
Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenesis of Diabetic Retinopathy
3. NADPH Oxidase
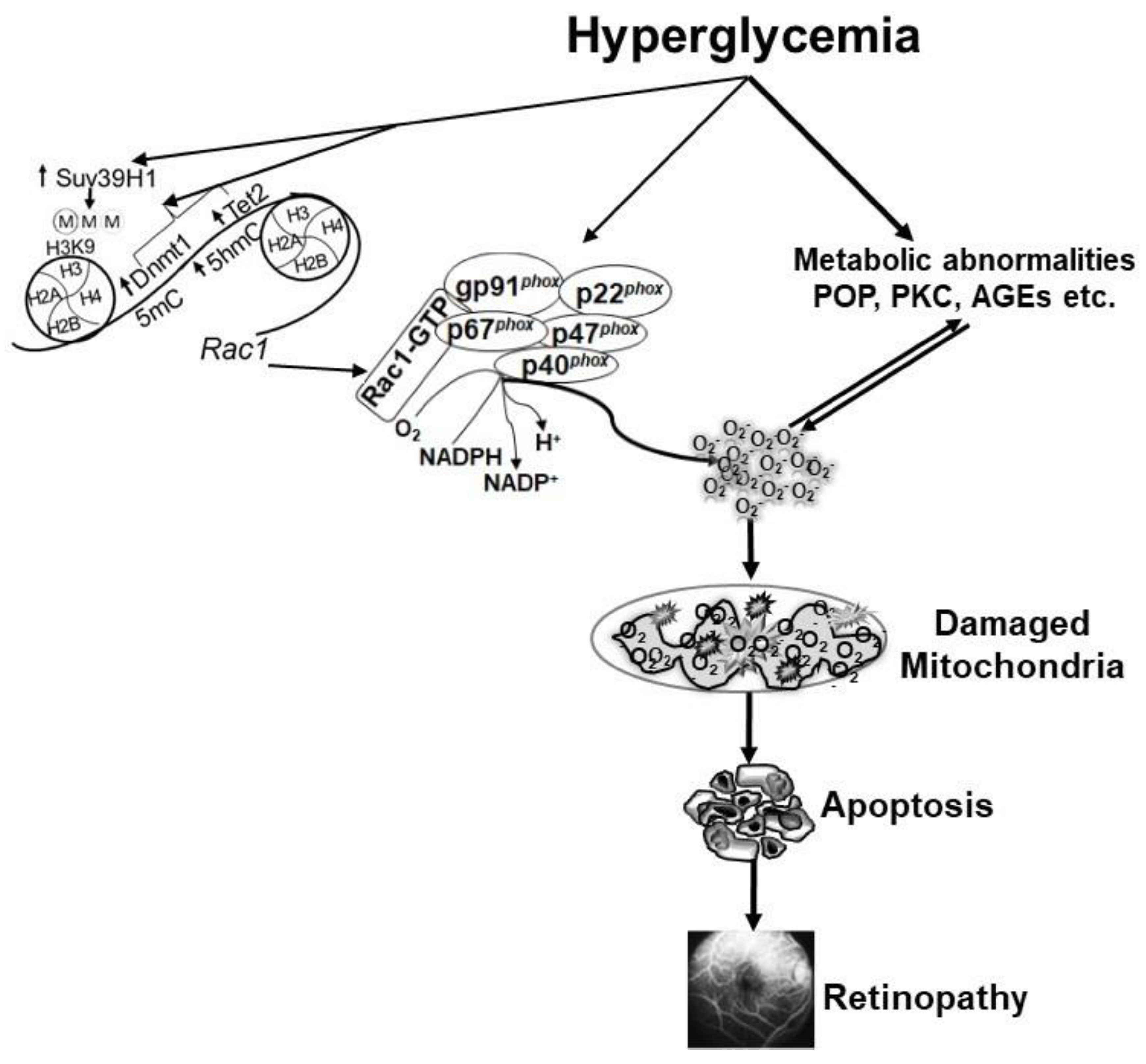
4. NADPH Oxidase and Diabetic Retinopathy
4.1. Functional Activation of Nox2
4.2. Transcriptional Activation of Rac1
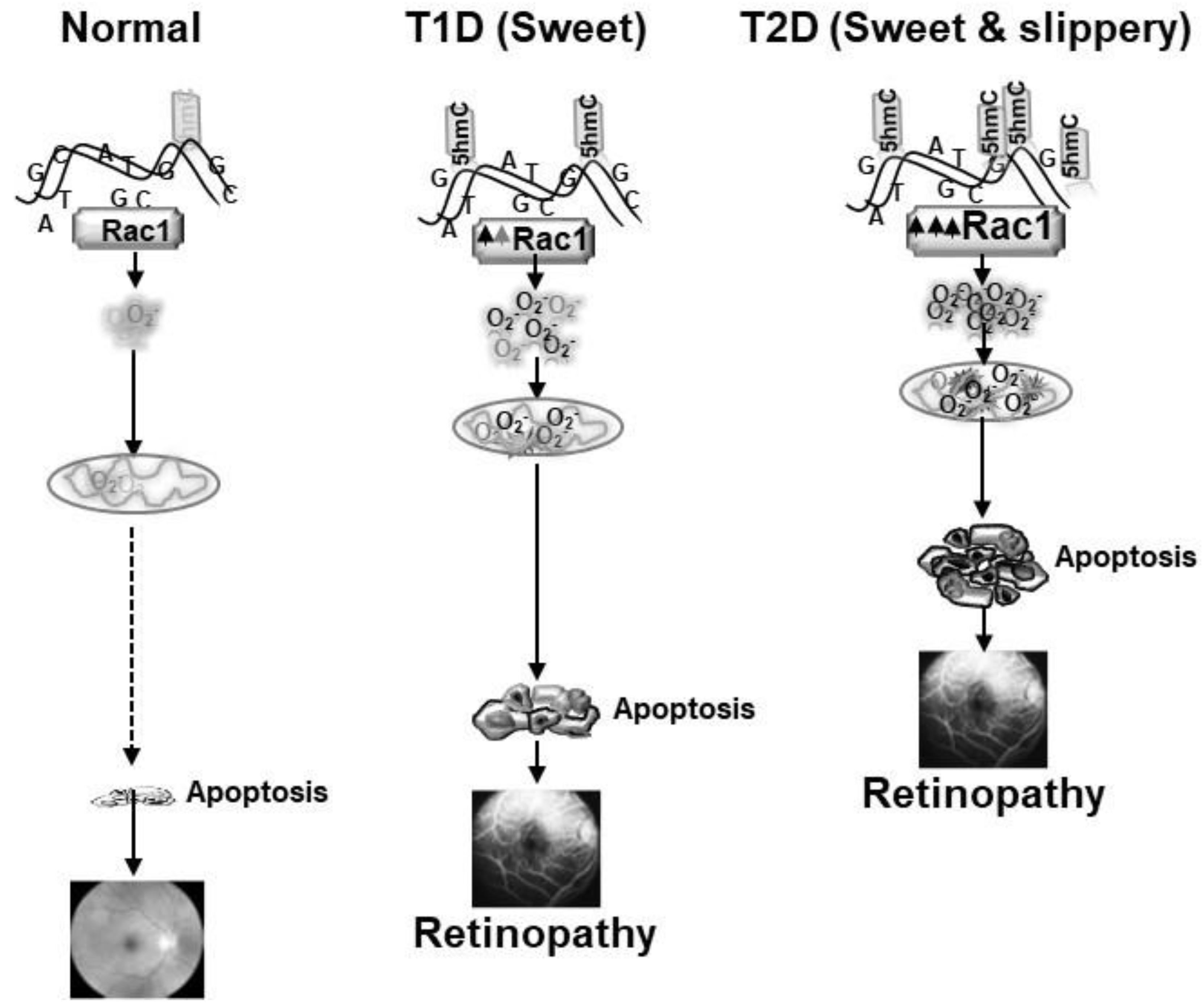
5. Sweet and Slippery Road: NADPH Oxidase and Retinopathy in Type 1 and Type 2 Diabetic Patients
6. Therapeutic Implications
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Anjana, R.M.; Pradeepa, R.; Das, A.K.; Deepa, M.; Bhansali, A.; Joshi, S.R.; Joshi, P.P.; Dhandhania, V.K.; Rao, P.V.; Sudha, V.; et al. Physical activity and inactivity patterns in India—Results from the ICMR-INDIAB study (Phase-1) [ICMR-INDIAB-5]. Int. J. Behav. Nutr. Phys. Act. 2014, 11, 26. [Google Scholar] [CrossRef]
- Popkin, B.M. Nutrition Transition and the Global Diabetes Epidemic. Curr. Diabetes Rep. 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Saaddine, J.B.; Honeycutt, A.A.; Narayan, K.M.V.; Zhang, X.; Klein, R.; Boyle, J.P. Projection of Diabetic Retinopathy and Other Major Eye Diseases Among People with Diabetes Mellitus: United States, 2005–2050. Arch. Ophthalmol. 2008, 126, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic Retinopathy and Systemic Factors. Middle East Afr. J. Ophthalmol. 2015, 22, 151–156. [Google Scholar] [CrossRef]
- Giuliari, G.P. Diabetic retinopathy: Current and new treatment options. Curr. Diabetes Rev. 2012, 8, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Wong, J.-S. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int. 2000, 58, S113–S119. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Klein, M.L.; Ferris, F.L., 3rd; Remaley, N.A.; Murphy, R.P.; Chantry, K.; Hoogwerf, B.J.; Miller, D. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch. Ophthalmol. 1996, 114, 1079–1084. [Google Scholar] [CrossRef]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.-R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef]
- Kern, T.S.; Tang, J.; Mizutani, M.; Kowluru, R.A.; Nagaraj, R.H.; Romeo, G.; Podesta, F.; Lorenzi, M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: Comparison of diabetes and galactosemia. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3972–3978. [Google Scholar]
- Kowluru, R.A.; Odenbach, S. Effect of Long-Term Administration of α-Lipoic Acid on Retinal Capillary Cell Death and the Development of Retinopathy in Diabetic Rats. Diabetes 2004, 53, 3233–3238. [Google Scholar] [CrossRef]
- Tonade, D.; Kern, T.S. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog. Retin. Eye Res. 2020, 100919. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp. Mol. Pathol. 2003, 75, 95–108. [Google Scholar] [CrossRef]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Lorenzi, M. The Polyol Pathway as a Mechanism for Diabetic Retinopathy: Attractive, Elusive, and Resilient. Exp. Diabetes Res. 2007, 2007, 1–10. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- SanGiovanni, J.P.; Chew, E.Y. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog. Retin. Eye Res. 2005, 24, 87–138. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L. Abnormalities of Retinal Metabolism in Diabetes or Experimental Galactosemia. IV. Antioxidant Defense System. Free. Radic. Biol. Med. 1997, 22, 587–592. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L.; Armstrong, D. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. III. Effects of antioxidants. Diabetes 1996, 45, 1233–1237. [Google Scholar] [CrossRef]
- Kanwar, M.; Chan, P.-S.; Kern, T.S.; Kowluru, R.A. Oxidative Damage in the Retinal Mitochondria of Diabetic Mice: Possible Protection by Superoxide Dismutase. Investig. Opthalmology Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy, oxidative stress and antioxidants. Curr. Top. Nutrac. Res. 2005, 3, 209–218. [Google Scholar]
- Kowluru, R.A. Mitochondrial Stability in Diabetic Retinopathy: Lessons Learned from Epigenetics. Diabetes 2019, 68, 241–247. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.S.; Ushio–Fukai, M.; Malik, A.B. NADPH Oxidase-Dependent Signaling in Endothelial Cells: Role in Physiology and Pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Kowluru, A. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: Lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 2011, 81, 965–975. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1–RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef]
- Wertheimer, E.; Gutierrez-Uzquiza, A.; Rosemblit, C.; Lopez-Haber, C.; Sosa, M.S.; Kazanietz, M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell. Signal. 2012, 24, 353–362. [Google Scholar] [CrossRef] [PubMed]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Kowluru, R.A. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 2014, 88, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lérida, I.; Sánchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2011, 31, 534–551. [Google Scholar] [CrossRef]
- Sahajpal, N.; Kowluru, A.; Kowluru, R.A. The Regulatory Role of Rac1, a Small Molecular Weight GTPase, in the Development of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 965. [Google Scholar] [CrossRef]
- Fritz, G.; Henninger, C. Rho GTPases: Novel Players in the Regulation of the DNA Damage Response? Biomolecules 2015, 5, 2417–2434. [Google Scholar] [CrossRef]
- Tong, L.; Tergaonkar, V. Rho protein GTPases and their interactions with NFκB: Crossroads of inflammation and matrix biology. Biosci. Rep. 2014, 34, e00115. [Google Scholar] [CrossRef]
- Sandrock, K.; Bielek, H.; Schradi, K.; Schmidt, G.; Klugbauer, N. The Nuclear Import of the Small GTPase Rac1 is Mediated by the Direct Interaction with Karyopherin α2. Traffic 2010, 11, 198–209. [Google Scholar] [CrossRef]
- Michaelson, D.; Abidi, W.; Guardavaccaro, D.; Zhou, M.; Ahearn, I.; Pagano, M.; Philips, M.R. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 2008, 181, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Osborn-Heaford, H.L.; Ryan, A.J.; Murthy, S.; Racila, A.-M.; He, C.; Sieren, J.C.; Spitz, D.R.; Carter, A.B. Mitochondrial Rac1 GTPase Import and Electron Transfer from Cytochrome c Are Required for Pulmonary Fibrosis. J. Biol. Chem. 2012, 287, 3301–3312. [Google Scholar] [CrossRef]
- Kowluru, R.A. Retinopathy in a Diet-Induced Type 2 Diabetic Rat Model and Role of Epigenetic Modifications. Diabetes 2020, 69, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Engerman, R.L.; Kern, T.S. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. VI. Comparison of retinal and cerebral cortex metabolism, and effects of antioxidant therapy. Free Radic. Biol. Med. 1999, 26, 371–378. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Jirousek, M.R.; Stramm, L.; Farid, N.; Engerman, R.L.; Kern, T.S. Abnormalities of retinal metabolism in diabetes or experimental galactosemia: V. Relationship between protein kinase C and ATPases. Diabetes 1998, 47, 464–469. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.-S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, 93751. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling. Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef]
- Chan, E.; Liu, G.-S.; Dusting, G. Redox mechanisms in pathological angiogenesis in the retina: Roles for NADPH oxidase. Curr. Pharm. Des. 2015, 21, 5988–5998. [Google Scholar] [CrossRef]
- Mohammad, G.; Duraisamy, A.J.; Kowluru, A.; Kowluru, R.A. Functional Regulation of an Oxidative Stress Mediator, Rac1, in Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 8643–8655. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; DeRicco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Bhattacharjee, S.; Kowluru, R.A. Adaptor Protein p66Shc: A Link Between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2019, 30, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Racing to the plasma membrane: The long and complex work commute of Rac1 during cell signaling. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef]
- Mohammed, A.M.; Syeda, K.; Hadden, T.; Kowluru, A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: Attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem. Pharmacol. 2013, 85, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-Rac1 Axis Promotes Activation of p38 MAP Kinase in the Development of Diabetic Retinopathy: Evidence for a Requisite Role for Protein Palmitoylation. Cell. Physiol. Biochem. 2015, 36, 208–220. [Google Scholar] [CrossRef]
- Leal, E.C.; Martins, J.; Voabil, P.; Liberal, J.; Chiavaroli, C.; Bauer, J.; Cunha-Vaz, J.; Ambrósio, A.F. Calcium Dobesilate Inhibits the Alterations in Tight Junction Proteins and Leukocyte Adhesion to Retinal Endothelial Cells Induced by Diabetes. Diabetes 2010, 59, 2637–2645. [Google Scholar] [CrossRef]
- Mishra, M.; Flaga, J.; Kowluru, R.A. Molecular Mechanism of Transcriptional Regulation of Matrix Metalloproteinase-9 in Diabetic Retinopathy. J. Cell. Physiol. 2015, 231, 1709–1718. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab. Investig. 2010, 90, 1365–1372. [Google Scholar] [CrossRef]
- Zhang, X.; Lai, D.; Bao, S.; Hambly, B.; Gillies, M. Triamcinolone Acetonide Inhibits p38MAPK Activation and Neuronal Apoptosis in Early Diabetic Retinopathy. Curr. Mol. Med. 2013, 13, 946–958. [Google Scholar] [CrossRef]
- Elmasry, K.; Ibrahim, A.S.; Saleh, H.; Elsherbiny, N.; Elshafey, S.; Hussein, K.A.; Al-Shabrawey, M. Role of endoplasmic reticulum stress in 12/15-lipoxygenase-induced retinal microvascular dysfunction in a mouse model of diabetic retinopathy. Diabetologia 2018, 61, 1220–1232. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Elshafey, S.; Sellak, H.; Hussein, K.A.; El-Sherbiny, M.; Abdelsaid, M.; Rizk, N.; Beasley, S.; Tawfik, A.M.; Smith, S.B.; et al. A lipidomic screen of hyperglycemia-treated HRECs links 12/15-Lipoxygenase to microvascular dysfunction during diabetic retinopathy via NADPH oxidase. J. Lipid Res. 2015, 56, 599–611. [Google Scholar] [CrossRef]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic Retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S. Contributions of Inflammatory Processes to the Development of the Early Stages of Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Rana, I.; Armani, R.; Agrotis, A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: Implications for retinopathy. Clin. Sci. 2013, 124, 597–615. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M.; Kumar, B. Diabetic retinopathy and transcriptional regulation of a small molecular weight G-Protein, Rac1. Exp. Eye Res. 2016, 147, 72–77. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mishra, M.; Kowluru, A.; Kowluru, R.A. Epigenetics and Regulation of Oxidative Stress in Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2018, 59, 4831–4840. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E.; El-Osta, A. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nat. Cell Biol. 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3403–3418. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The Science of Change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef]
- Ling, C.; Groop, L. Epigenetics: A Molecular Link Between Environmental Factors and Type 2 Diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y. Epigenetic Cross-Talk between DNA Methylation and Histone Modifications in Human Cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef]
- Kondo, Y. Targeting histone methyltransferase EZH2 as cancer treatment. J. Biochem. 2014, 156, 249–257. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA Modifications: Function and Applications in Normal and Disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Shan, Y.; Mishra, M.; Shen, Y. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab. Investig. 2016, 96, 1040–1049. [Google Scholar] [CrossRef]
- Estève, P.-O.; Chin, H.G.; Benner, J.; Feehery, G.R.; Samaranayake, M.; Horwitz, G.A.; Jacobsen, S.E.; Pradhan, S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. USA 2009, 106, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Radhakrishnan, R.; Ghulam, M. Diabetic retinopathy and epigenetic modifications: Role of histone methylation and DNA methylation. Sci Rep. 2021, 2013. Under editorial review. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L.; Klein, R.; American Diabetes Association. Retinopathy in Diabetes. Diabetes Care 2003, 27 (Suppl. 1), S84–S87. [Google Scholar] [CrossRef] [PubMed]
- Amutha, A.; Pradeepa, R.; Chella, K.S.; Anjana, R.M.; Unnikrishnan, R.; Mohan, V. Lipid Profile in Childhood-and Youth-Onset Type 2 Diabetes and their Association with Microvascular Complications. J. Assoc. Phys. India 2017, 65, 42–47. [Google Scholar]
- Kumar, B.; Kowluru, A.; Kowluru, R.A. Lipotoxicity Augments Glucotoxicity-Induced Mitochondrial Damage in the Development of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2015, 56, 2985–2992. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M.; Kowluru, A.; Kumar, B. Hyperlipidemia and the development of diabetic retinopathy: Comparison between type 1 and type 2 animal models. Metabolism 2016, 65, 1570–1581. [Google Scholar] [CrossRef]
- Meng, W.; Shah, K.P.; Pollack, S.; Toppila, I.; Hebert, H.L.; McCarthy, M.I.; Groop, L.; Ahlqvist, E.; Lyssenko, V.; Agardh, E.; et al. A genome-wide association study suggests new evidence for an association of the NADPH Oxidase 4 (NOX4) gene with severe diabetic retinopathy in type 2 diabetes. Acta Ophthalmol. 2018, 96, e811–e819. [Google Scholar] [CrossRef]
- Appukuttan, B.; Ma, Y.; Stempel, A.; Ashander, L.M.; Deliyanti, D.; Wilkinson-Berka, J.L.; Smith, J.R. Effect of NADPH oxidase 1 and 4 blockade in activated human retinal endothelial cells. Clin. Exp. Ophthalmol. 2018, 46, 652–660. [Google Scholar] [CrossRef]
- Deliyanti, D.; AlRashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef]
- Gautam, J.; Ku, J.-M.; Regmi, S.C.; Jeong, H.; Wang, Y.; Banskota, S.; Park, M.-H.; Nam, T.-G.; Jeong, B.-S.; Kim, J.-A. Dual Inhibition of NOX2 and Receptor Tyrosine Kinase by BJ-1301 Enhances Anticancer Therapy Efficacy via Suppression of Autocrine-Stimulatory Factors in Lung Cancer. Mol. Cancer Ther. 2017, 16, 2144–2156. [Google Scholar] [CrossRef]
- Payapilly, A.; Malliri, A. Compartmentalisation of RAC1 signalling. Curr. Opin. Cell Biol. 2018, 54, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kleniewska, P.; Piechota, A.; Skibska, B.; Gorąca, A. The NADPH Oxidase Family and its Inhibitors. Arch. Immunol. Ther. Exp. 2012, 60, 277–294. [Google Scholar] [CrossRef]
- Joshi, S.; Peck, A.B.; Khan, S.R. NADPH Oxidase as a Therapeutic Target for Oxalate Induced Injury in Kidneys. Oxid. Med. Cell. Longev. 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel Competitive Inhibitor of NAD(P)H Oxidase Assembly Attenuates Vascular O2−and Systolic Blood Pressure in Mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.Y.-C.; Chen, T.-H.; Garg, S.J.; Sun, C.-C.; Kang, J.-H.; Wu, W.-C.; Hung, M.-J.; Lai, C.-C.; Cherng, W.-J.; Hwang, Y.-S. Association of Statin Therapy with Prevention of Vision-Threatening Diabetic Retinopathy. JAMA Ophthalmol. 2019, 137, 363. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L. Angiotensin and diabetic retinopathy. Int. J. Biochem. Cell Biol. 2006, 38, 752–765. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Robak, T. New nucleoside analogs for patients with hematological malignancies. Expert Opin. Investig. Drugs 2011, 20, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark. Insights 2019, 14, 1177271919846454. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int. J. Cardiol. 2017, 227, 66–82. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowluru, R.A. Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants 2021, 10, 783. https://doi.org/10.3390/antiox10050783
Kowluru RA. Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants. 2021; 10(5):783. https://doi.org/10.3390/antiox10050783
Chicago/Turabian StyleKowluru, Renu A. 2021. "Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road" Antioxidants 10, no. 5: 783. https://doi.org/10.3390/antiox10050783
APA StyleKowluru, R. A. (2021). Diabetic Retinopathy and NADPH Oxidase-2: A Sweet Slippery Road. Antioxidants, 10(5), 783. https://doi.org/10.3390/antiox10050783