
Dual Mechanisms of Cardiac Action Potential Prolongation by 4-Oxo-Nonenal Increasing the Risk of Arrhythmia; Late Na+ Current Induction and hERG K+ Channel Inhibition

Abstract
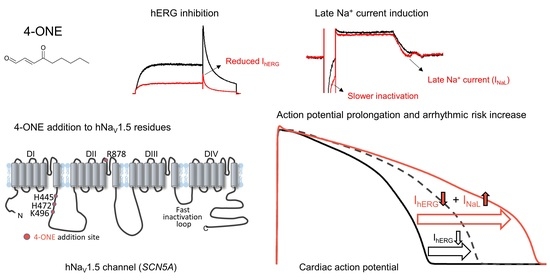
1. Introduction
2. Materials and Methods
2.1. Cell Preparation
2.2. Electrophysiological Recording
2.3. In Silico Simulation
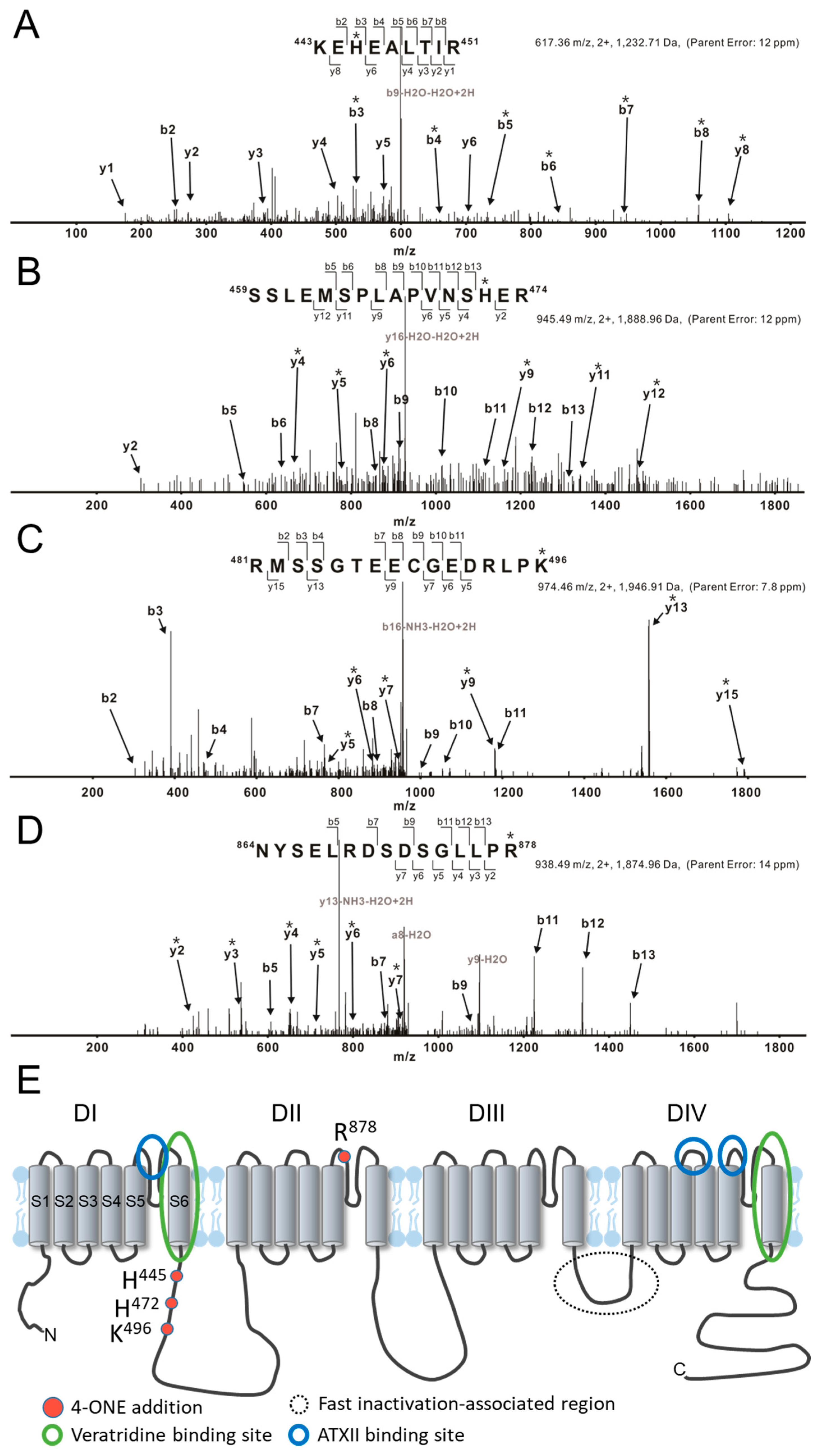
2.4. Tandem Mass Spectrometry
2.5. Chemicals
2.6. Statistical Analysis
3. Results
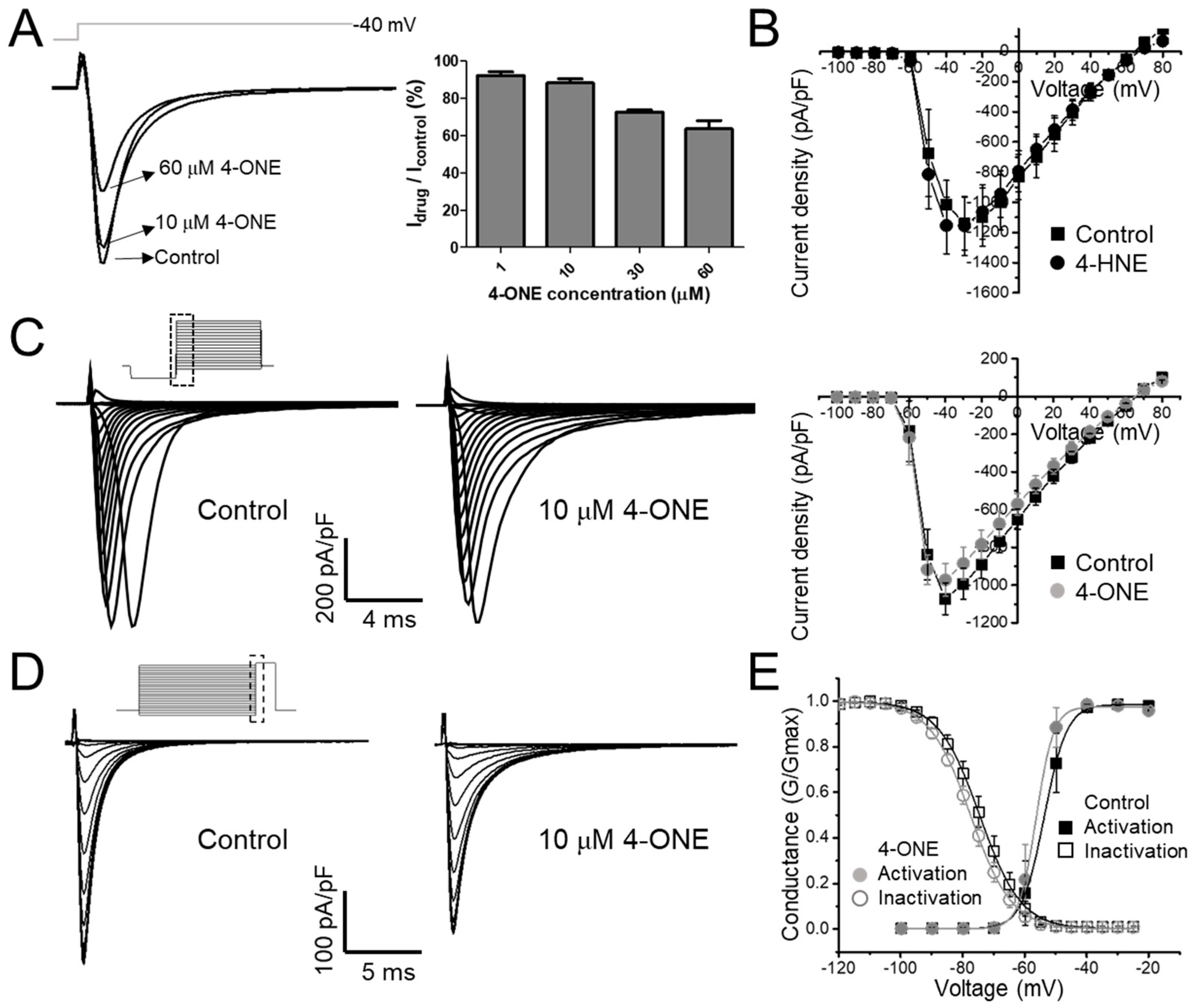
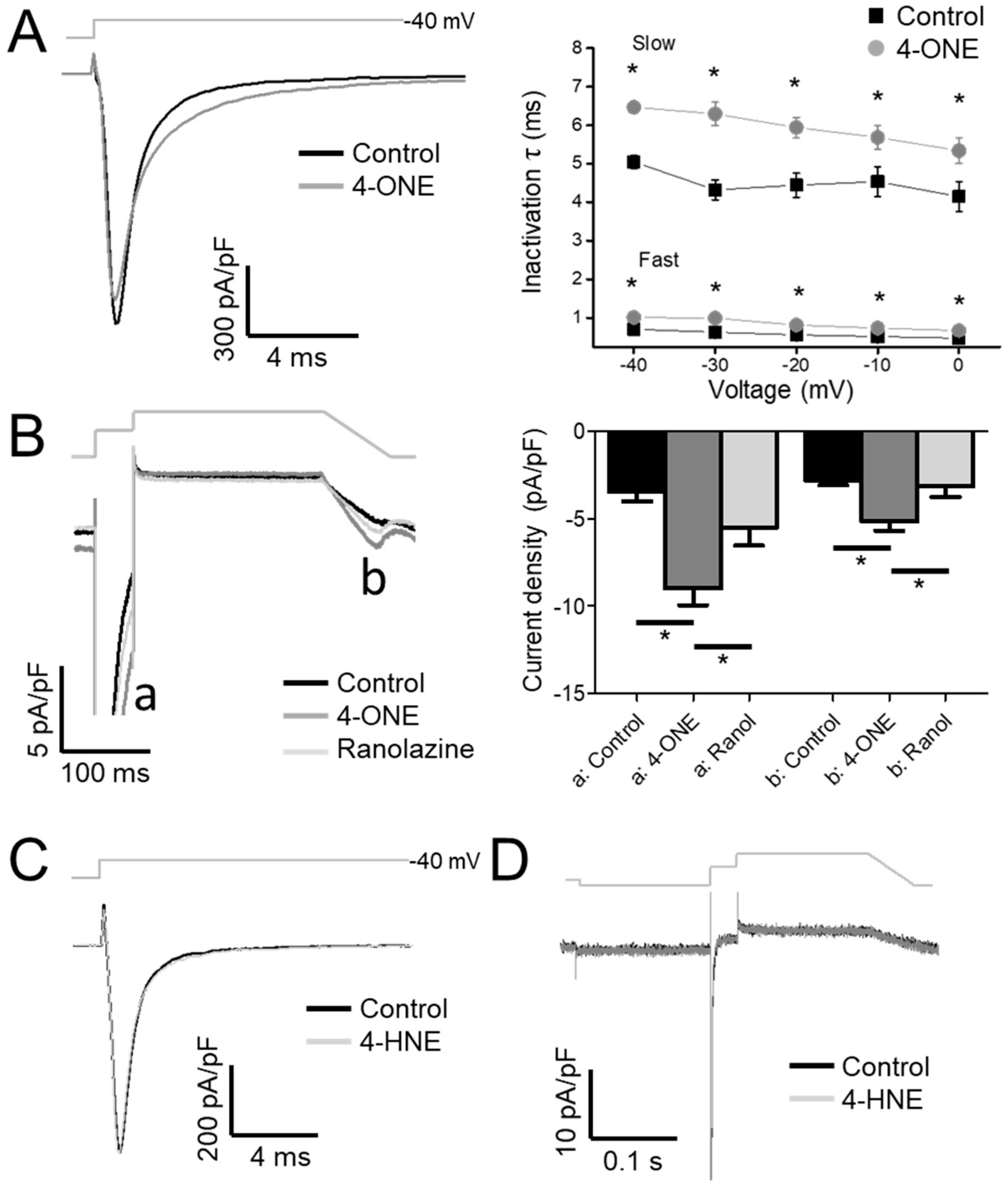
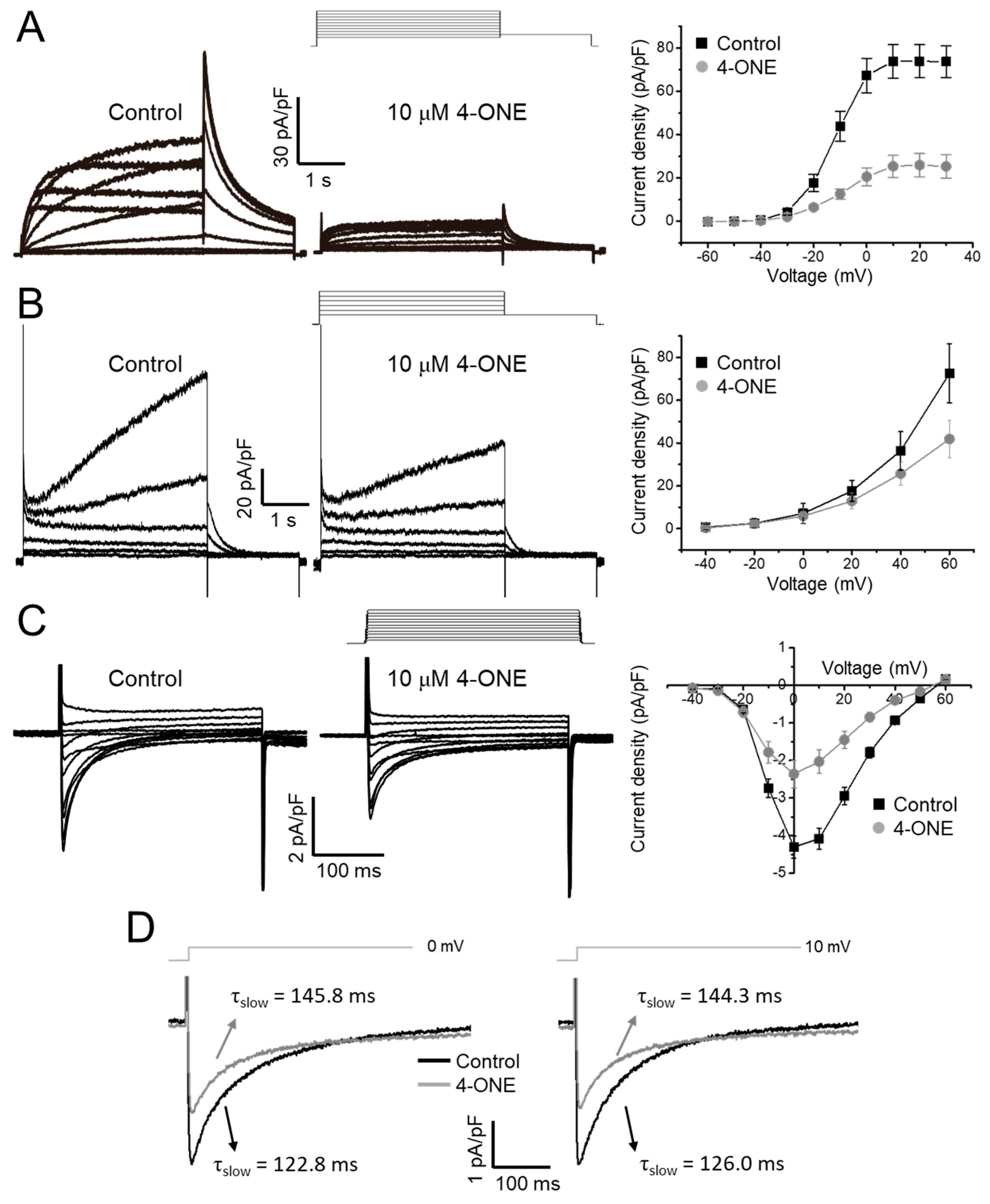
3.1. Slowed hNaV1.5 Inactivation and INaL Induction by 4-ONE
3.2. Multiple Effects of 4-ONE on IKr, IKs, and ICa,L
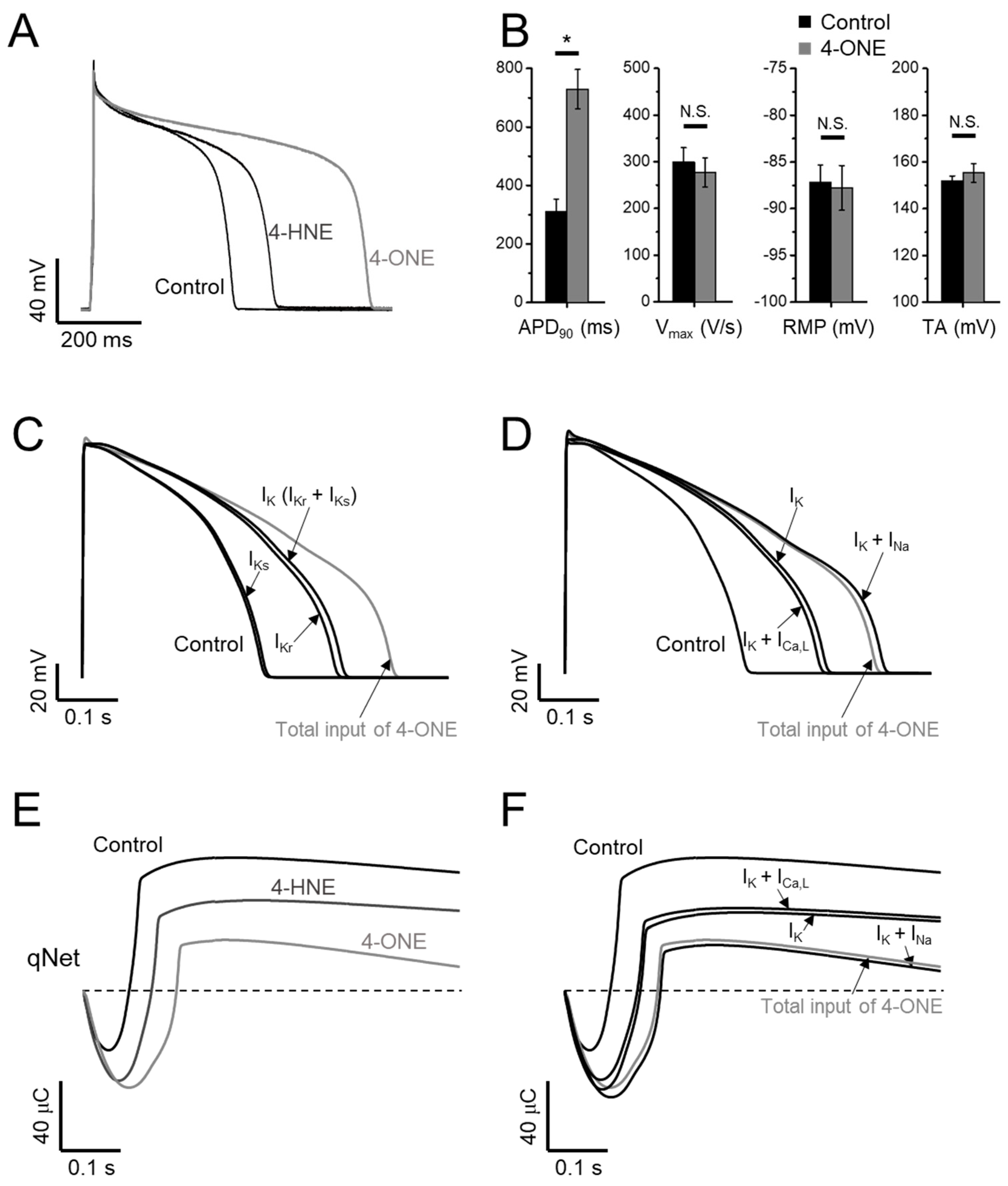
3.3. APD Prolongation and Increased Risk of Arrhythmia by 4-ONE
4. Discussion
4.1. INaL and Inactivation of NaV1.5
4.2. Pathophysiological Implication of 4-ONE and INaL
4.3. Application of CiPA in Silico Model
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blair, I.A. Endogenous glutathione adducts. Curr. Drug. Metab. 2006, 7, 853–872. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R. Advanced lipoxidation end-products. Chem. Biol. Interact. 2011, 192, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef]
- Kim, C.E.; Lee, S.J.; Seo, K.W.; Park, H.M.; Yun, J.W.; Bae, J.U.; Bae, S.S.; Kim, C.D. Acrolein increases 5-lipoxygenase expression in murine macrophages through activation of ERK pathway. Toxicol. Appl. Pharmacol. 2010, 245, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, C.E.; Yun, M.R.; Seo, K.W.; Park, H.M.; Yun, J.W.; Shin, H.K.; Bae, S.S.; Kim, C.D. 4-Hydroxynonenal enhances MMP-9 production in murine macrophages via 5-lipoxygenase-mediated activation of ERK and p38 MAPK. Toxicol. Appl. Pharmacol. 2010, 242, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, R.; Yu, L.; Zhang, Y.; Ren, J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011, 32, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Gentile, F.; Pizzimenti, S.; Canuto, R.A.; Daga, M.; Arcaro, A.; Cetrangolo, G.P.; Lepore, A.; Ferretti, C.; Dianzani, C.; et al. Mitochondrial dysfunction in cancer and neurodegenerative diseases: Spotlight on fatty acid oxidation and lipoperoxidation products. Antioxidants 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef] [PubMed]
- Asselin, C.; Ducharme, A.; Ntimbane, T.; Ruiz, M.; Fortier, A.; Guertin, M.C.; Lavoie, J.; Diaz, A.; Levy, E.; Tardif, J.C.; et al. Circulating levels of linoleic acid and HDL-cholesterol are major determinants of 4-hydroxynonenal protein adducts in patients with heart failure. Redox Biol. 2014, 2, 148–155. [Google Scholar] [CrossRef]
- Giam, B.; Chu, P.Y.; Kuruppu, S.; Smith, A.I.; Horlock, D.; Kiriazis, H.; Du, X.J.; Kaje, D.M.; Rajapakse, N.W. N-acetylcysteine attenuates the development of cardiac fibrosis and remodeling in a mouse model of heart failure. Physiol. Rep. 2016, 4, e12757. [Google Scholar] [CrossRef]
- Gupta, R.C.; Singh-Gupta, V.; Zhang, K.F.; Xu, J.; Sabbah, H.N. Elamipretide (Bendavia (TM)) restores 4-hydroxy-2-nonenal protein adducts and aldehyde dehydrogenase-2 activity and mRNA expression in left ventricular myocardium of dogs with advanced heart failure. Circulation 2016, 134, A12949. [Google Scholar]
- Choi, S.W.; Choi, S.W.; Jeon, Y.K.; Moon, S.H.; Zhang, Y.H.; Kim, S.J. Suppression of hERG K(+) current and cardiac action potential prolongation by 4-hydroxynonenal via dual mechanisms. Redox Biol. 2018, 19, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Liu, N.; Priori, S.G. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 2009, 6, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L.; Roden, D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.M.; Amin, A.S. Clinical spectrum of SCN5A mutations: Long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.K.; Saint, D.A.; Gage, P.W. Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. 1996, 497, 337–347. [Google Scholar] [CrossRef]
- Undrovinas, A.I.; Maltsev, V.A.; Sabbah, H.N. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol. Life Sci. 1999, 55, 494–505. [Google Scholar] [CrossRef]
- Valdivia, C.R.; Chu, W.W.; Pu, J.L.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef]
- Zhang, W.H.; Liu, J.; Xu, G.; Yuan, Q.; Sayre, L.M. Model studies on protein side chain modification by 4-oxo-2-nonenal. Chem. Res. Toxicol. 2003, 16, 512–523. [Google Scholar] [CrossRef]
- Doorn, J.A.; Petersen, D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem. Biol. Interact. 2003, 143, 93–100. [Google Scholar] [CrossRef]
- Lee, S.H.; Blair, I.A. Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol. 2000, 13, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Clark, T.E.; McAlexander, M.A.; Nassenstein, C.; Sheardown, S.A.; Wilson, S.; Thornton, J. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J. Physiol. 2008, 586, 3447–3459. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ridder, B.J.; Han, X.; Wu, W.W.; Sheng, J.; Tran, P.N.; Wu, M.; Randolph, A.; Johnstone, R.H.; Mirams, G.R.; et al. Assessment of an in silico mechanistic model for proarrhythmia risk prediction under the CiPA initiative. Clin. Pharmacol. Ther. 2019, 105, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Chang, K.C.; Beattie, K.A.; Sheng, J.; Tran, P.N.; Wu, W.W.; Wu, M.; Strauss, D.G.; Colatsky, T.; Li, Z. Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Front. Physiol. 2017, 8, 616. [Google Scholar] [CrossRef]
- Jian, W.; Lee, S.H.; Mesaros, C.; Oe, T.; Silva Elipe, M.V.; Blair, I.A. A novel 4-oxo-2(E)-nonenal-derived endogenous thiadiazabicyclo glutathione adduct formed during cellular oxidative stress. Chem. Res. Toxicol. 2007, 20, 1008–1018. [Google Scholar] [CrossRef]
- Doorn, J.A.; Petersen, D.R. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 2002, 15, 1445–1450. [Google Scholar] [CrossRef]
- Clancy, C.E.; Tateyama, M.; Kass, R.S. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J. Clin. Investig. 2002, 110, 1251–1262. [Google Scholar] [CrossRef]
- Tian, X.L.; Yong, S.L.; Wan, X.; Wu, L.; Chung, M.K.; Tchou, P.J.; Rosenbaum, D.S.; Van Wagoner, D.R.; Kirsch, G.E.; Wang, Q. Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc. Res. 2004, 61, 256–267. [Google Scholar] [CrossRef]
- Kistamás, K.; Hézső, T.; Horváth, B.; Nánási, P.P. Late sodium current and calcium homeostasis in arrhythmogenesis. Channels 2021, 15, 1–19. [Google Scholar] [CrossRef]
- Mangold, K.E.; Brumback, B.D.; Angsutararux, P.; Voelker, T.L.; Zhu, W.; Kang, P.W.; Moreno, J.D.; Silva, J.R. Mechanisms and models of cardiac sodium channel inactivation. Channels 2017, 11, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Wilde, A.A.M.; Lodder, E.M. The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene 2015, 573, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, S.; Chen, Q.; Wan, X.; Shen, J.; Hoeltge, G.A.; Timur, A.A.; Keating, M.T.; Kirsch, G.E. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J. Med. Genet. 2004, 41, e66. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.; Greer-Short, A.; Satroplus, T.; Patel, N.; Nassal, D.; Mohler, P.J.; Hund, T.J. CaMKII-dependent late Na+ current increases electrical dispersion and arrhythmia in ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H794–H801. [Google Scholar] [CrossRef]
- Hallaq, H.; Wang, D.W.; Kunic, J.D.; George, A.L., Jr.; Wells, K.S.; Murray, K.T. Activation osf protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H782–H789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Ma, J.; Zhang, P.; Wan, W.; Kong, L.; Wu, L. Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflugers Arch. 2012, 463, 513–522. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Polak, J. Hypoxia. 4. Hypoxia and ion channel function. Am. J. Physiol. Cell Physiol. 2011, 300, C951–C967. [Google Scholar] [CrossRef] [PubMed]
- Ahern, G.P.; Hsu, S.F.; Klyachko, V.A.; Jackson, M.B. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 2000, 275, 28810–28815. [Google Scholar] [CrossRef]
- Denac, H.; Mevissen, M.; Scholtysik, G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 453–479. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Maier, L.S. Role of ranolazine in angina, heart failure, arrhythmias, and diabetes. Pharmacol. Therapeutics 2012, 133, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Khera, S.; Kolte, D.; Aronow, W.S.; Iwai, S. Antiarrhythmic properties of ranolazine: A review of the current evidence. Int. J. Cardiol. 2015, 187, 66–74. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | 4-HNE | 4-ONE | |
|---|---|---|---|
| ths (INav) | |||
| thL (INaL) | 200.0 | 400.0 | |
| tfcaf (ICa,L) | |||
| tfcas (ICa,L) | |||
| Conductance for IKr | 1.0 | 0.6 | 0.4 |
| Conductance for IKs | 1.0 | 0.8 | 0.7 |
| Conductance for ICa,L | 1.0 | 1.0 | 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.-W.; Yin, M.-Z.; Park, N.-K.; Woo, J.-H.; Kim, S.-J. Dual Mechanisms of Cardiac Action Potential Prolongation by 4-Oxo-Nonenal Increasing the Risk of Arrhythmia; Late Na+ Current Induction and hERG K+ Channel Inhibition. Antioxidants 2021, 10, 1139. https://doi.org/10.3390/antiox10071139
Choi S-W, Yin M-Z, Park N-K, Woo J-H, Kim S-J. Dual Mechanisms of Cardiac Action Potential Prolongation by 4-Oxo-Nonenal Increasing the Risk of Arrhythmia; Late Na+ Current Induction and hERG K+ Channel Inhibition. Antioxidants. 2021; 10(7):1139. https://doi.org/10.3390/antiox10071139
Chicago/Turabian StyleChoi, Seong-Woo, Ming-Zhe Yin, Na-Kyeong Park, Joo-Han Woo, and Sung-Joon Kim. 2021. "Dual Mechanisms of Cardiac Action Potential Prolongation by 4-Oxo-Nonenal Increasing the Risk of Arrhythmia; Late Na+ Current Induction and hERG K+ Channel Inhibition" Antioxidants 10, no. 7: 1139. https://doi.org/10.3390/antiox10071139
APA StyleChoi, S.-W., Yin, M.-Z., Park, N.-K., Woo, J.-H., & Kim, S.-J. (2021). Dual Mechanisms of Cardiac Action Potential Prolongation by 4-Oxo-Nonenal Increasing the Risk of Arrhythmia; Late Na+ Current Induction and hERG K+ Channel Inhibition. Antioxidants, 10(7), 1139. https://doi.org/10.3390/antiox10071139