
MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes

 , , ,
, , ,  ,
,  and
and 
Abstract
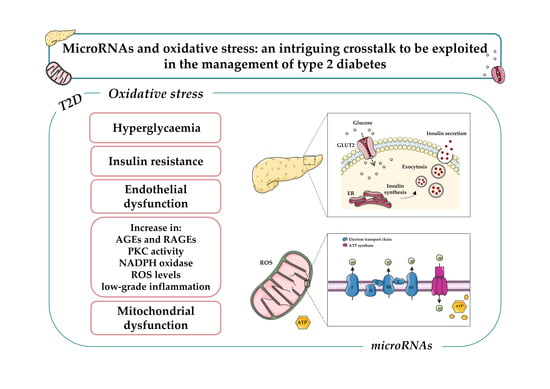
1. Introduction
2. Survey Methodology
Study Selection Criteria
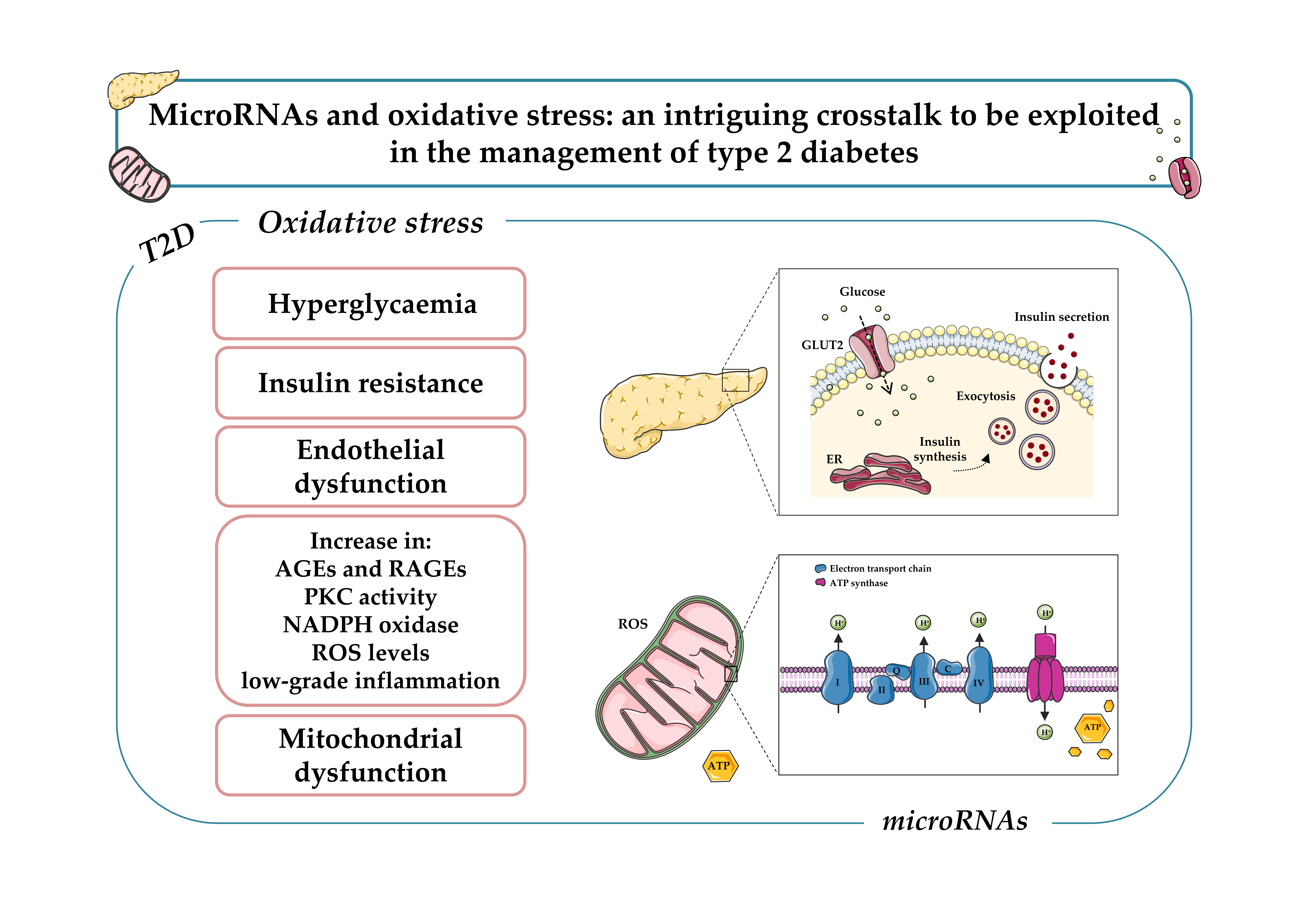
3. Oxidative Stress and Diabetes
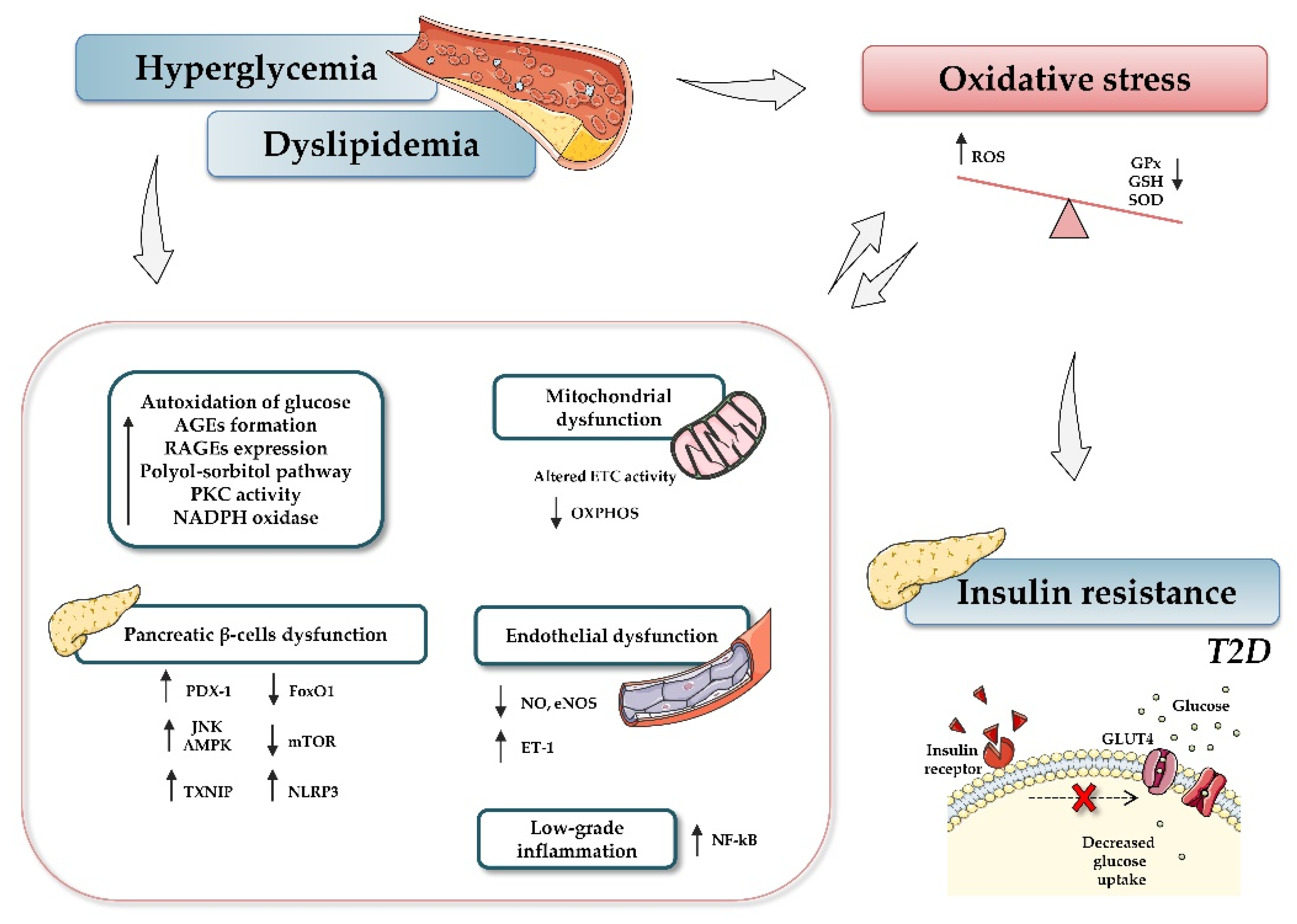
4. MiRNAs
5. Oxidative Stress, miRNAs, and Diabetes
5.1. MiRNAs, Hyperglycemia-Induced ROS and Endothelial Dysfunction
5.2. MiRNAs and AGE-Induced Oxidative Stress
5.3. MiRNAs and Pancreatic β-Cell Function
5.4. MitomiR
6. MiRNAs, Therapeutic Targets, and Delivery Systems
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diabetes Atlas. 2020. Available online: https://diabetesatlas.org/en (accessed on 3 March 2021).
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Folli, F.; Corradi, D.; Fanti, P.; Davalli, A.; Paez, A.; Giaccari, A.; Perego, C.; Muscogiuri, G. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: Avenues for a mechanistic-based therapeutic approach. Curr. Diabetes Rev. 2011, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Chikezie, P.C.; Ojiako, O.A.; Ogbuji, A.C. Oxidative stress in diabetes mellitus. Int. J. Biol. Chem. 2015, 9, 92–109. [Google Scholar] [CrossRef]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic Complications of Diabetes Mellitus: A Mini Review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef]
- Victor, V.M.; Rocha, M.; Herance, R.; Hernandez-Mijares, A. Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr. Pharm. Des. 2011, 17, 3947–3958. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; Garcia, M.L.; Silva, A.M.; et al. Sugar-lowering drugs for type 2 diabetes mellitus and metabolic syndrome-review of classical and new compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the seed supports microRNA targeting specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef]
- Regazzi, R. MicroRNAs as therapeutic targets for the treatment of diabetes mellitus and its complications. Expert Opin. Ther. Targets 2018, 22, 153–160. [Google Scholar] [CrossRef]
- Wu, B.; Miller, D. Involvement of microRNAs in diabetes and its complications. Methods Mol. Biol. 2017, 1617, 225–239. [Google Scholar] [CrossRef]
- Baroukh, N.; Ravier, M.A.; Loder, M.K.; Hill, E.V.; Bounacer, A.; Scharfmann, R.; Rutter, G.A.; Van Obberghen, E. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J. Biol. Chem. 2007, 282, 19575–19588. [Google Scholar] [CrossRef]
- LaPierre, M.P.; Stoffel, M. MicroRNAs as stress regulators in pancreatic beta cells and diabetes. Mol. Metab. 2017, 6, 1010–1023. [Google Scholar] [CrossRef]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef]
- Siasos, G.; Paschou, S.A.; Tousoulis, D. Mitochondria and diabetes. Ann. Transl. Med. 2020, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative stress in diabetes: Implications for vascular and other complications. Int. J. Mol. Sci. 2013, 14, 21525–21550. [Google Scholar] [CrossRef]
- Brubaker, P.L.; Drucker, D.J. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004, 145, 2653–2659. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Bierhaus, A.; Hofmann, M.A.; Ziegler, R.; Nawroth, P.P. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovas. Res. 1998, 37, 586–600. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, N.; Suematsu, M.; Kaseda, K.; Matsui, T. Advanced glycation end products: A molecular target for vascular complications in diabetes. Mol. Med. 2015, 21, S32–S40. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Hori, O.; Stern, D.; Schmidt, A.M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J. Clin. Investig. 1996, 97, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; D’Ardes, D.; Davi, G. Oxidative stress in chronic vascular disease: From prediction to prevention. Vasc. Pharmacol. 2015, 74, 23–37. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 2009, 150, 2569–2576. [Google Scholar] [CrossRef]
- Wang, X.; Yu, S.; Wang, C.Y.; Wang, Y.; Liu, H.X.; Cui, Y.; Zhang, L.D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev. Biol. Anim. 2015, 51, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.E.; Prasad, C.; Vijayagopal, P.; Juma, S.; Imrhan, V. Advanced glycation end products, inflammation, and chronic metabolic diseases: Links in a chain? Crit. Rev. Food Sci. Nutr. 2016, 56, 989–998. [Google Scholar] [CrossRef]
- Li, Y.; Chang, Y.; Ye, N.; Chen, Y.; Zhang, N.; Sun, Y. Advanced glycation end products-induced mitochondrial energy metabolism dysfunction alters proliferation of human umbilical vein endothelial cells. Mol. Med. Rep. 2017, 15, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- de Paula, M.L.A.; Villela, A.M.R.; Negri, M.M.; Kanaan, S.; Weide, L.d.C.C. Role of advanced glycation end products related to the onset of diabetic kidney disease complications. Clin. Biomed. Res. 2017, 37. [Google Scholar] [CrossRef]
- Lin, N.; Zhang, H.; Su, Q. Advanced glycation end-products induce injury to pancreatic beta cells through oxidative stress. Diabetes Metab. 2012, 38, 250–257. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Biden, T.J. Protein kinase C function in muscle, liver, and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes 2008, 57, 1774–1783. [Google Scholar] [CrossRef]
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443. [Google Scholar] [CrossRef]
- Domingueti, C.P.; Dusse, L.M.; Carvalho, M.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complic. 2016, 30, 738–745. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann. N. Y. Acad. Sci. 2002, 967, 146–157. [Google Scholar] [CrossRef]
- Cazzolli, R.; Mitchell, T.W.; Burchfield, J.G.; Pedersen, D.J.; Turner, N.; Biden, T.J.; Schmitz-Peiffer, C. Dilinoleoyl-phosphatidic acid mediates reduced IRS-1 tyrosine phosphorylation in rat skeletal muscle cells and mouse muscle. Diabetologia 2007, 50, 1732–1742. [Google Scholar] [CrossRef][Green Version]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and beta-cell dysfunction. Oxid. Med. Cell Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Rebelato, E.; Abdulkader, F.; Krause, M.; Carpinelli, A.; Curi, R. Reactive oxygen and nitrogen species generation, antioxidant defenses, and beta-cell function: A critical role for amino acids. J. Endocrinol. 2012, 214, 11–20. [Google Scholar] [CrossRef]
- Kim-Muller, J.Y.; Fan, J.; Kim, Y.J.; Lee, S.A.; Ishida, E.; Blaner, W.S.; Accili, D. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic beta cells in diabetic mice. Nat. Commun. 2016, 7, 12631. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Szkudelska, K. The relevance of AMP-activated protein kinase in insulin-secreting beta cells: A potential target for improving beta cell function? J. Physiol. Biochem. 2019, 75, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yamamoto, T.; Hisatome, I.; Li, Y.; Cheng, W.; Sun, N.; Cai, B.; Huang, T.; Zhu, Y.; Li, Z.; et al. Uric acid induces oxidative stress and growth inhibition by activating adenosine monophosphate-activated protein kinase and extracellular signal-regulated kinase signal pathways in pancreatic beta cells. Mol. Cell Endocrinol. 2013, 375, 89–96. [Google Scholar] [CrossRef]
- Blandino-Rosano, M.; Chen, A.Y.; Scheys, J.O.; Alejandro, E.U.; Gould, A.P.; Taranukha, T.; Elghazi, L.; Cras-Meneur, C.; Bernal-Mizrachi, E. mTORC1 signaling and regulation of pancreatic beta-cell mass. Cell Cycle 2012, 11, 1892–1902. [Google Scholar] [CrossRef]
- Chau, G.C.; Im, D.U.; Kang, T.M.; Bae, J.M.; Kim, W.; Pyo, S.; Moon, E.Y.; Um, S.H. mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J. Cell Biol. 2017, 216, 2091–2105. [Google Scholar] [CrossRef]
- Maedler, K.; Ardestani, A. mTORC in beta cells: More than only recognizing comestibles. J. Cell Biol. 2017, 216, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef]
- Sokolova, M.; Sahraoui, A.; Hoyem, M.; Ogaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, H.; Li, X.; Zha, Y.; Hou, W. Hydroxysafflor yellow A attenuates high glucose-induced pancreatic beta-cells oxidative damage via inhibiting JNK/c-jun signaling pathway. Biochem. Biophys. Res. Commun. 2018, 505, 353–359. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Hayes, H.L.; Moss, L.G.; Schisler, J.C.; Haldeman, J.M.; Zhang, Z.; Rosenberg, P.B.; Newgard, C.B.; Hohmeier, H.E. Pdx-1 activates islet alpha- and beta-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol. Cell Biol. 2013, 33, 4017–4029. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Miyoshi, T.; Siomi, H. Many ways to generate microRNA-like small RNAs: Non-canonical pathways for microRNA production. Mol. Genet. Genom. 2010, 284, 95–103. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, L.; Esguerra, J.L. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiol. 2014, 211, 273–284. [Google Scholar] [CrossRef]
- Chen, H.; Lan, H.Y.; Roukos, D.H.; Cho, W.C. Application of microRNAs in diabetes mellitus. J. Endocrinol. 2014, 222, R1–R10. [Google Scholar] [CrossRef]
- Golbidi, S.; Ebadi, S.A.; Laher, I. Antioxidants in the treatment of diabetes. Curr. Diabetes Rev. 2011, 7, 106–125. [Google Scholar] [CrossRef] [PubMed]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative stress-induced miR-200c disrupts the regulatory loop among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.D.; Liu, W.L.; Zeng, K.; Lei, H.Y.; Zhang, Q.G.; Zhou, S.Q.; Xu, S.Y. Advanced glycation end products activate the miRNA/RhoA/ROCK2 pathway in endothelial cells. Microcirculation 2014, 21, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-induced microRNA-34a causes diabetic endothelial dysfunction by downregulating Sirtuin1. Arter. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Yang, K.Y. Berberine Alleviates oxidative stress in islets of diabetic mice by inhibiting miR-106b expression and up-regulating SIRT1. J. Cell Biochem. 2017, 118, 4349–4357. [Google Scholar] [CrossRef]
- Li, L.M.; Hou, D.X.; Guo, Y.L.; Yang, J.W.; Liu, Y.; Zhang, C.Y.; Zen, K. Role of microRNA-214-targeting phosphatase and tensin homolog in advanced glycation end product-induced apoptosis delay in monocytes. J. Immunol. 2011, 186, 2552–2560. [Google Scholar] [CrossRef]
- Muratsu-Ikeda, S.; Nangaku, M.; Ikeda, Y.; Tanaka, T.; Wada, T.; Inagi, R. Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS ONE 2012, 7, e41462. [Google Scholar] [CrossRef]
- Fleissner, F.; Jazbutyte, V.; Fiedler, J.; Gupta, S.K.; Yin, X.; Xu, Q.; Galuppo, P.; Kneitz, S.; Mayr, M.; Ertl, G.; et al. Short communication: Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a microRNA-21-dependent mechanism. Circ. Res. 2010, 107, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar] [CrossRef]
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818. [Google Scholar] [CrossRef]
- Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Shapira, T.; Mandelbaum, A.D.; Rivkin, N.; Nir, T.; Lennox, K.A.; Behlke, M.A.; Dor, Y.; et al. miRNAs control insulin content in pancreatic beta-cells via downregulation of transcriptional repressors. EMBO J. 2011, 30, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Mohan, R.; Ozcan, S.; Tang, X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic beta-cells. J. Biol. Chem. 2012, 287, 31155–31164. [Google Scholar] [CrossRef]
- Matsuoka, T.A.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving Mafa expression in diabetic islet beta-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657. [Google Scholar] [CrossRef] [PubMed]
- Latreille, M.; Hausser, J.; Stutzer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a regulates pancreatic beta cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Plaisance, V.; Abderrahmani, A.; Perret-Menoud, V.; Jacquemin, P.; Lemaigre, F.; Regazzi, R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J. Biol. Chem. 2006, 281, 26932–26942. [Google Scholar] [CrossRef]
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes 2014, 63, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Baseler, W.A.; Thapa, D.; Jagannathan, R.; Dabkowski, E.R.; Croston, T.L.; Hollander, J.M. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am. J. Physiol. Cell Physiol. 2012, 303, C1244–C1251. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef]
- Bienertova-Vasku, J.; Sana, J.; Slaby, O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013, 336, 1–7. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 modulates the cellular energy metabolism shift during H2O2-induced oxidative stress by repressing ISCU in H9c2 cardiomyocytes. Cell Physiol. Biochem. 2017, 43, 383–394. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Tagliabue, E.; Prattichizzo, F.; Micheloni, S.; Sangalli, E.; Specchia, C.; Uccellatore, A.C.; Lupini, S.; Spinetti, G.; et al. Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naive T2D. Cardiovasc. Diabetol. 2019, 18, 18. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc. Diabetol. 2018, 17, 105. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, Y.; Guo, S.; Xiao, L.; Wu, L.; Wang, Z.; Liang, C.; Yao, R.; Zhang, Y. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3322–3338. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627. [Google Scholar] [CrossRef]
- Yamakuchi, M. MicroRNA Regulation of SIRT1. Front. Physiol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Kassan, M.; Vikram, A.; Li, Q.; Kim, Y.R.; Kumar, S.; Gabani, M.; Liu, J.; Jacobs, J.S.; Irani, K. MicroRNA-204 promotes vascular endoplasmic reticulum stress and endothelial dysfunction by targeting Sirtuin1. Sci. Rep. 2017, 7, 9308. [Google Scholar] [CrossRef]
- Comer, F.I.; Parent, C.A. PI 3-kinases and PTEN: How opposites chemoattract. Cell 2002, 109, 541–544. [Google Scholar] [CrossRef]
- Iguchi, H.; Urashima, Y.; Inagaki, Y.; Ikeda, Y.; Okamura, M.; Tanaka, T.; Uchida, A.; Yamamoto, T.T.; Kodama, T.; Sakai, J. SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic beta-cell proliferation. J. Biol. Chem. 2007, 282, 19052–19061. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Liu, C.; Naji, A.; Stoffers, D.A. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes 2013, 62, 887–895. [Google Scholar] [CrossRef]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The emerging role of MitomiRs in the pathophysiology of human disease. Adv. Exp. Med. Biol. 2015, 888, 123–154. [Google Scholar] [CrossRef]
- Bandiera, S.; Mategot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Chen, Y.; Leong, K.W. MicroRNA delivery for regenerative medicine. Adv. Drug Deliv. Rev. 2015, 88, 108–122. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, S.; Abreu-Goodger, C.; Enright, A.J. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods 2008, 5, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Liu, H.; Huang, Y. Survey of computational algorithms for microRNA target prediction. Curr. Genom. 2009, 10, 478–492. [Google Scholar] [CrossRef]
- Grieco, G.E.; Brusco, N.; Licata, G.; Nigi, L.; Formichi, C.; Dotta, F.; Sebastiani, G. Targeting microRNAs as a therapeutic strategy to reduce oxidative stress in diabetes. Int. J. Mol. Sci. 2019, 20, 6358. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Chen, H.; Wei, M.; Chen, X.; Zhang, Y.; Cao, L.; Yuan, P.; Wang, F.; Yang, G.; Ma, J. Gold nanoparticle-based miR155 antagonist macrophage delivery restores the cardiac function in ovariectomized diabetic mouse model. Int. J. Nanomed. 2017, 12, 4963–4979. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Feng, Y.; Xie, Y.; Shao, Y.; Wu, M.; Deng, X.; Yuan, W.E.; Chen, Y.; Shi, X. Nanoparticle-microRNA-146a-5p polyplexes ameliorate diabetic peripheral neuropathy by modulating inflammation and apoptosis. Nanomedicine 2019, 17, 188–197. [Google Scholar] [CrossRef]
- Fu, P.P.; Xia, Q.; Hwang, H.M.; Ray, P.C.; Yu, H. Mechanisms of nanotoxicity: Generation of reactive oxygen species. J. Food Drug Anal. 2014, 22, 64–75. [Google Scholar] [CrossRef]
- Panahi, Y.; Farshbaf, M.; Mohammadhosseini, M.; Mirahadi, M.; Khalilov, R.; Saghfi, S.; Akbarzadeh, A. Recent advances on liposomal nanoparticles: Synthesis, characterization and biomedical applications. Artif. Cells Nanomed. Biotechnol. 2017, 45, 788–799. [Google Scholar] [CrossRef]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as nanocarriers for nucleic acid and drug delivery in cancer therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef]
- Akhtar, S.; Chandrasekhar, B.; Yousif, M.H.; Renno, W.; Benter, I.F.; El-Hashim, A.Z. Chronic administration of nano-sized PAMAM dendrimers in vivo inhibits EGFR-ERK1/2-ROCK signaling pathway and attenuates diabetes-induced vascular remodeling and dysfunction. Nanomedicine 2019, 18, 78–89. [Google Scholar] [CrossRef]
- Akhtar, S.; Yousif, M.H.; Dhaunsi, G.S.; Sarkhouh, F.; Chandrasekhar, B.; Attur, S.; Benter, I.F. Activation of ErbB2 and downstream signalling via Rho Kinases and ERK1/2 contributes to diabetes-induced vascular dysfunction. PLoS ONE 2013, 8, e67813. [Google Scholar] [CrossRef] [PubMed]
- Gray, W.D.; Wu, R.J.; Yin, X.; Zhou, J.; Davis, M.E.; Luo, Y. Dendrimeric bowties featuring hemispheric-selective decoration of ligands for microRNA-based therapy. Biomacromolecules 2013, 14, 101–109. [Google Scholar] [CrossRef]
- Dzmitruk, V.; Apartsin, E.; Ihnatsyeu-Kachan, A.; Abashkin, V.; Shcharbin, D.; Bryszewska, M. Dendrimers show promise for siRNA and microRNA therapeutics. Pharmaceutics 2018, 10, 126. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Cell-type-specific, Aptamer-functionalized agents for targeted disease therapy. Mol. Ther. Nucleic Acids 2014, 3, e169. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef]
- Trajkovski, M.; Hausser, J.; Soutschek, J.; Bhat, B.; Akin, A.; Zavolan, M.; Heim, M.H.; Stoffel, M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011, 474, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Fu, T.; Seok, S.; Kim, D.H.; Yu, E.; Lee, K.W.; Kang, Y.; Li, X.; Kemper, B.; Kemper, J.K. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 2013, 12, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Baitzel, C.; Konner, A.C.; Nicholls, H.T.; Vogt, M.C.; Herrmanns, K.; Scheja, L.; Haumaitre, C.; Wolf, A.M.; Knippschild, U.; et al. Obesity-induced overexpression of miR-802 impairs glucose metabolism through silencing of Hnf1b. Nature 2013, 494, 111–115. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| miRNA | Up/DownRegulation | Cell Type/Tissue | Target Gene | Effects | Reference |
|---|---|---|---|---|---|
| miR-200 family members | Up-regulation | HUVECs; in vivo mouse pancreatic β-cells | SIRT1 PRDX2 | Reduction of endothelial cell growth; elevated ROS production and apoptosis | [62] |
| miR-200b/miR-200c | Down-regulation | HUVECs | ROCK2 | Elevated ROS production and apoptosis | [63,64] |
| miR-34 | Up-regulation | Type 2 diabetic db/db mice | SIRT1 | Vascular cells senescence | [65] |
| miR-106b | Up-regulation | Mouse pancreatic β-cell line NIT-1 cells | SIRT1 | Increased oxidative stress | [66] |
| miR-204 | Up-regulation | HUVECs; C57/Bl6 mice | SIRT1 CHOP ATF6 | Endothelial dysfunction and vascular endoplasmic reticulum stress | [65] |
| miR-214 | Down-regulation | THP-1 cells | PTEN | Apoptosis and development of inflammatory responses | [67] |
| miR-205 | Down-regulation | Human renal tubular HK-2 cells | EGLN2 | Elevated ROS production and suppression of antioxidant enzymes | [68] |
| miR-21 | Up-regulation | Human APCs | FoxO1 SOD2 | Reduction of NO bioavailability and increased intracellular ROS levels | [69] |
| miR-375 | Down-regulation | MIN6 cells; INS-1E cells; miR-375−/−, Lep−/− (375/ob) mice; diabetic GK rats | Aifm1 Pdk1 MTPN | Attenuation of insulin release; reduction of β-cell mass and proliferation | [70,71] |
| miR-182 miR-148 miR-2 6miR-24 | Down-regulation | MIN6 cells; isolated primary islets | Sox 6Bhlhe22 | Down-regulation of insulin mRNA levels and insulin promoter activity | [72] |
| miR-30d | Down-regulation | Islets isolated from db/db mice | MAP4K4 MafA | Inhibition of insulin production and release | [73,74] |
| miR-7 | Up-regulation | Transgenic mice; human islets from mildly T2D, obese non-diabetic, and control subjects | mTOR | β-cell differentiation; impaired insulin release | [75] |
| miR-9 | Up-regulation | MIN6 and dissociated islet cells | Onecut 2 | Inhibition of glucose-stimulated insulin exocytosis | [76] |
| miR-23a | |||||
| miR-149 | Down-regulation | Transgenic mice; skeletal muscle from HFD-fed obese mice | SIRT-1 PGC-1α | Altered mitochondrial function and biogenesis | [77] |
| miR-141 | Down-regulation | Cardiac myocytes from diabetic mice | Slc25a3 | Alteration of mitochondrial function (ETC-complex V) | [78] |
| miR-338 miR-210 miR-181c | Down-regulation | Cardiac myocytes from diabetic rats | COX1 COXIV | Alteration of mitochondrial function (ETC-complex IV) | [79,80] |
| miR-210 | Down-regulation | H9c2 cardiomyocytes | ISCU COX10 | Alteration of mitochondrial function (ETC-complex III) and up-regulation of glycolysis | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezza, T.; de Marañón, A.M.; Canet, F.; Díaz-Pozo, P.; Marti, M.; D’Ocon, P.; Apostolova, N.; Rocha, M.; Víctor, V.M. MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes. Antioxidants 2021, 10, 802. https://doi.org/10.3390/antiox10050802
Vezza T, de Marañón AM, Canet F, Díaz-Pozo P, Marti M, D’Ocon P, Apostolova N, Rocha M, Víctor VM. MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes. Antioxidants. 2021; 10(5):802. https://doi.org/10.3390/antiox10050802
Chicago/Turabian StyleVezza, Teresa, Aranzazu M. de Marañón, Francisco Canet, Pedro Díaz-Pozo, Miguel Marti, Pilar D’Ocon, Nadezda Apostolova, Milagros Rocha, and Víctor M. Víctor. 2021. "MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes" Antioxidants 10, no. 5: 802. https://doi.org/10.3390/antiox10050802
APA StyleVezza, T., de Marañón, A. M., Canet, F., Díaz-Pozo, P., Marti, M., D’Ocon, P., Apostolova, N., Rocha, M., & Víctor, V. M. (2021). MicroRNAs and Oxidative Stress: An Intriguing Crosstalk to Be Exploited in the Management of Type 2 Diabetes. Antioxidants, 10(5), 802. https://doi.org/10.3390/antiox10050802