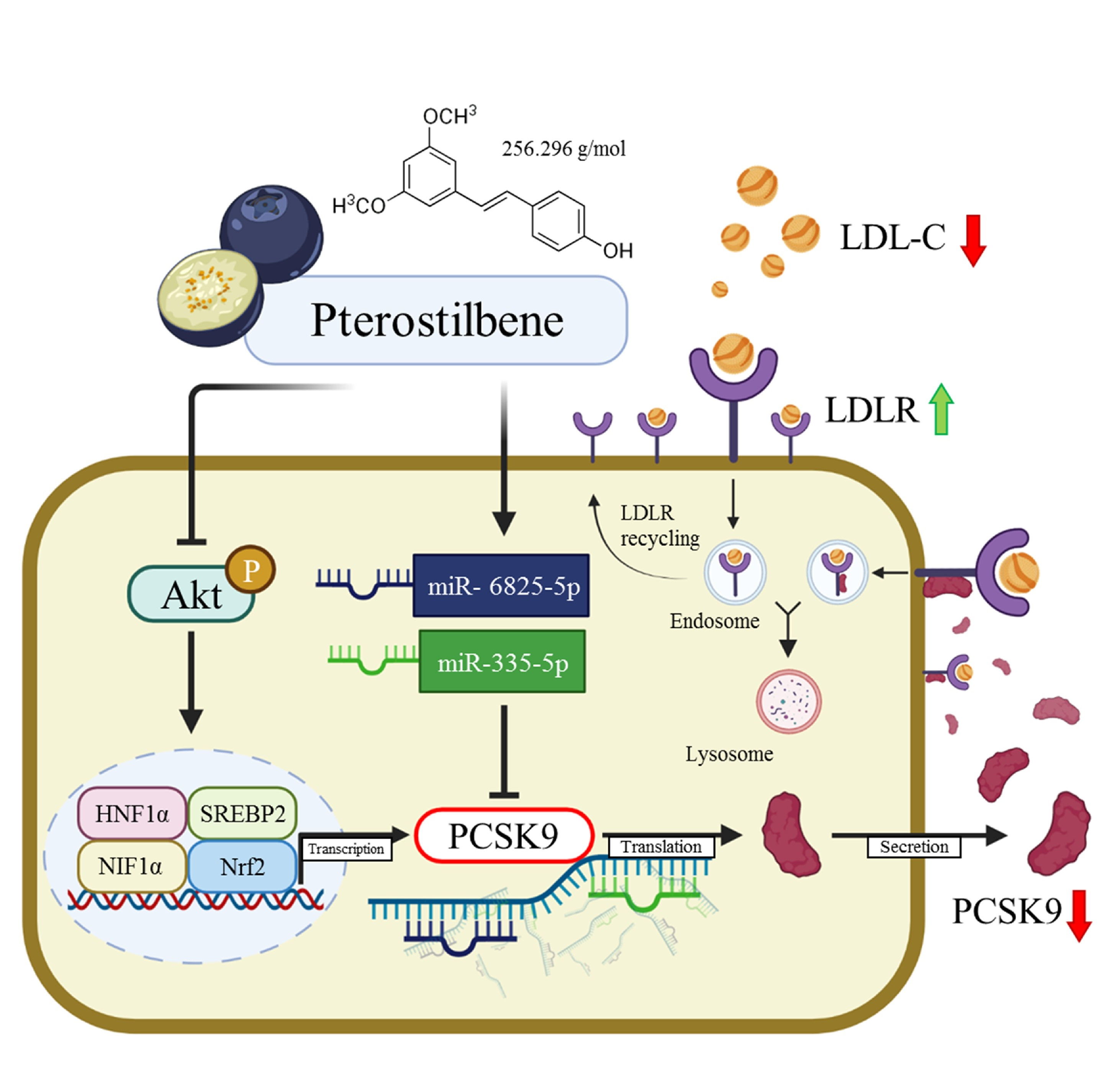
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Cells and Cell Culture
2.3. Western Blot Analysis
2.4. Sulforhodamine B Cell Viability Assay
2.5. shSREBP2 (Short Hairpin RNA against SREBP2) Knockdown Procedure
2.6. RT-qPCR of hsa-miR-335 and hsa-miR-6825 Assay
2.7. Real-Time Reverse Transcriptase PCR
2.8. Flow Cytometry Detection of Cell-Surface LDLR
2.9. Intracellular LDL Uptake Evaluation
2.10. Immunocytochemistry Assessment of Intracellular LDL Density
2.11. Plasmid Transfection and PCSK9 Promoter/Luciferase Reported Assay
2.12. Statistical Analysis
3. Results
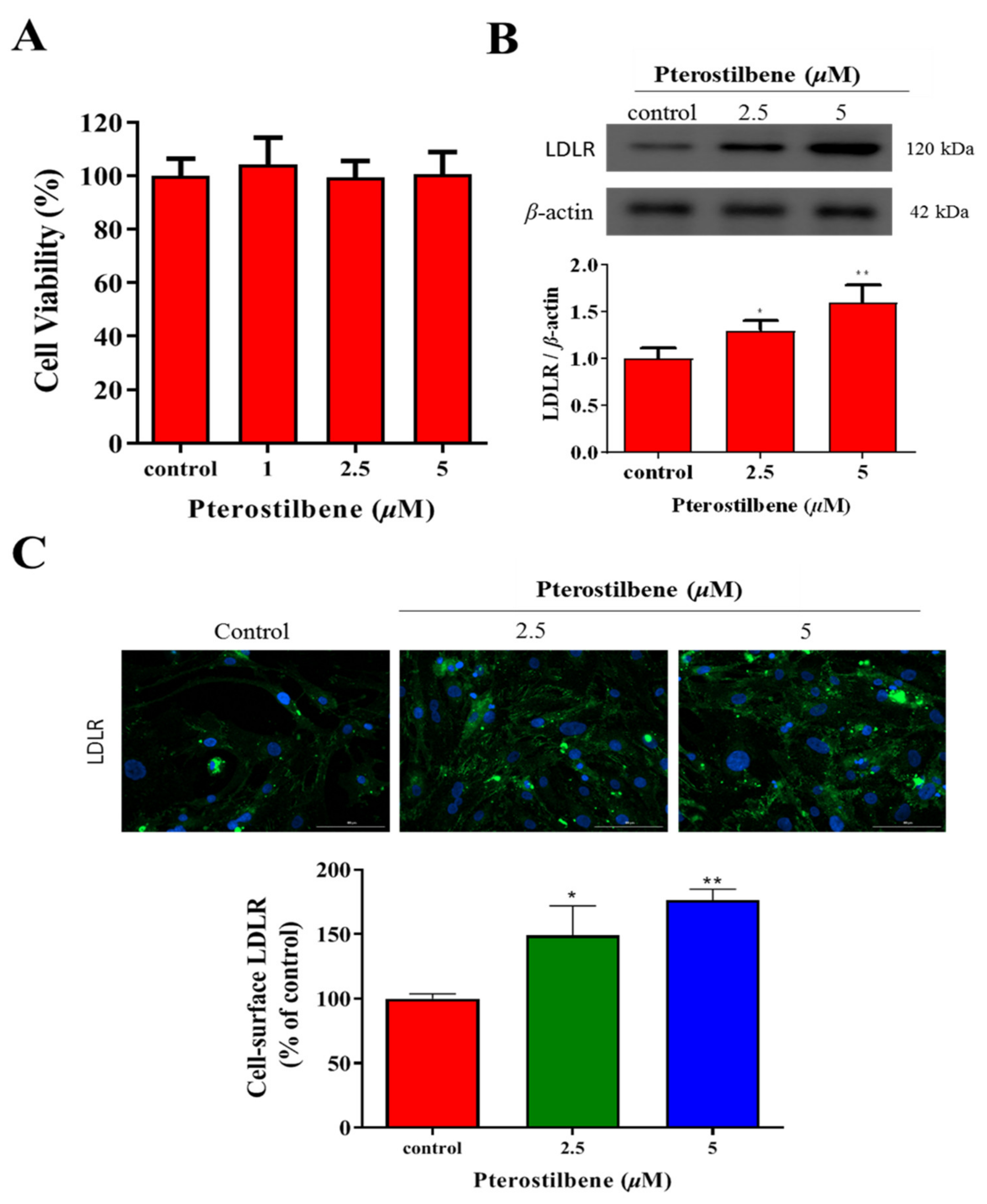
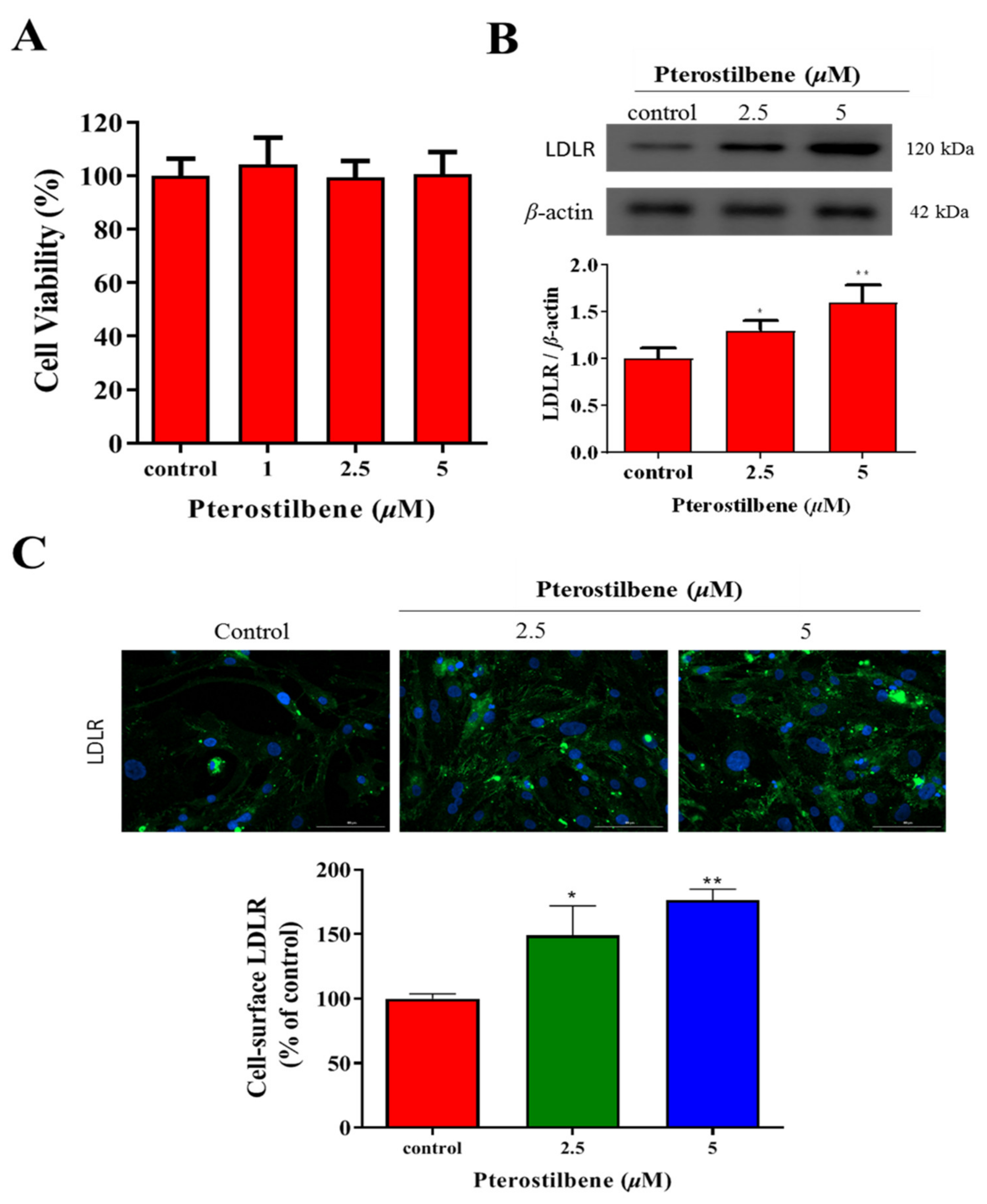
3.1. Pterostilbene Exhibits No Apparent Cytotoxicity but Significantly Enhances LDLR Expression
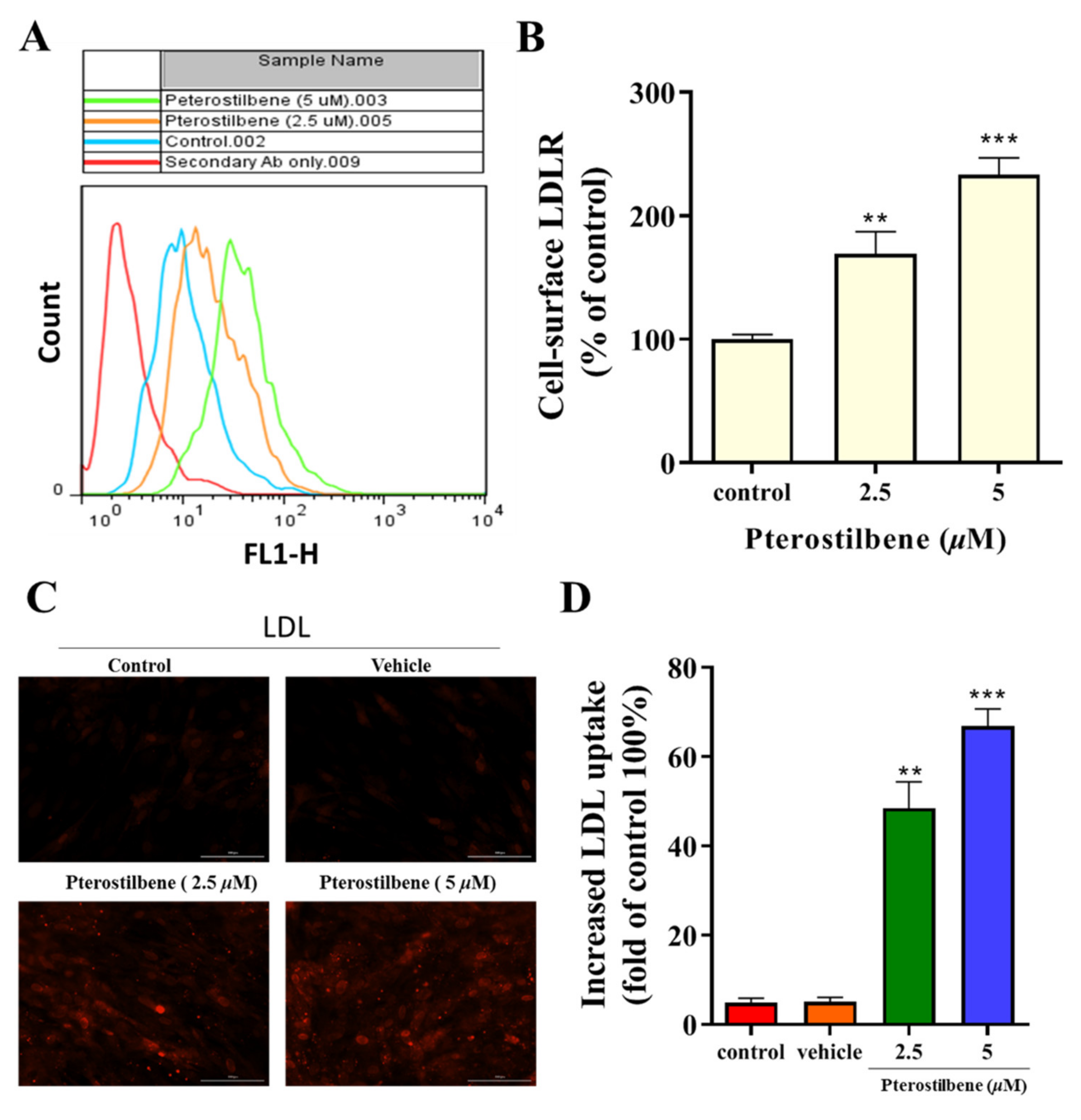
3.2. Pterostilbene Enhances the Cell-Surface LDLR Expression and the Uptake of LDL
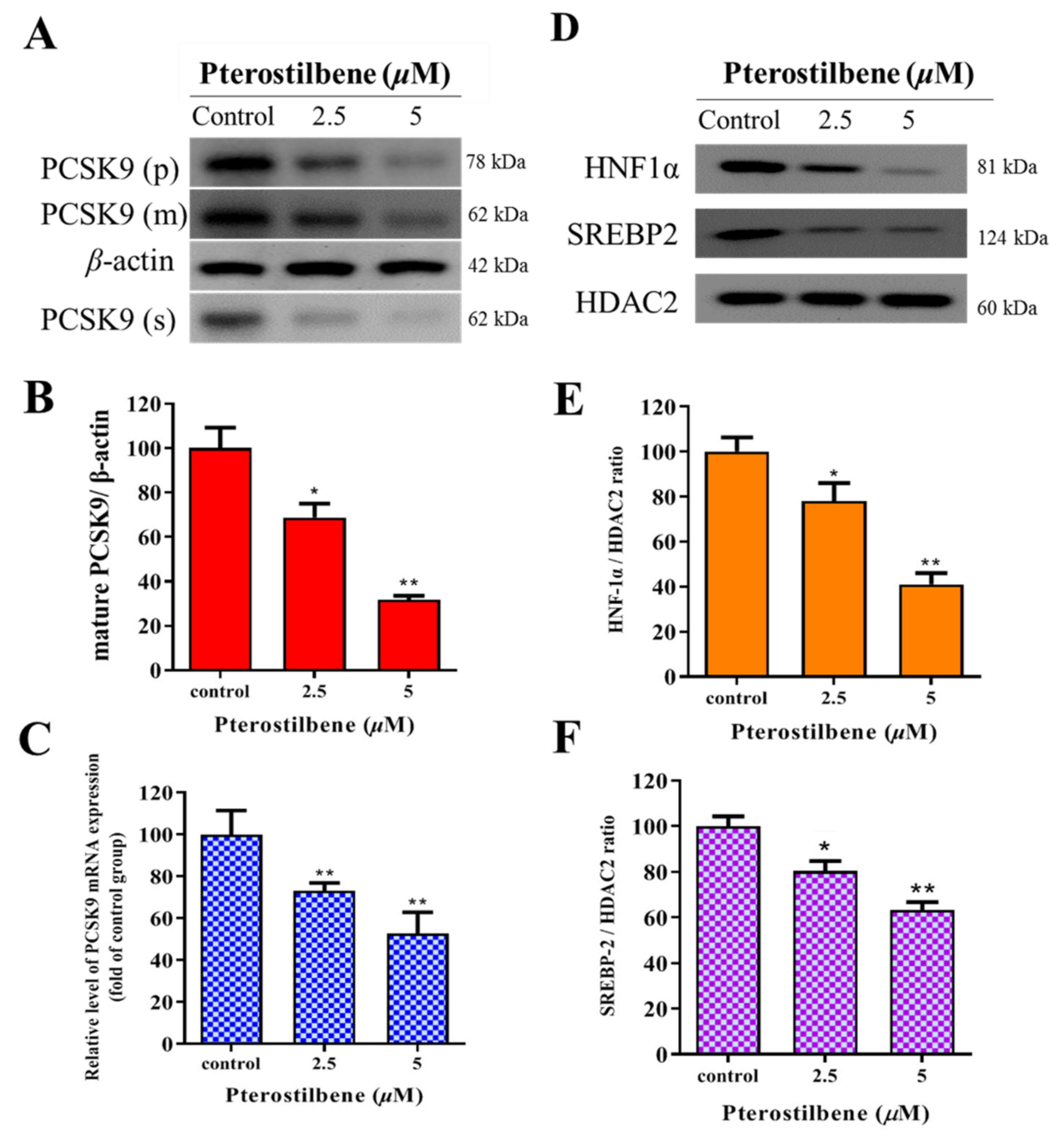
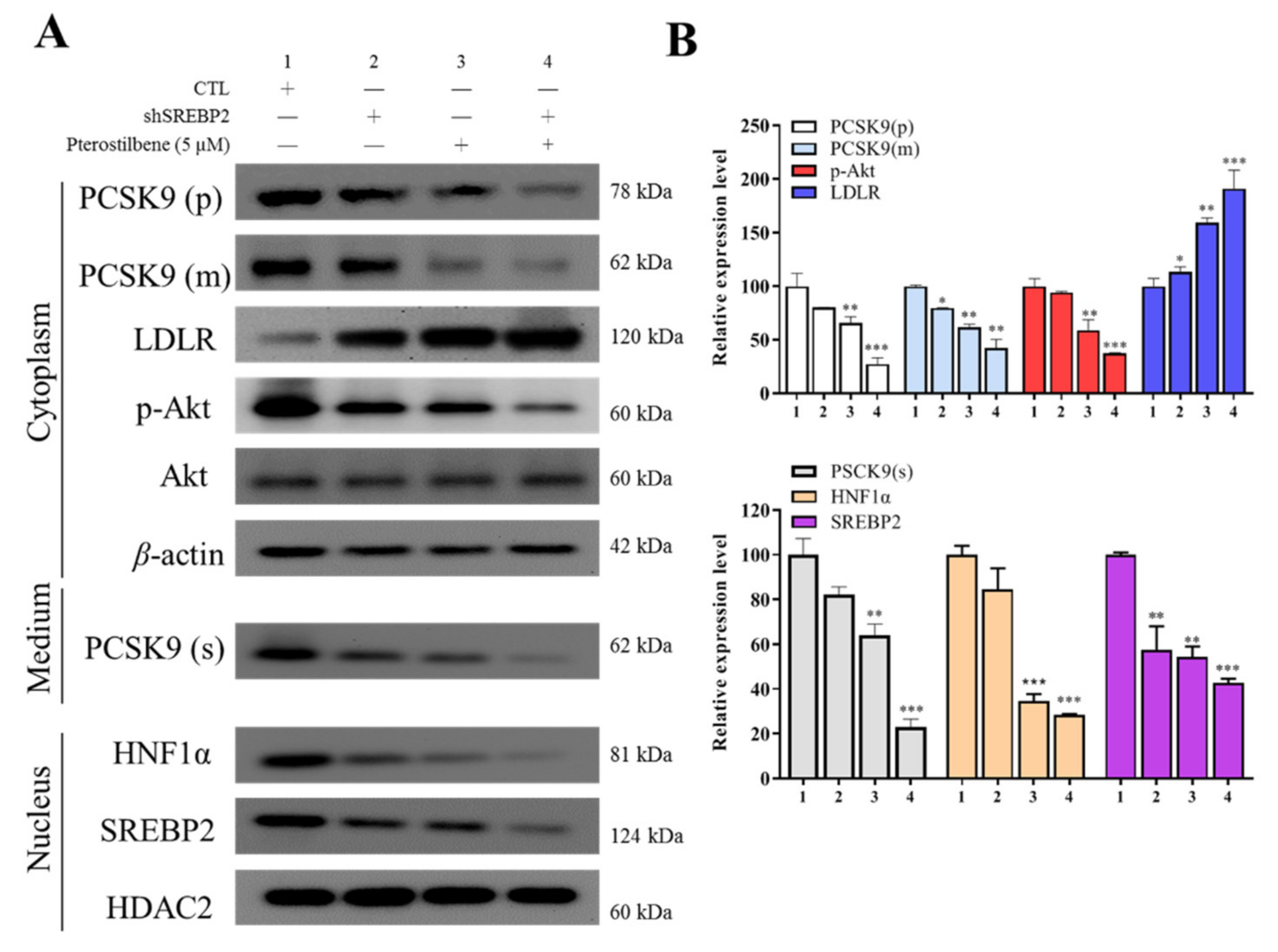
3.3. Pterostilbene Suppresses PCSK9/HNF1α/SREBP2 Signaling
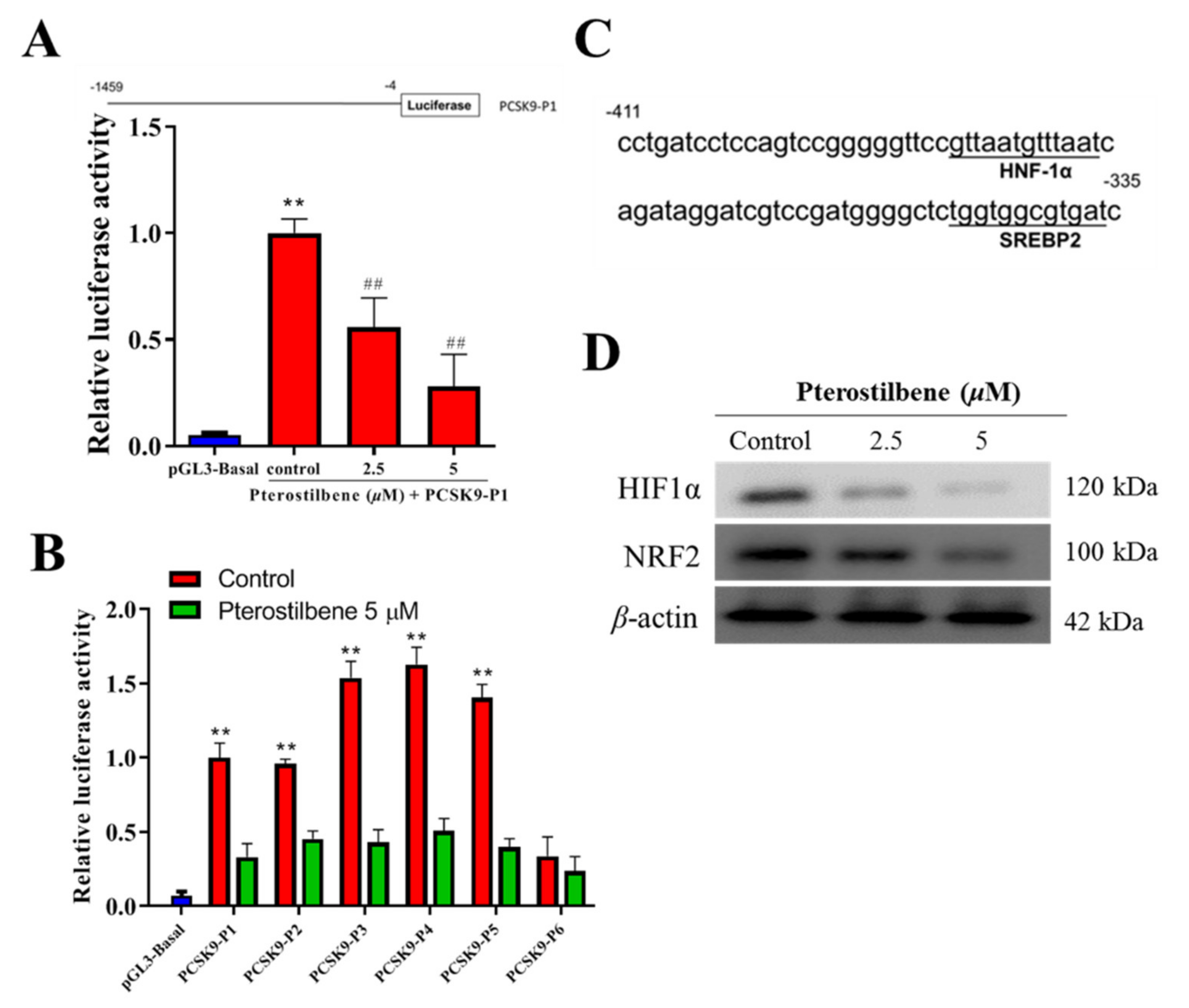
3.4. Pterostilbene Suppresses the PCSK9 Promoter Activity and Hyperlipidemia-Associated Transcription Factors
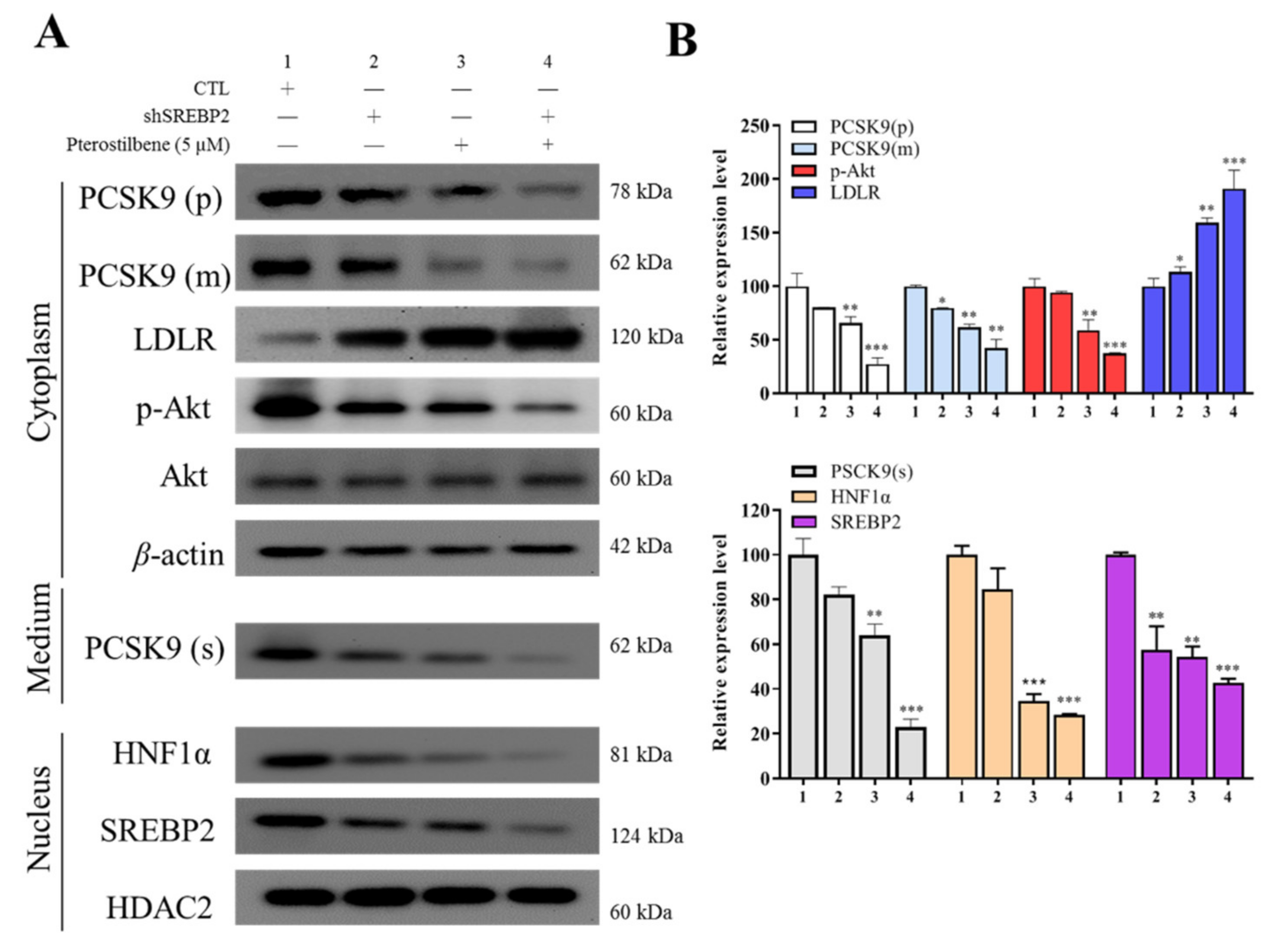
3.5. Pterostilbene as Compared to shSREBP2 Induced Higher Protein Expression of LDLR and Lower Nuclear Accumulation of HNF-1α and SREBP2
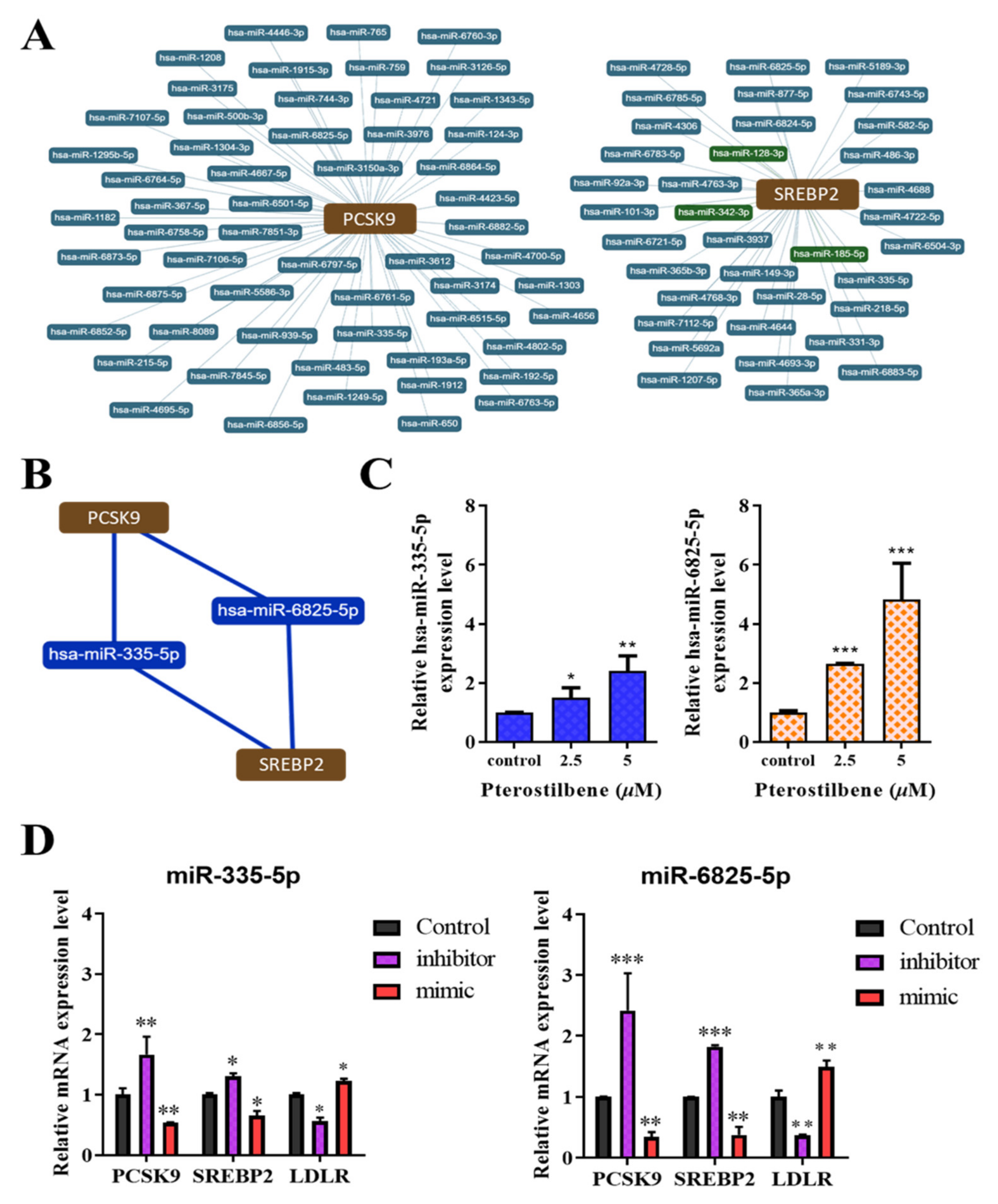
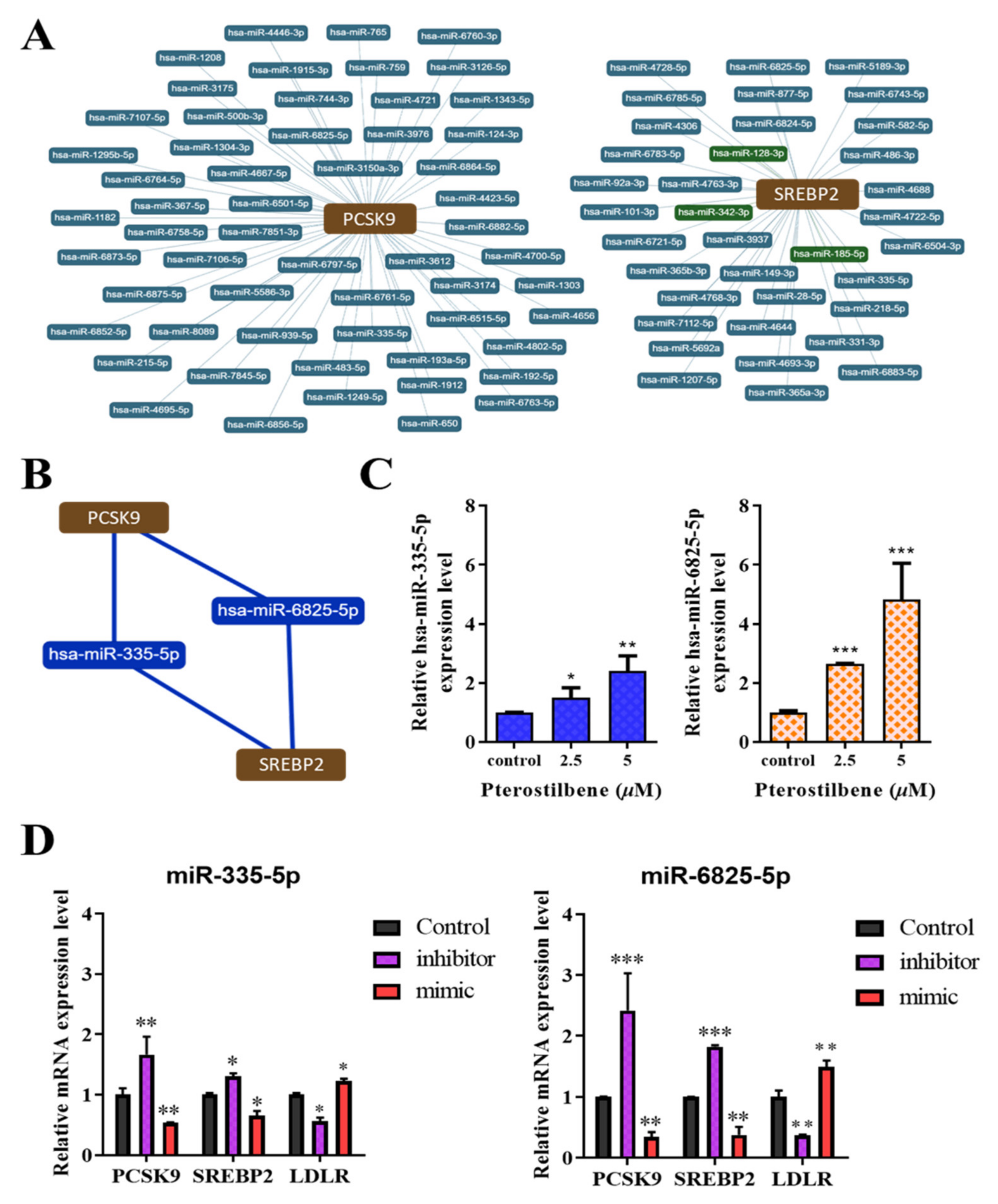
3.6. Pterostilbene Exerts Epigenetic Regulation of PCSK9, SREBP2, and LDLR mRNA Expression through hsa-miR-335 and hsa-miR-6825, Which Mediate the PCSK9/SREBP2 Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, Y.S.; Li, T.D.; Zeng, Z.H. Mechanisms underlying direct actions of hyperlipidemia on myocardium: An updated review. Lipids Health Dis. 2020, 19, 23. [Google Scholar] [CrossRef] [Green Version]
- Pekkanen, J.; Linn, S.; Heiss, G.; Suchindran, C.M.; Leon, A.; Rifkind, B.M.; Tyroler, H.A. Ten-Year Mortality from Cardiovascular Disease in Relation to Cholesterol Level among Men with and without Preexisting Cardiovascular Disease. N. Engl. J. Med. 1990, 322, 1700–1707. [Google Scholar] [CrossRef]
- Agrawal, S.; Zaritsky, J.J.; Fornoni, A.; Smoyer, W.E. Dyslipidaemia in nephrotic syndrome: Mechanisms and treatment. Nat. Rev. Nephrol. 2018, 14, 57–70. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [Green Version]
- Kahn, E.; Baarine, M.; Pelloux, S.; Riedinger, J.M.; Frouin, F.; Tourneur, Y.; Lizard, G. Iron nanoparticles increase 7-ketocholesterol-induced cell death, inflammation, and oxidation on murine cardiac HL1-NB cells. Int. J. Nanomed. 2010, 5, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Wexler, D.; Roth, A.; Barak, T.; Sheps, D.; Keren, G. Usefulness of anti-oxidized LDL antibody determination for assessment of clinical control in patients with heart failure. Eur. J. Heart Fail. 2006, 8, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michos, E.D.; McEvoy, J.W.; Blumenthal, R.S. Lipid Management for the Prevention of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2019, 381, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory pro-protein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.; Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Li, L.; Rohrbach, S.; Schulz, R.; Schlüter, K.-D. Autocrine effects of PCSK9 on cardiomyocytes. Basic Res. Cardiol. 2020, 115, 1–13. [Google Scholar] [CrossRef]
- Leucker, T.M.; Weiss, R.G.; Schär, M.; Bonanno, G.; Mathews, L.; Jones, S.R.; Brown, T.T.; Moore, R.; Afework, Y.; Gerstenblith, G.; et al. Coronary Endothelial Dysfunction Is Associated with Elevated Serum PCSK9 Levels in People With HIV Independent of Low-Density Lipoprotein Cholesterol. J. Am. Heart Assoc. 2018, 7, e009996. [Google Scholar] [CrossRef] [Green Version]
- De Jongh, S.; Lilien, M.R.; op’t Roodt, J.; Stroes, E.S.; Bakker, H.D.; Kastelein, J.J. Early statin therapy restores endothelial function in children with familial hypercholesterolemia. J. Am. Coll. Cardiol. 2002, 40, 2117–2121. [Google Scholar] [CrossRef] [Green Version]
- Giunzioni, I.; Tavori, H.; Covarrubias, R.; Major, A.S.; Ding, L.; Zhang, Y.; DeVay, R.M.; Hong, L.; Fan, D.; Predazzi, I.M.; et al. Local effects of human PCSK9 on the atherosclerotic lesion. J. Pathol. 2016, 238, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, N.; Tibolla, G.; Pirillo, A.; Cipollone, F.; Mezzetti, A.; Pacia, S.; Corsini, A.; Catapano, A.L. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 2012, 220, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, T.C.; Knobel, J.; Burkert-Rettenmaier, S.; Li, X.; Meyer, I.S.; Jungmann, A.; Sicklinger, F.; Backs, J.; Lasitschka, F.; Müller, O.J.; et al. Secretome Analysis of Cardiomyocytes Identifies PCSK6 (Proprotein Convertase Subtilisin/Kexin Type 6) as a Novel Player in Cardiac Remodeling After Myocardial Infarction. Circulation 2020, 141, 1628–1644. [Google Scholar] [CrossRef]
- Adorni, M.P.; Zimetti, F.; Lupo, M.G.; Ruscica, M.; Ferri, N. Naturally Occurring PCSK9 Inhibitors. Nutrients 2020, 12, 1440. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Macchi, C.; Banach, M.; Corsini, A.; Sirtori, C.R.; Ferri, N.; Ruscica, M. Changes in circulating pro-protein convertase subtilisin/kexin type 9 levels—Experimental and clinical approaches with lipid-lowering agents. Eur. J. Prev. Cardiol. 2019, 26, 930–949. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.K.; Santos, R.D. PCSK9 Inhibition with Monoclonal Antibodies: Modern Management of Hypercholesterolemia. J. Clin. Pharmacol. 2016, 57, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Kapetanovic, I.M.; Muzzio, M.; Huang, Z.; Thompson, T.N.; McCormick, D.L. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother. Pharmacol. 2011, 68, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Lu, S.; Zhong, J.; Huang, K.; Zhang, S. Protective Effects of Pterostilbene Against Myocardial Ischemia/Reperfusion Injury in Rats. Inflammation 2017, 40, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Mccormack, D.; McFadden, D. A Review of Pterostilbene Antioxidant Activity and Disease Modification. Oxidative Med. Cell. Longev. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mayne, J.; Dewpura, T.; Raymond, A.; Cousins, M.; Chaplin, A.; Lahey, K.A.; LaHaye, S.A.; Mbikay, M.; Ooi, T.C.; Chrétien, M. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Dong, B.; Park, S.W.; Lee, H.S.; Chen, W.; Liu, J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J. Biol. Chem. 2009, 284, 28885–28895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, K.S.; Bowen, K.J.; Tindall, A.M.; Sullivan, V.K.; Johnston, E.A.; Fleming, J.A.; Kris-Etherton, P.M. The Effect of Inflammation and Insulin Resistance on Lipid and Lipoprotein Responsiveness to Dietary Intervention. Curr Dev. Nutr. 2020, 4, nzaa160. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Żak, J.; Malinowski, B.; Popek, G.; Grześk, G. PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 2017, 8, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Belanger, A.J.; Luo, Z.; Vincent, K.A.; Akita, G.Y.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-inducible factor 1 mediates hypox-ia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated re-ceptor alpha/retinoid X receptor. Biochem. Biophys. Res. Commun. 2007, 364, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Zhao, Y.-Z.; Chen, M.-T.; Zhang, F.-L.; Liu, X.-L.; Wang, Y.; Liu, C.-Z.; Yu, J.; Zhang, J.-W. Hypoxia-inducible factor-1 (HIF-1) promotes LDL and VLDL uptake through inducing VLDLR under hypoxia. Biochem. J. 2012, 441, 675–683. [Google Scholar] [CrossRef]
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.-I.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262. [Google Scholar] [CrossRef]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protocol 2016, 6, e1984. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Sodroski, C.; Cha, H.; Li, Q.; Liang, T.J. Infection of Hepatocytes with HCV Increases Cell Surface Levels of Heparan Sulfate Proteoglycans, Uptake of Cholesterol and Lipoprotein, and Virus Entry by Up-regulating SMAD6 and SMAD7. Gastroenterology 2017, 152, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, X.; Zhang, X.; Su, C.; Yang, M.; He, W.; Du, Y.; Si, S.; Wang, L.; Hong, B. A small-molecule inhibitor of PCSK9 transcription ameliorates ather-osclerosis through the modulation of FoxO1/3 and HNF1α. EBioMedicine 2020, 52, 102650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, M.J.; Fernández, M.; Picó, Y.; Mañes, J.; Asensi, M.; Carda, C.; Asensio, G.; Estrela, J.M. Dietary administration of high doses of pterostilbene and quercetin to mice is not toxic. J. Agric. Food Chem. 2009, 57, 3180–3186. [Google Scholar] [CrossRef]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Wang, P.; Sang, S. Metabolism and pharmacokinetics of resveratrol and pterostilbene. BioFactors 2018, 44, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Haza, A.I. Selective apoptotic effects of piceatannol and myricetin in human cancer cells. J. Appl. Toxicol. 2012, 32, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Chalal, M.; Klinguer, A.; Echairi, A.; Meunier, P.; Vervandier-Fasseur, D.; Adrian, M. Antimicrobial Activity of Resveratrol Analogues. Molecules 2014, 19, 7679–7688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrano-Padial, C.; Prieto, A.; Puerto, M.; Pichardo, S. Toxicological Evaluation of Piceatannol, Pterostilbene, and ε-Viniferin for Their Potential Use in the Food Industry: A Review. Foods 2021, 10, 592. [Google Scholar] [CrossRef]
- Tolba, M.F.; Abdel-Rahman, S.Z. Pterostilbine, an active component of blueberries, sensitizes colon cancer cells to 5-fluorouracil cytotoxicity. Sci. Rep. 2015, 5, 15239. [Google Scholar] [CrossRef] [Green Version]
- Yilmazer, A. Cancer cell lines involving cancer stem cell populations respond to oxidative stress. Biotechnol. Rep. 2017, 17, 24–30. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, Y.; Hu, W.; Li, X.; Yang, X.; Zhou, X.; Yin, Z.; Kong, D.; Yao, Z.; Hajjar, D.P.; et al. Peroxisome Prolifera-tor-activated receptor γ activation by ligands and dephosphorylation induces proprotein convertase subtilisin kexin type 9 and low density lipoprotein receptor expression. J. Biol. Chem. 2012, 287, 23667–23677. [Google Scholar] [CrossRef] [Green Version]
- Rimando, A.M.; Nagmani, R.; Feller, D.R.; Yokoyama, W. Pterostilbene, a New Agonist for the Peroxisome Proliferator-Activated Receptor α-Isoform, Lowers Plasma Lipoproteins and Cholesterol in Hypercholesterolemic Hamsters. J. Agric. Food Chem. 2005, 53, 3403–3407. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Wu, M.; Li, H.; Kraemer, F.; Adeli, K.; Seidah, N.; Park, S.W.; Liu, J. Strong induction of PCSK9 gene expression through HNF1α and SREBP2: Mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J. Lipid Res. 2010, 51, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2–Related Factor 2 Promotes Atherosclerosis by Effects on Plasma Lipoproteins and Cholesterol Transport That Overshadow Antioxidant Protection. Arter. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Číž, M.; Dvořáková, A.; Skočková, V.; Kubala, L. The Role of Dietary Phenolic Compounds in Epigenetic Modulation Involved in Inflammatory Processes. Antioxidants 2020, 9, 691. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, T.; Wu, M.-P.; Sheu, J.-R.; Hsia, C.-W.; Bhavan, P.S.; Manubolu, M.; Chung, C.-L.; Hsia, C.-H. Involvement of antioxidant defenses and nf-κb/erk signaling in anti-inflammatory effects of pterostilbene, a natural analogue of resveratrol. Appl. Sci. 2021, 11, 4666. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Perrot, N.; Puri, R. Therapeutic Agents Targeting Cardiometabolic Risk for Preventing and Treating Atherosclerotic Cardiovascular Diseases. Clin. Pharmacol. Ther. 2018, 104, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Shende, V.R.; Wu, M.; Singh, A.B.; Dong, B.; Kan, C.F.K.; Liu, J. Reduction of circulating PCSK9 and LDL-C levels by liver-specific knockdown of HNF1α in normolipidemic mice. J. Lipid Res. 2015, 56, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.-Y.; Ding, X.-Q.; Gu, T.-T.; Guo, W.-J.; Jiao, R.-Q.; Song, L.; Sun, Y.; Pan, Y.; Kong, L.-D. Pterostilbene Improves Hepatic Lipid Accumulation via the MiR-34a/Sirt1/SREBP-1 Pathway in Fructose-Fed Rats. J. Agric. Food Chem. 2020, 68, 1436–1446. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-K.; Yeh, C.-T.; Kuo, K.-T.; Yadav, V.K.; Fong, I.-H.; Kounis, N.G.; Hu, P.; Hung, M.-Y. Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression. Antioxidants 2021, 10, 1280. https://doi.org/10.3390/antiox10081280
Lin Y-K, Yeh C-T, Kuo K-T, Yadav VK, Fong I-H, Kounis NG, Hu P, Hung M-Y. Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression. Antioxidants. 2021; 10(8):1280. https://doi.org/10.3390/antiox10081280
Chicago/Turabian StyleLin, Yen-Kuang, Chi-Tai Yeh, Kuang-Tai Kuo, Vijesh Kumar Yadav, Iat-Hang Fong, Nicholas G. Kounis, Patrick Hu, and Ming-Yow Hung. 2021. "Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression" Antioxidants 10, no. 8: 1280. https://doi.org/10.3390/antiox10081280
APA StyleLin, Y.-K., Yeh, C.-T., Kuo, K.-T., Yadav, V. K., Fong, I.-H., Kounis, N. G., Hu, P., & Hung, M.-Y. (2021). Pterostilbene Increases LDL Metabolism in HL-1 Cardiomyocytes by Modulating the PCSK9/HNF1α/SREBP2/LDLR Signaling Cascade, Upregulating Epigenetic hsa-miR-335 and hsa-miR-6825, and LDL Receptor Expression. Antioxidants, 10(8), 1280. https://doi.org/10.3390/antiox10081280