
Overexpression of MnSOD Protects against Cold Storage-Induced Mitochondrial Injury but Not against OMA1-Dependent OPA1 Proteolytic Processing in Rat Renal Proximal Tubular Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cold Storage/Rewarming Model
2.2. SiRNA Transfection
2.3. Protein Lysates and Western Blotting
2.4. Measurement of NRK Cell Cytotoxicity
2.5. High-Resolution Respirometry
2.6. Statistical Analysis
3. Results
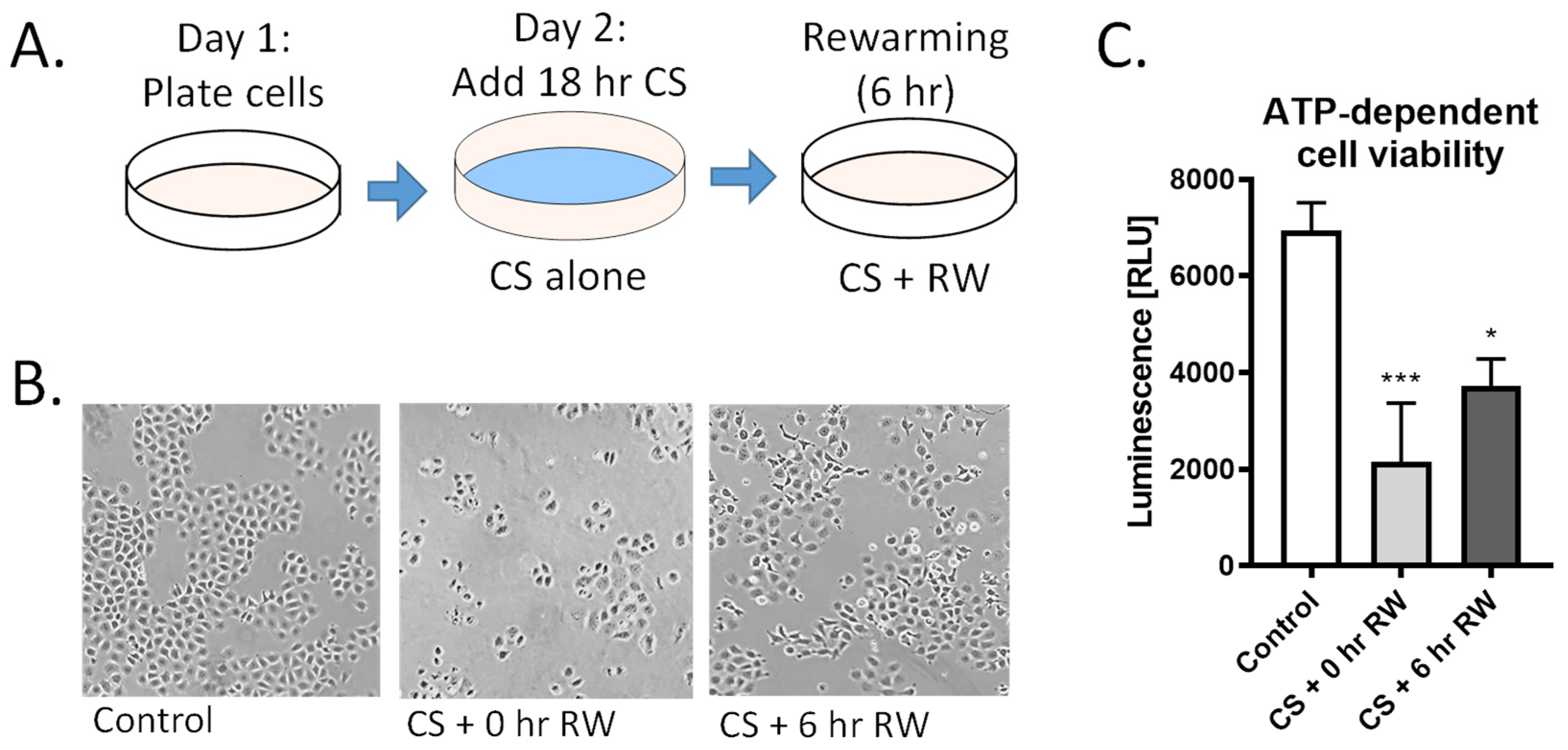
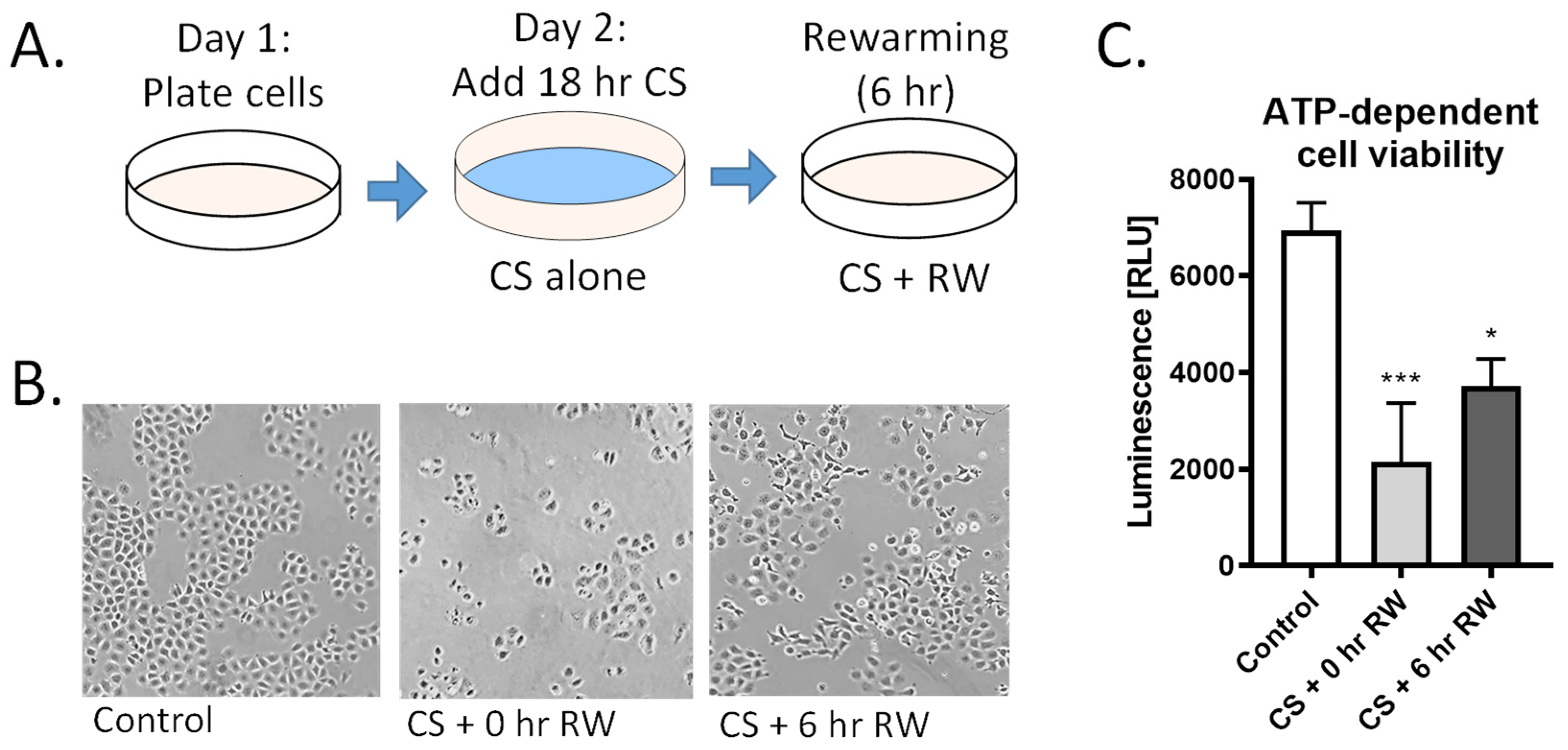
3.1. CS and CS + RW Induces NRK Cell Injury
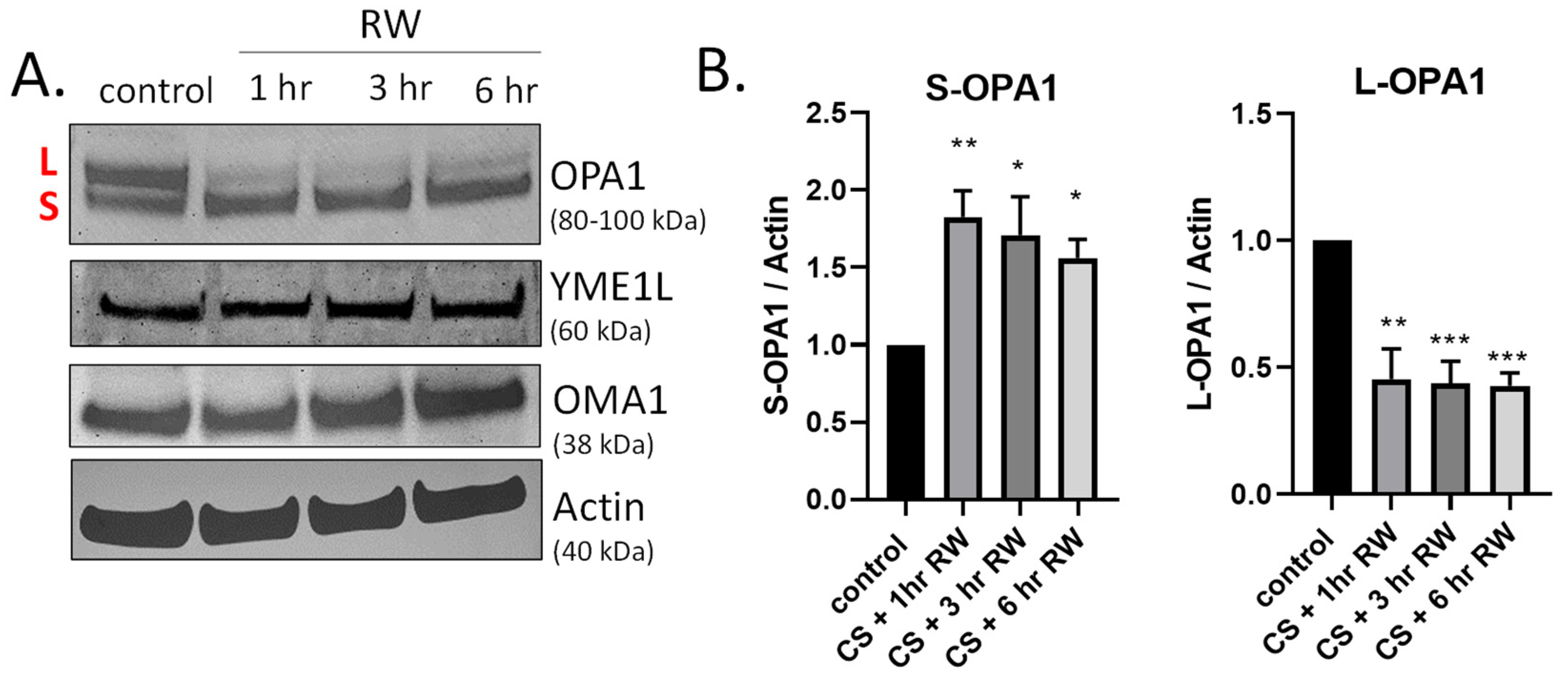
3.2. Rapid Proteolytic Processing of OPA1 Occurs during CS + RW in NRK Cells
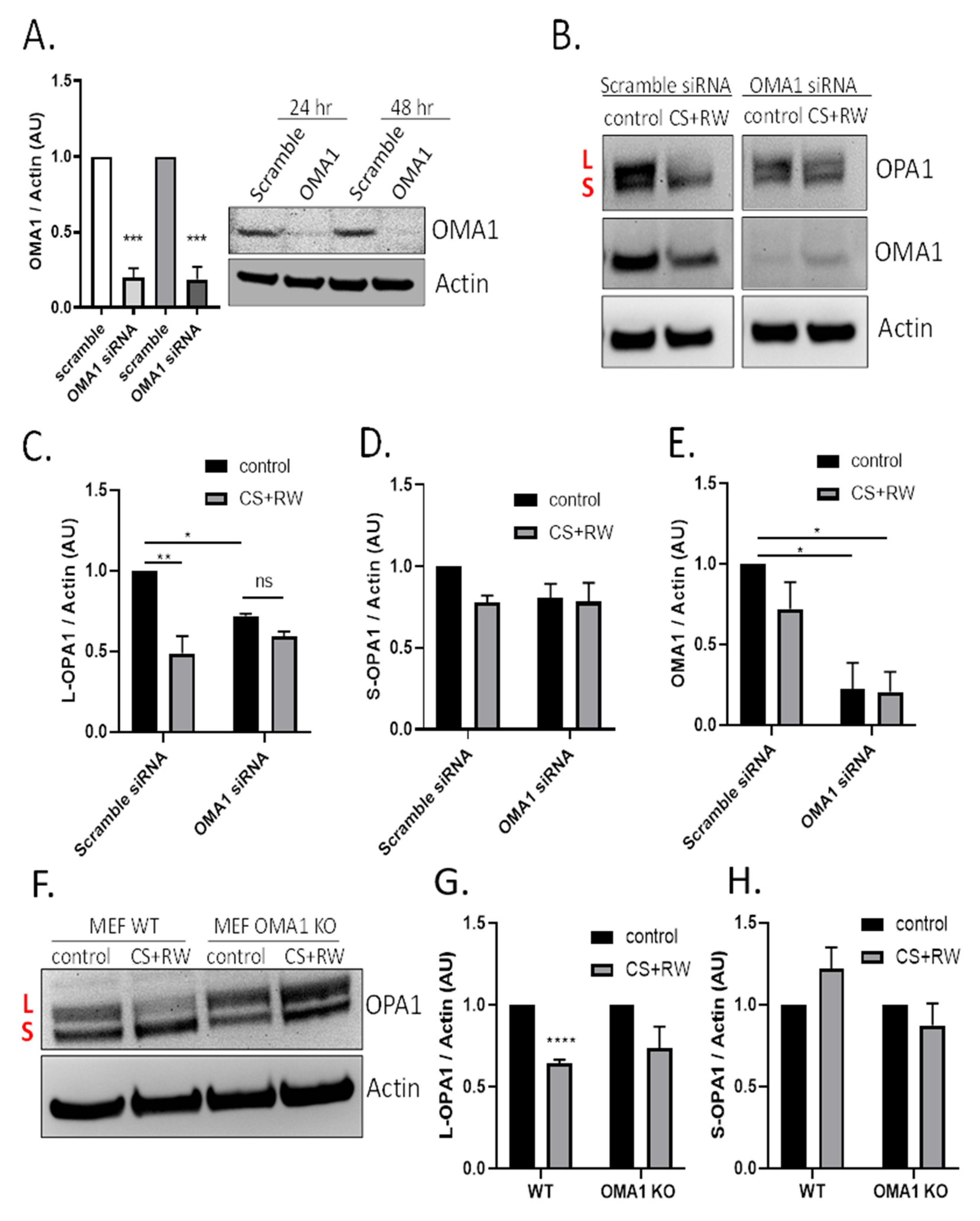
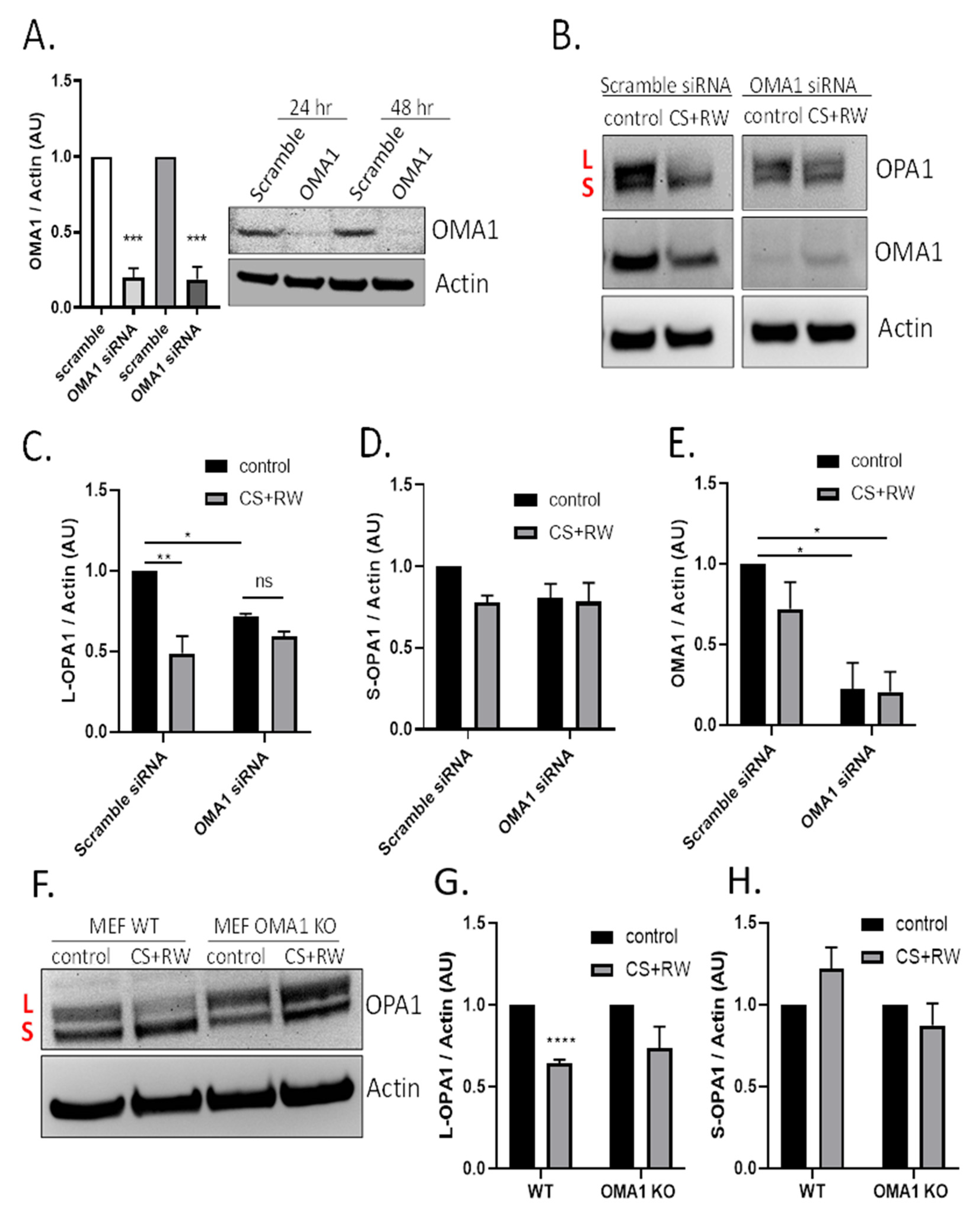
3.3. CS + RW Induces OMA1-Dependent OPA1 Proteolytic Processing in NRK Cells
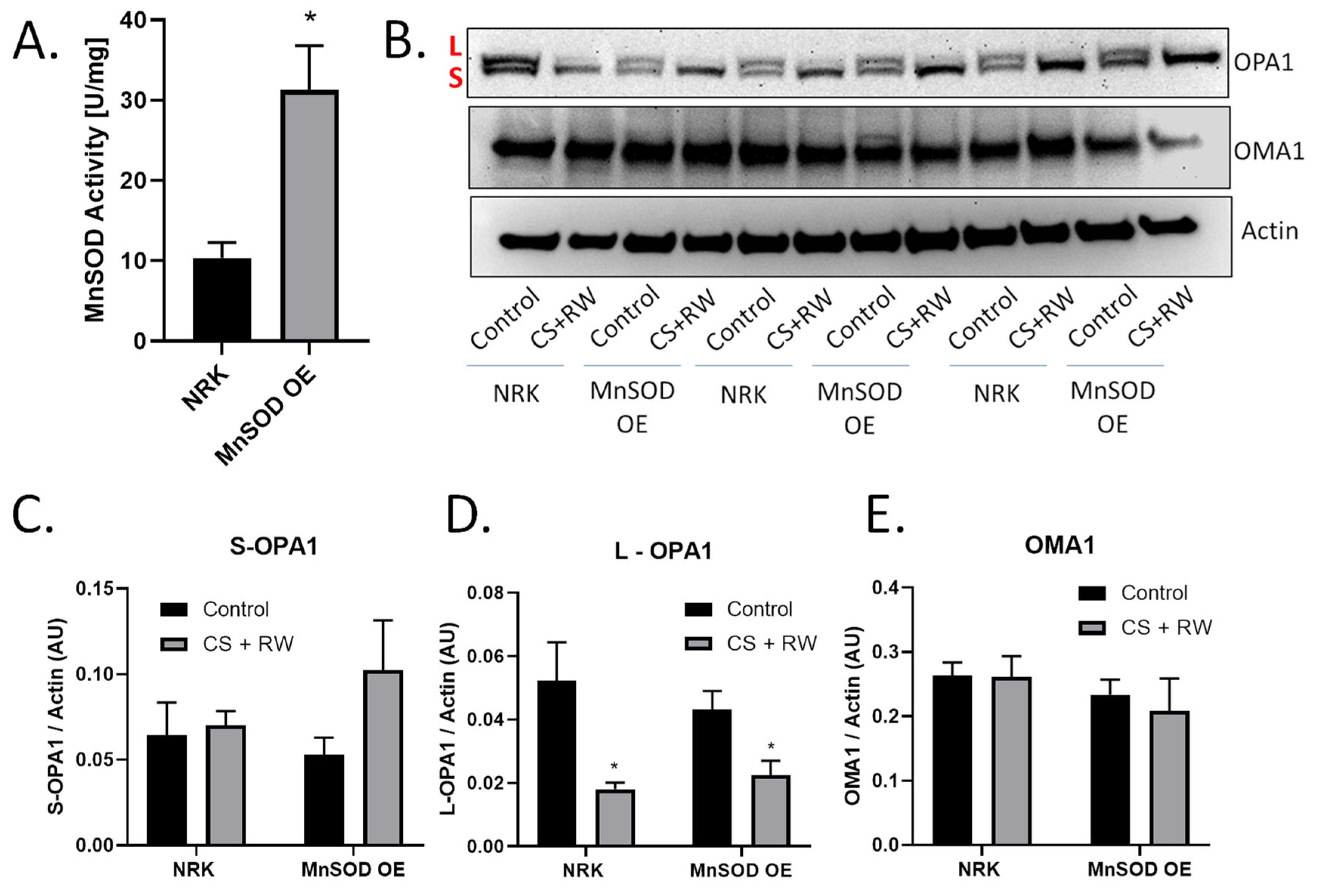
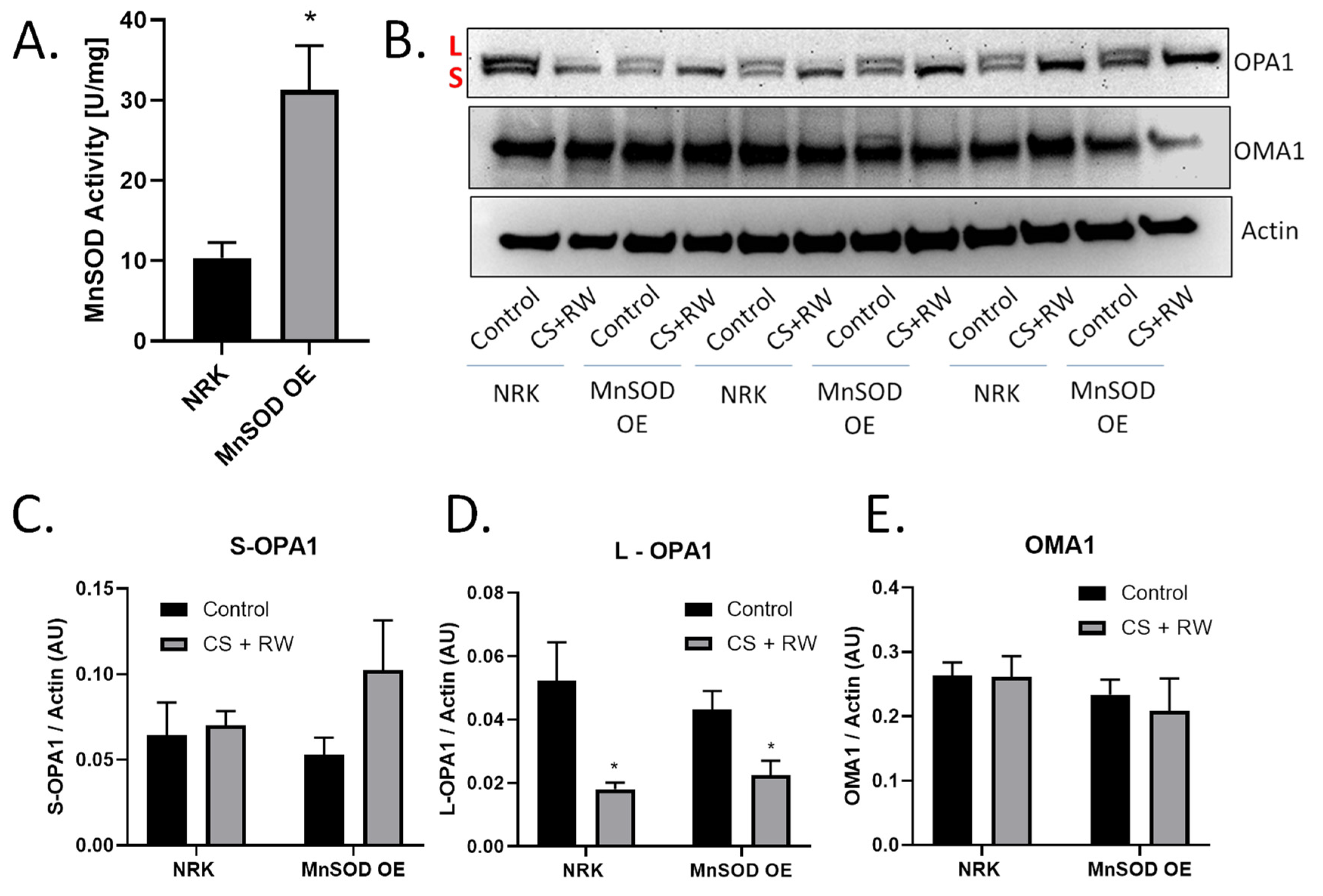
3.4. MnSOD Overexpression Does Not Prevent OMA1-Dependent OPA1 Proteolytic Processing during CS + RW
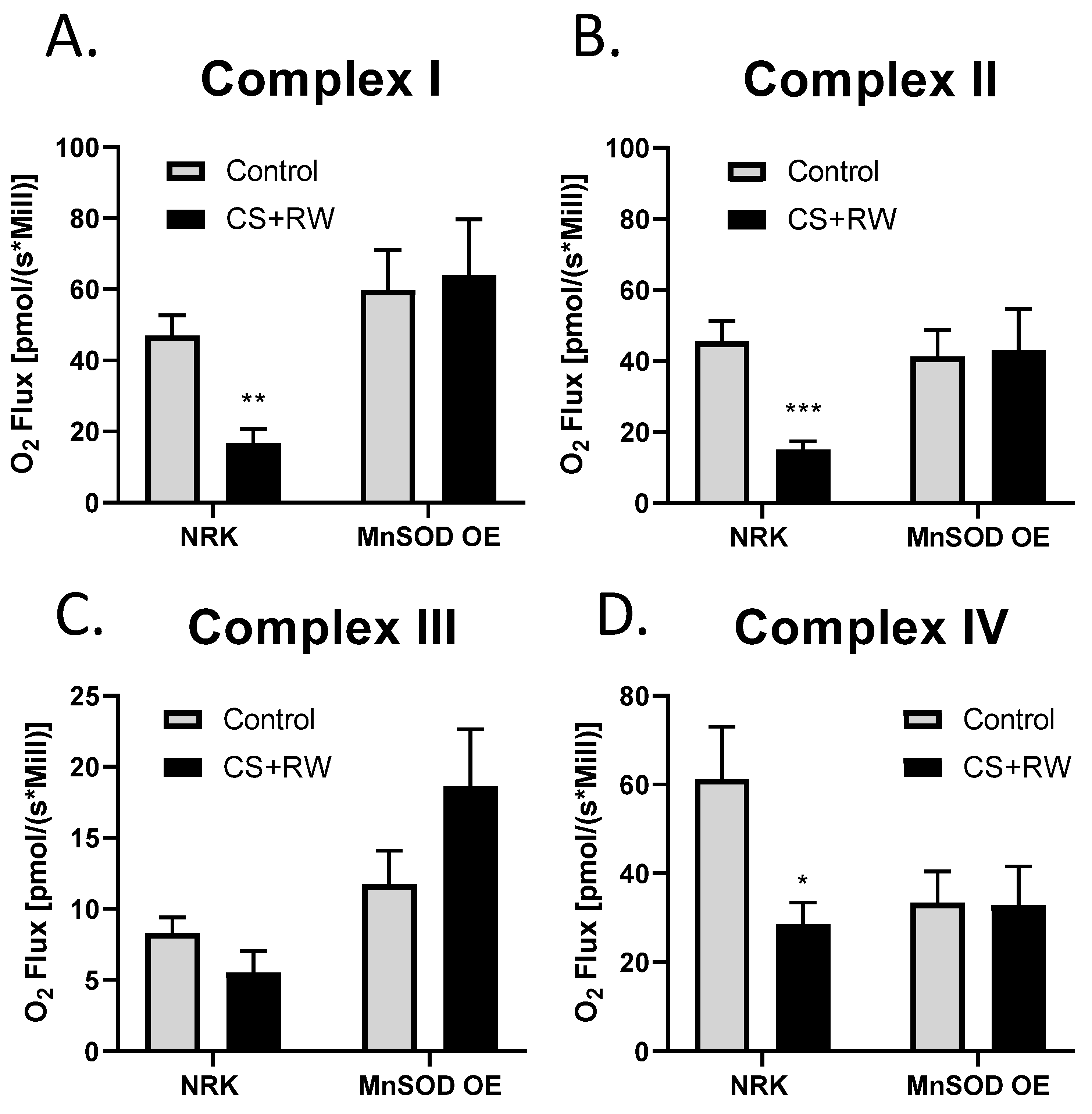
3.5. MnSOD Overexpression Attenuates CS + RW-Induced Mitochondrial Respiratory Dysfunction
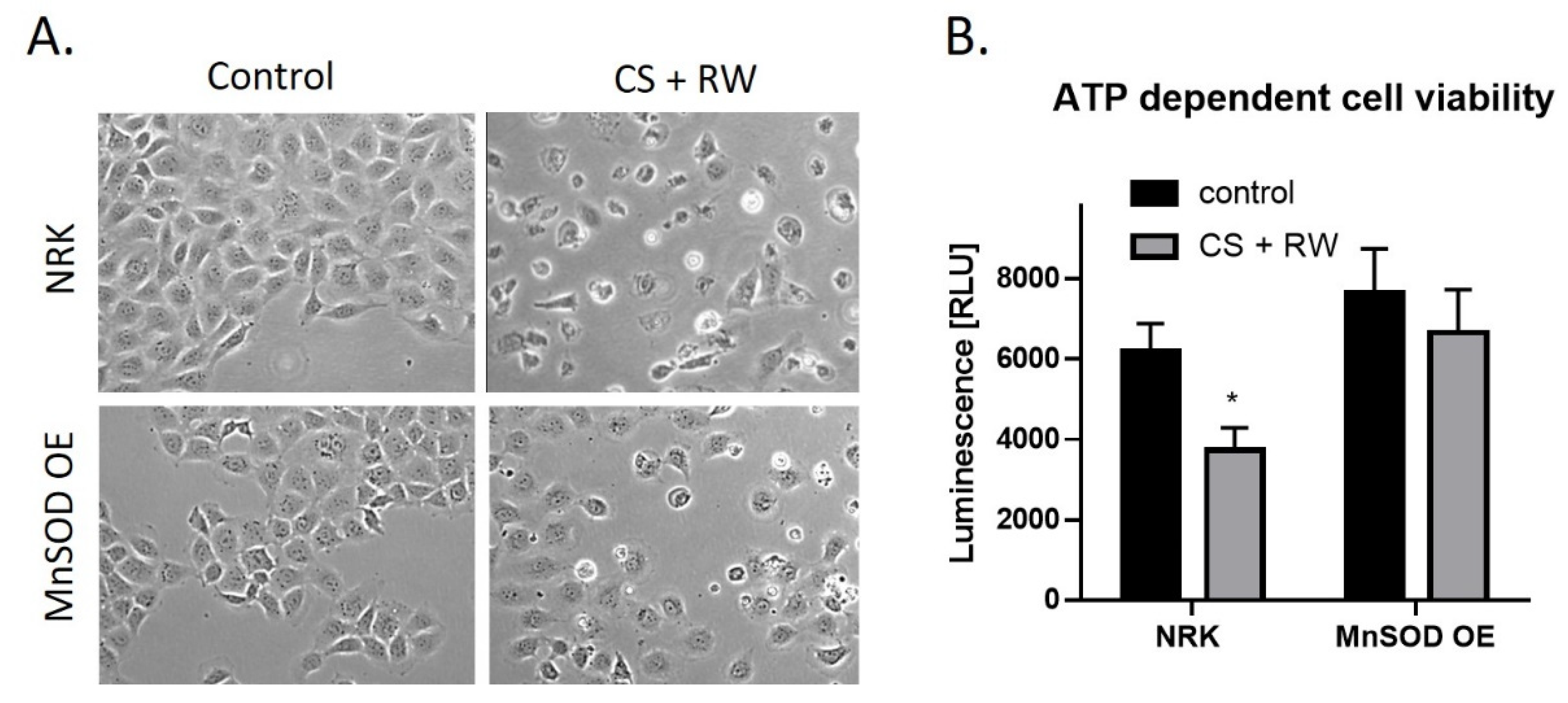
3.6. Overexpression of MnSOD Attenuates CS + RW-Induced Cell Injury
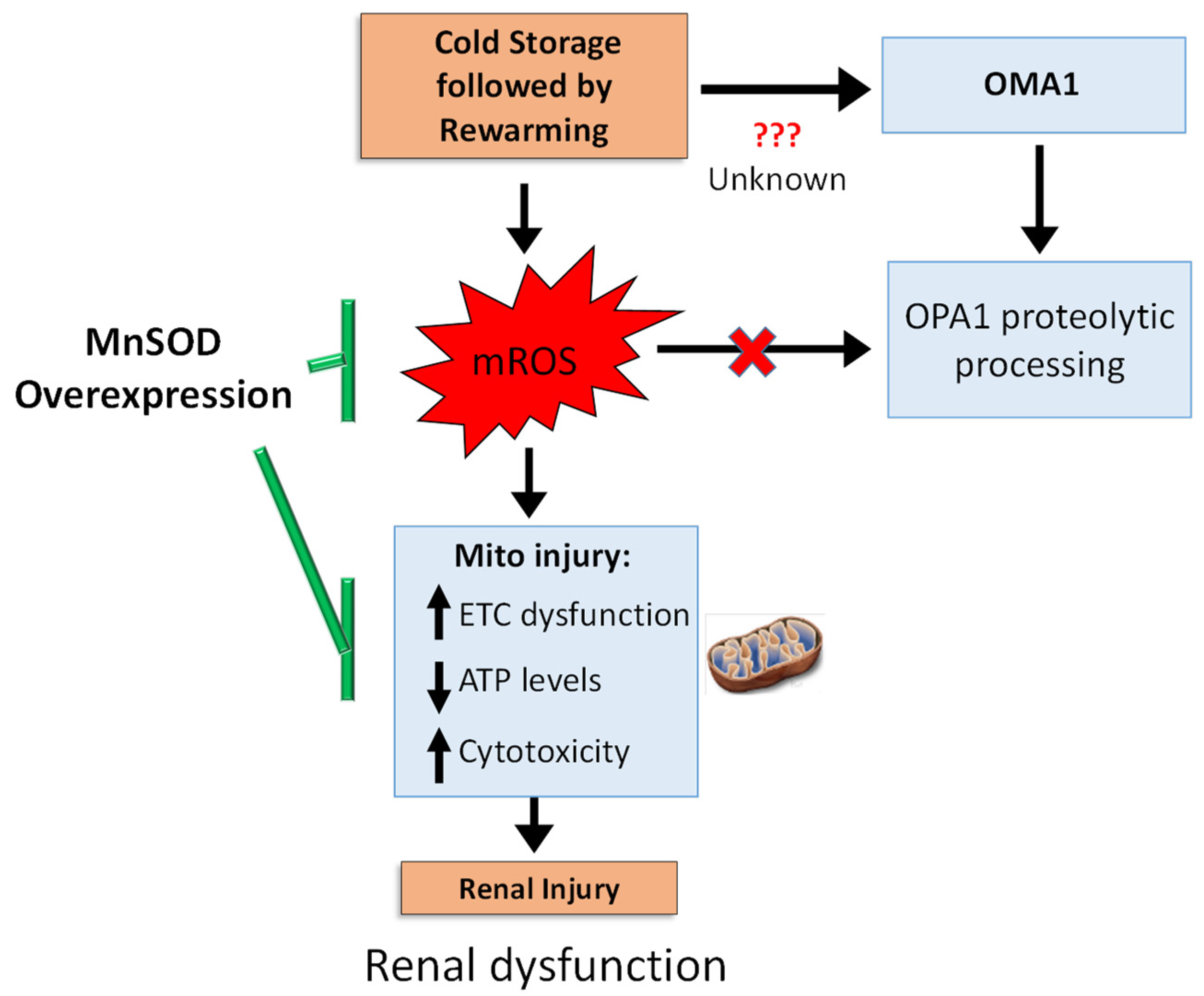
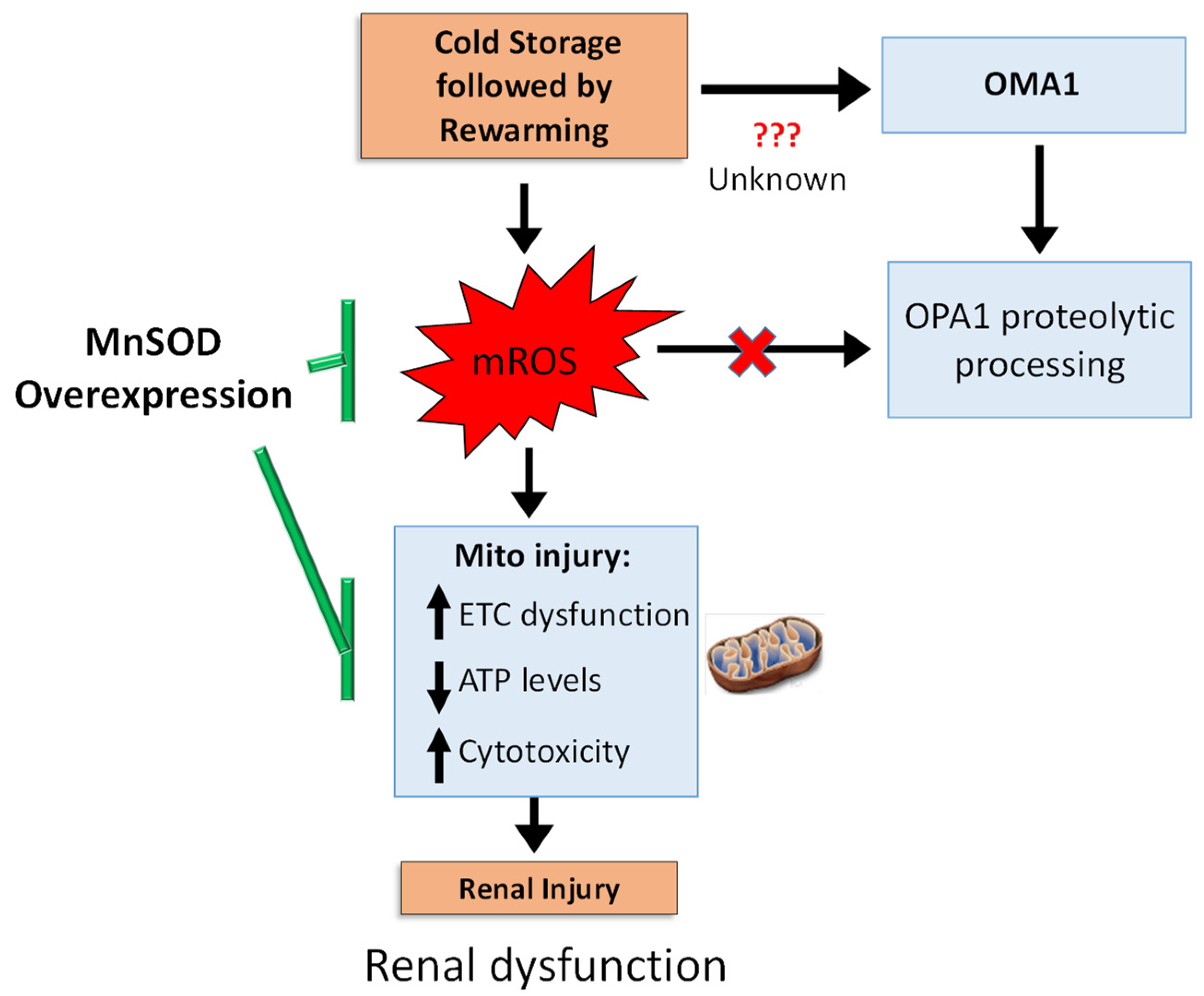
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS | cold storage |
| mROS | mitochondrial reactive oxygen species |
| MnSOD | manganese superoxide dismutase |
| CS + RW | cold storage followed by rewarming |
| NRK cells | normal rat kidney proximal tubular cells |
| siRNA | small interfering RNA |
| HRR | high-resolution respirometry |
| ESKD | end-stage kidney disease |
| S-OPA1 | short OPA1 |
| L-OPA1 | long OPA1 |
References
- Seng, J.J.B.; Tan, J.Y.; Yeam, C.T.; Htay, H.; Foo, W.Y.M. Factors affecting medication adherence among pre-dialysis chronic kidney disease patients: A systematic review and meta-analysis of literature. Int. Urol. Nephrol. 2020, 52, 903–916. [Google Scholar] [CrossRef]
- Southard, J.H.; Lutz, M.F.; Ametani, M.S.; Belzer, F.O. Stimulation of ATP synthesis in hypothermically perfused dog kidneys by adenosine and PO4. Cryobiology 1984, 21, 13–19. [Google Scholar] [CrossRef]
- Southard, J.H.; Ametani, M.S.; Lutz, M.F.; Belzer, F.O. Effects of hypothermic perfusion of kidneys on tissue and mitochondrial phospholipids. Cryobiology 1984, 21, 20–24. [Google Scholar] [CrossRef]
- Southard, J.H.; Van der Laan, N.C.; Lutz, M.; Pavlock, G.S.; Belzer, J.P.; Belzer, F.O. Comparison of the effect of temperature on kidney cortex mitochondria from rabbit, dog, pig, and human: Arrhenius plots of ADP-stimulated respiration. Cryobiology 1983, 20, 395–400. [Google Scholar] [CrossRef]
- Maathuis, M.H.; Leuvenink, H.G.; Ploeg, R.J. Perspectives in organ preservation. Transplantation 2007, 83, 1289–1298. [Google Scholar] [CrossRef]
- Salahudeen, A.K. Cold ischemic injury of transplanted kidneys: New insights from experimental studies. Am. J. Physiol. Renal Physiol. 2004, 287, F181–F187. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.K. Free radicals in kidney disease and transplantation. Saudi J. Kidney Dis. Transpl. 1999, 10, 137–143. [Google Scholar]
- Salahudeen, A.K. Cold ischemic injury of transplanted organs: Some new strategies against an old problem. Am. J. Transplant. 2004, 4, 1. [Google Scholar] [CrossRef]
- Salahudeen, A.K. Consequences of cold ischemic injury of kidneys in clinical transplantation. J. Investig. Med. 2004, 52, 296–298. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Haider, N.; May, W. Cold ischemia and the reduced long-term survival of cadaveric renal allografts. Kidney Int. 2004, 65, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.K.; Huang, H.; Joshi, M.; Moore, N.A.; Jenkins, J.K. Involvement of the mitochondrial pathway in cold storage and rewarming-associated apoptosis of human renal proximal tubular cells. Am. J. Transplant. 2003, 3, 273–280. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Huang, H.; Patel, P.; Jenkins, J.K. Mechanism and prevention of cold storage-induced human renal tubular cell injury. Transplantation 2000, 70, 1424–1431. [Google Scholar] [CrossRef] [Green Version]
- Faure, J.P.; Hauet, T.; Han, Z.; Goujon, J.M.; Petit, I.; Mauco, G.; Eugene, M.; Carretier, M.; Papadopoulos, V. Polyethylene glycol reduces early and long-term cold ischemia-reperfusion and renal medulla injury. J. Pharmacol. Exp. Ther. 2002, 302, 861–870. [Google Scholar] [CrossRef]
- Hauet, T.; Mothes, D.; Goujon, J.M.; Carretier, M.; Eugene, M. Protective effect of polyethylene glycol against prolonged cold ischemia and reperfusion injury: Study in the isolated perfused rat kidney. J. Pharmacol. Exp. Ther. 2001, 297, 946–952. [Google Scholar]
- Mitchell, T.; Saba, H.; Laakman, J.; Parajuli, N.; MacMillan-Crow, L.A. Role of mitochondrial-derived oxidants in renal tubular cell cold-storage injury. Free Radic. Biol. Med. 2010, 49, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, N.; Campbell, L.H.; Marine, A.; Brockbank, K.G.; Macmillan-Crow, L.A. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLoS ONE 2012, 7, e48590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.; Rotaru, D.; Saba, H.; Smith, R.A.; Murphy, M.P.; MacMillan-Crow, L.A. The mitochondria-targeted antioxidant mitoquinone protects against cold storage injury of renal tubular cells and rat kidneys. J. Pharmacol. Exp. Ther. 2011, 336, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.B.; Blaszak, R.T.; Parajuli, N. Targeting Mitochondria during Cold Storage to Maintain Proteasome Function and Improve Renal Outcome after Transplantation. Int. J. Mol. Sci. 2020, 21, 3506. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, N.; Shrum, S.; Tobacyk, J.; Harb, A.; Arthur, J.M.; MacMillan-Crow, L.A. Renal cold storage followed by transplantation impairs expression of key mitochondrial fission and fusion proteins. PLoS ONE 2017, 12, e0185542. [Google Scholar]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Arnaune-Pelloquin, L.; Emorine, L.J.; Mils, V.; Daloyau, M.; Hamel, C.; Amati-Bonneau, P.; et al. Mitochondrial dynamics and disease, OPA1. Biochim. Biophys. Acta 2006, 1763, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Lo, S.; MacMillan-Crow, L.A.; Parajuli, N. Renal cold storage followed by transplantation impairs proteasome function and mitochondrial protein homeostasis. Am. J. Physiol. Renal. Physiol. 2019, 316, F42–F53. [Google Scholar] [CrossRef]
- Shrum, S.; Rusch, N.J.; MacMillan-Crow, L.A. Specific BK Channel Activator NS11021 Protects Rat Renal Proximal Tubular Cells from Cold Storage-Induced Mitochondrial Injury In Vitro. Biomolecules 2019, 9, 825. [Google Scholar] [CrossRef] [Green Version]
- Cruthirds, D.L.; Saba, H.; MacMillan-Crow, L.A. Overexpression of manganese superoxide dismutase protects against ATP depletion-mediated cell death of proximal tubule cells. Arch. Biochem. Biophys. 2005, 437, 96–105. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Shrum, S.; Tobacyk, J.; Lo, S.; Parajuli, N.; MacMillan-Crow, L.A. The BK activator NS11021 partially protects rat kidneys from cold storage and transplantation-induced mitochondrial and renal injury. Arch. Biochem. Biophys. 2020, 688, 108410. [Google Scholar] [CrossRef]
- Tobacyk, J.; Parajuli, N.; Shrum, S.; Crow, J.P.; MacMillan-Crow, L.A. The first direct activity assay for the mitochondrial protease OMA1. Mitochondrion 2019, 46, 1–5. [Google Scholar] [CrossRef]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal. Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [Green Version]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [Green Version]
- Del Dotto, V.; Fogazza, M.; Lenaers, G.; Rugolo, M.; Carelli, V.; Zanna, C. OPA1: How much do we know to approach therapy? Pharmacol. Res. 2018, 131, 199–210. [Google Scholar] [CrossRef]
- Guillery, O.; Malka, F.; Landes, T.; Guillou, E.; Blackstone, C.; Lombes, A.; Belenguer, P.; Arnoult, D.; Rojo, M. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol. Cell 2008, 100, 315–325. [Google Scholar] [CrossRef]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal. Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef] [Green Version]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Bohovych, I.; Donaldson, G.; Christianson, S.; Zahayko, N.; Khalimonchuk, O. Stress-triggered activation of the metalloprotease Oma1 involves its C-terminal region and is important for mitochondrial stress protection in yeast. J. Biol. Chem. 2014, 289, 13259–13272. [Google Scholar] [CrossRef] [Green Version]
- Silic-Benussi, M.; Scattolin, G.; Cavallari, I.; Minuzzo, S.; Del Bianco, P.; Francescato, S.; Basso, G.; Indraccolo, S.; D’Agostino, D.M.; Ciminale, V. Selective killing of human T-ALL cells: An integrated approach targeting redox homeostasis and the OMA1/OPA1 axis. Cell Death Dis. 2018, 9, 822. [Google Scholar] [CrossRef]
- Bohovych, I.; Dietz, J.V.; Swenson, S.; Zahayko, N.; Khalimonchuk, O. Redox Regulation of the Mitochondrial Quality Control Protease Oma1. Antioxid. Redox Signal. 2019, 31, 429–443. [Google Scholar] [CrossRef]
- Shrum, S.; MacMillan-Crow, L.A.; Parajuli, N. Cold Storage Exacerbates Renal and Mitochondrial Dysfunction Following Transplantation. J. Kidney 2016, 2, 114. [Google Scholar]
- Karhumaki, P.; Tiitinen, S.L.; Turpeinen, H.; Parkkinen, J. Inhibition of ERK1/2 activation by phenolic antioxidants protects kidney tubular cells during cold storage. Transplantation 2007, 83, 948–953. [Google Scholar] [CrossRef]
- Bartels-Stringer, M.; Kramers, C.; Wetzels, J.F.; Russel, F.G.; Groot, H.; Rauen, U. Hypothermia causes a marked injury to rat proximal tubular cells that is aggravated by all currently used preservation solutions. Cryobiology 2003, 47, 82–91. [Google Scholar] [CrossRef]
- Ahlenstiel, T.; Burkhardt, G.; Kohler, H.; Kuhlmann, M.K. Improved cold preservation of kidney tubular cells by means of adding bioflavonoids to organ preservation solutions. Transplantation 2006, 81, 231–239. [Google Scholar] [CrossRef]
- Bartels-Stringer, M.; Verpalen, J.T.; Wetzels, J.F.; Russel, F.G.; Kramers, C. Iron chelation or anti-oxidants prevent renal cell damage in the rewarming phase after normoxic, but not hypoxic cold incubation. Cryobiology 2007, 54, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Candas, D.; Li, J.J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef] [Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Cruthirds, D.L.; Novak, L.; Akhi, K.M.; Sanders, P.W.; Thompson, J.A.; MacMillan-Crow, L.A. Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Arch. Biochem. Biophys. 2003, 412, 27–33. [Google Scholar] [CrossRef]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep. 2015, 16, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta Bioenerg. 2018, 1859, 263–269. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Sheu, S.S.; Yoon, Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J. Biol. Chem. 2020, 295, 6543–6560. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Li, Q.; Liu, S.; An, X.; Huang, Z.; Zhang, B.; Yuan, Y.; Xing, C. Protective effect of hyperoside against renal ischemia-reperfusion injury via modulating mitochondrial fission, oxidative stress, and apoptosis. Free Radic. Res. 2019, 53, 727–736. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobacyk, J.; KC, G.; MacMillan-Crow, L.A. Overexpression of MnSOD Protects against Cold Storage-Induced Mitochondrial Injury but Not against OMA1-Dependent OPA1 Proteolytic Processing in Rat Renal Proximal Tubular Cells. Antioxidants 2021, 10, 1272. https://doi.org/10.3390/antiox10081272
Tobacyk J, KC G, MacMillan-Crow LA. Overexpression of MnSOD Protects against Cold Storage-Induced Mitochondrial Injury but Not against OMA1-Dependent OPA1 Proteolytic Processing in Rat Renal Proximal Tubular Cells. Antioxidants. 2021; 10(8):1272. https://doi.org/10.3390/antiox10081272
Chicago/Turabian StyleTobacyk, Julia, Grishma KC, and Lee Ann MacMillan-Crow. 2021. "Overexpression of MnSOD Protects against Cold Storage-Induced Mitochondrial Injury but Not against OMA1-Dependent OPA1 Proteolytic Processing in Rat Renal Proximal Tubular Cells" Antioxidants 10, no. 8: 1272. https://doi.org/10.3390/antiox10081272
APA StyleTobacyk, J., KC, G., & MacMillan-Crow, L. A. (2021). Overexpression of MnSOD Protects against Cold Storage-Induced Mitochondrial Injury but Not against OMA1-Dependent OPA1 Proteolytic Processing in Rat Renal Proximal Tubular Cells. Antioxidants, 10(8), 1272. https://doi.org/10.3390/antiox10081272