
Molecular Mechanisms Associated with ROS-Dependent Angiogenesis in Lower Extremity Artery Disease

 , , , , , ,
, , , , , , 
 and
and 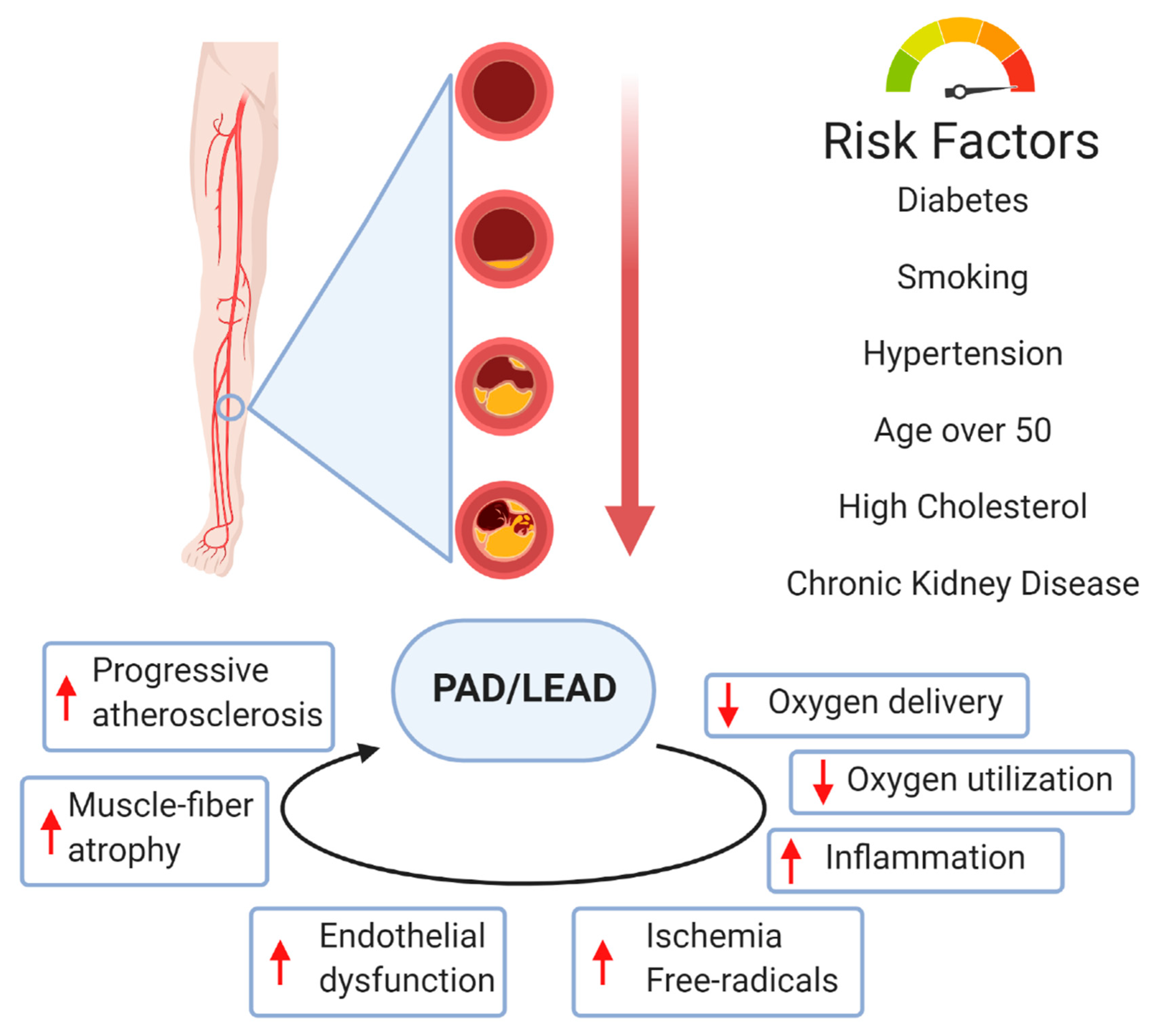{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Peripheral Arterial Disease
3. Molecular Basis of Vessel Disease
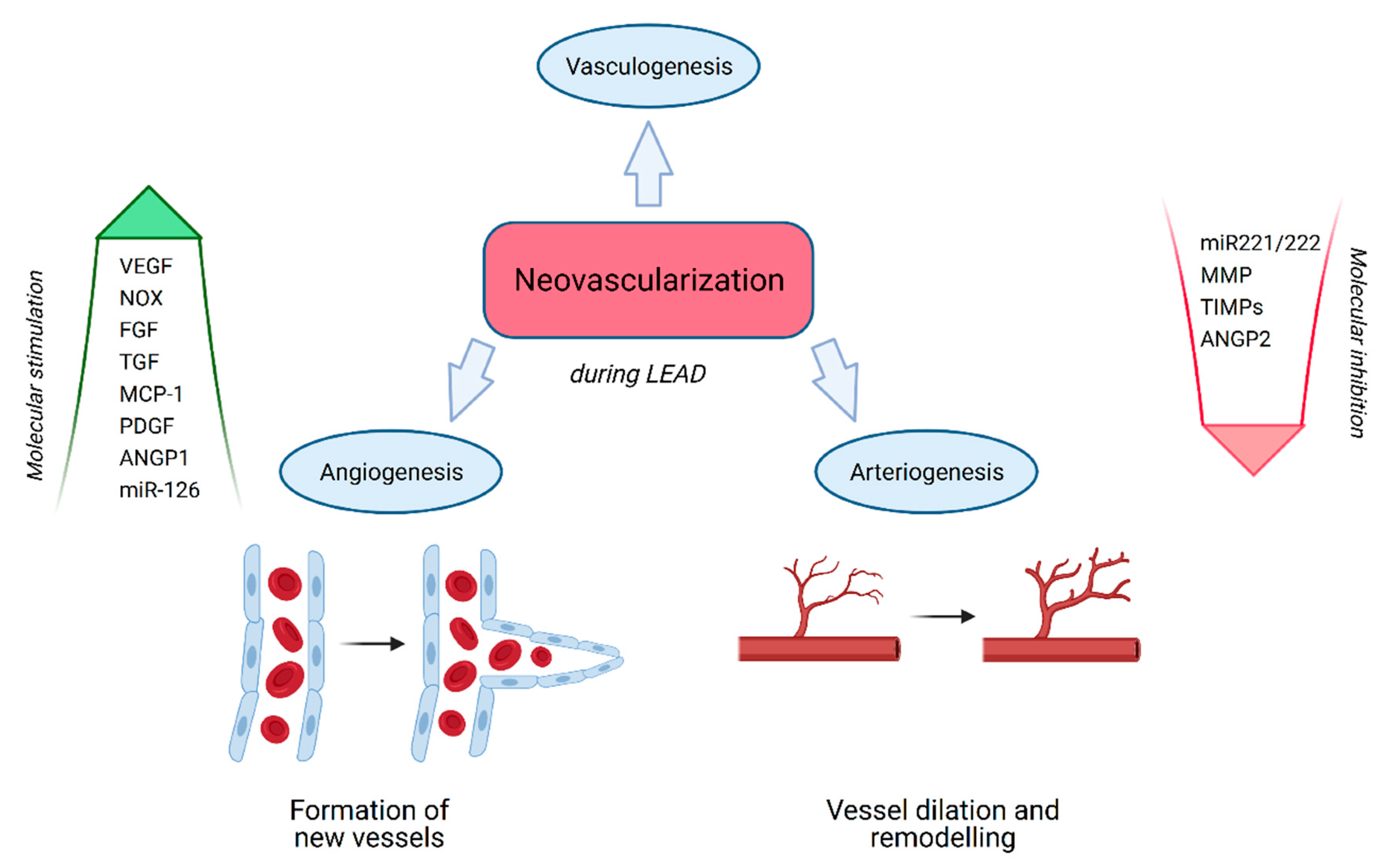
3.1. Neovascularization
3.2. Vasculogenesis
3.3. Angiogenesis
3.4. Arteriogenesis
4. Tissue Regeneration and Repair by Neovascularization
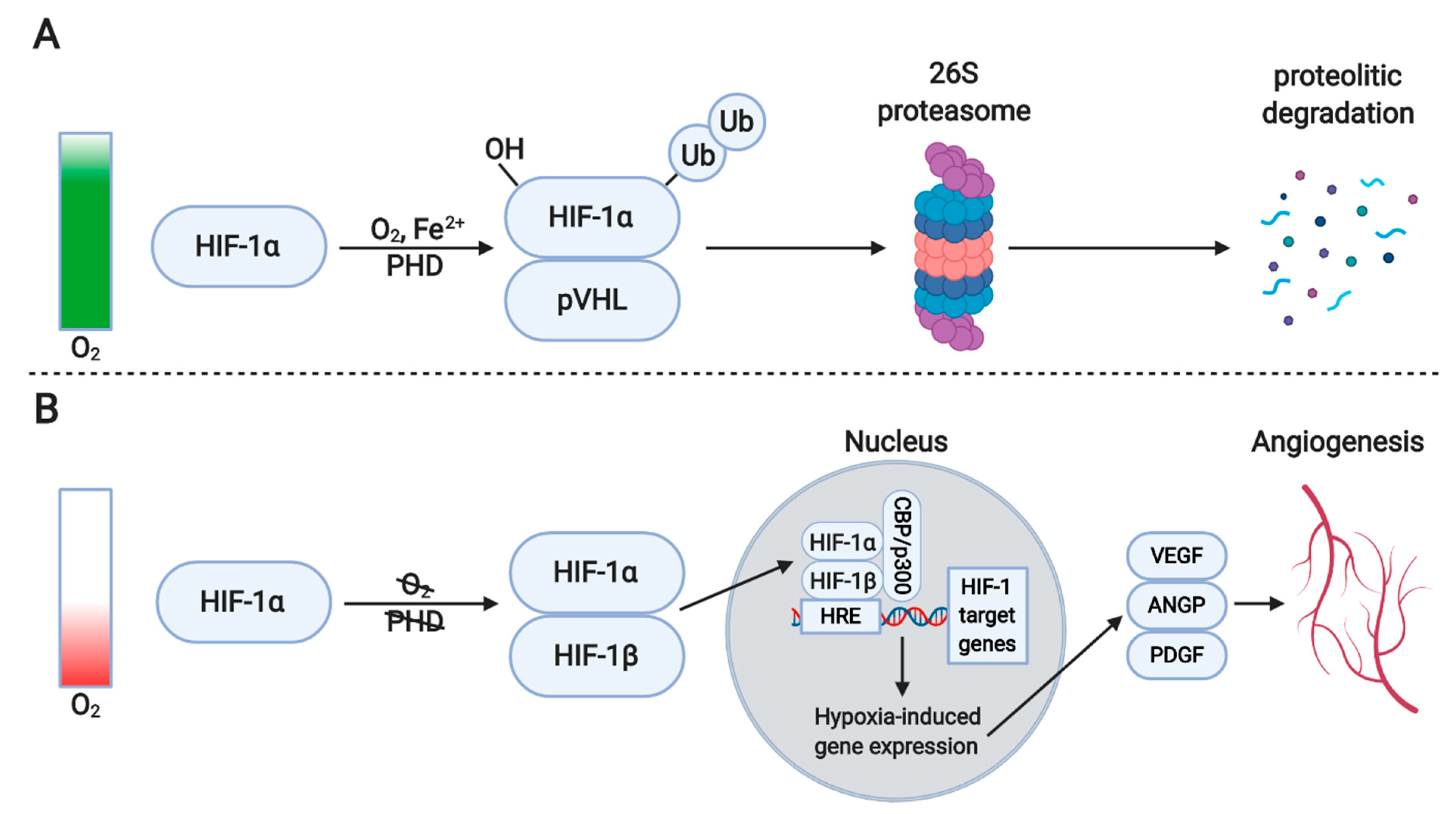
4.1. Hypoxia
4.2. Inflammation
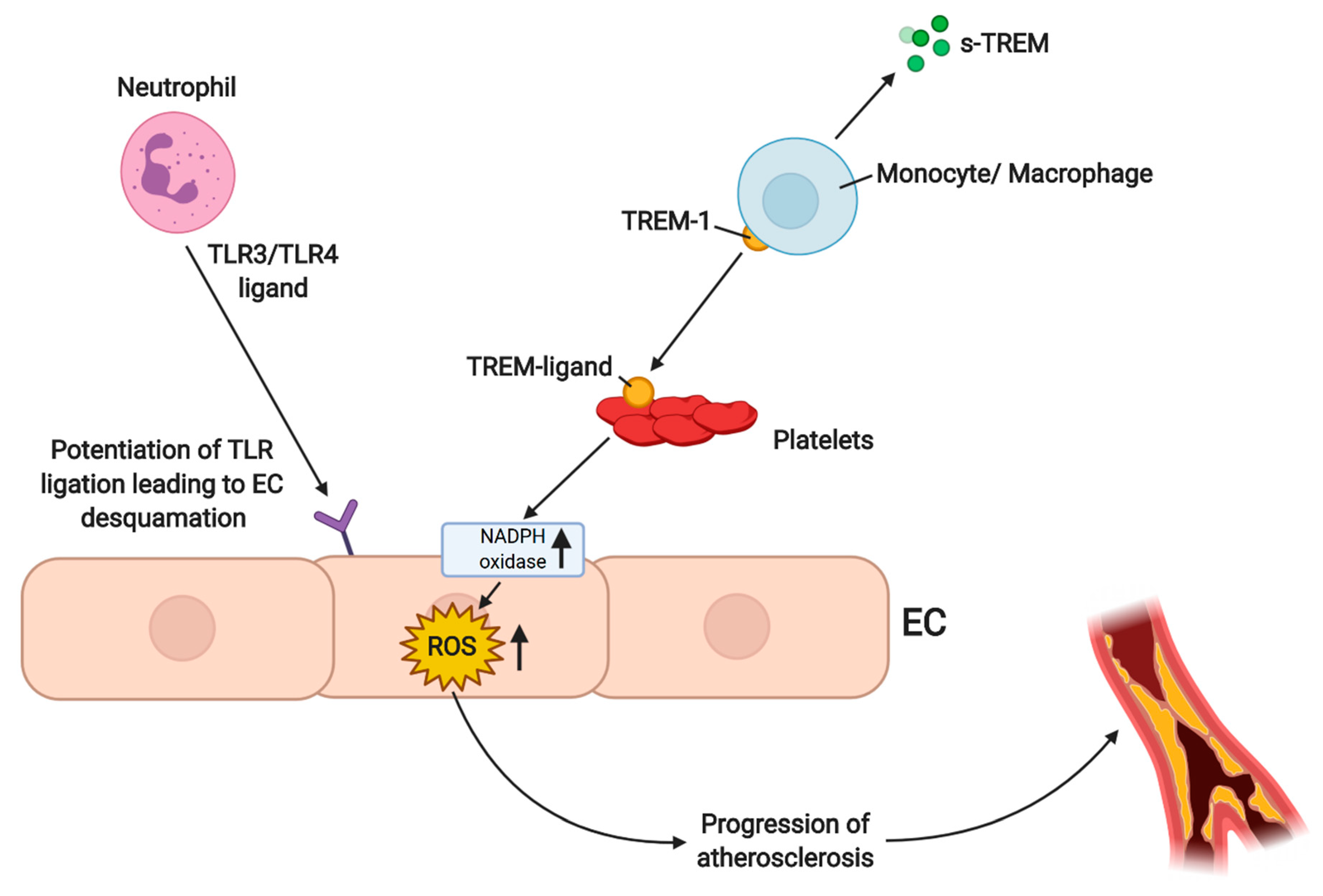
4.3. ROS-Dependent Angiogenesis in PAD/LEAD
4.4. Angiogenic and Arteriogenic Growth Factors
4.4.1. VEGF
4.4.2. FGF
4.4.3. Angiopoietin
4.4.4. Non-Coding RNAs in Neovascularization
4.4.5. Vasculogenesis and Lymphangiogenesis
4.4.6. Arteriogenesis
5. Mechanisms of Neovascularization and Cell-Based Therapies
5.1. Endothelial Cells (ECs)
5.2. Endothelial Cell Sprouting
5.3. Stem Cell Therapy and Angiogenesis
5.4. Molecular Association with Traditional Therapies in LEAD
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonham, C.A.; Kuehlmann, B.; Gurtner, G.C. Impaired Neovascularization in Aging. Adv. Wound Care 2020, 9, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Mechanisms of angiogenesis and lymphangiogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97. [Google Scholar] [CrossRef]
- Rizzi, A.; Benagiano, V.; Ribatti, D. Angiogenesis versus arteriogenesis. Rom. J. Morphol. Embryol. 2017, 58, 15–19. [Google Scholar]
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.A.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Sartipy, F.; Sigvant, B.; Lundin, F.; Wahlberg, E. Ten Year Mortality in Different Peripheral Arterial Disease Stages: A Population Based Observational Study on Outcome. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 529–536. [Google Scholar] [CrossRef]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.R. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67. [Google Scholar] [CrossRef]
- De Weerd, M.; Greving, J.P.; De Jong, A.W.F.; Buskens, E.; Bots, M.L. Prevalence of asymptomatic carotid artery stenosis according to age and sex systematic review and metaregression analysis. Stroke 2009, 40, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Jude, E.B.; Oyibo, S.O.; Chalmers, N.; Boulton, A.J.M. Peripheral arterial disease in diabetic and nondiabetic patients: A comparison of severity and outcome. Diabetes Care 2001, 24, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Criqui, M.H.; Denenberg, J.O.; Knoke, J.D.; Ridker, P.M.; Fronek, A. Risk factors for progression of peripheral arterial disease in large and small vessels. Circulation 2006, 113, 2623–2629. [Google Scholar] [CrossRef]
- Stone, P.A.; Yacoub, M. Inflammatory biomarkers in peripheral arterial disease. Semin. Vasc. Surg. 2014, 27, 148–151. [Google Scholar] [CrossRef]
- Signorelli, S.S.; Marino, E.; Scuto, S.; Di Raimondo, D. Pathophysiology of peripheral arterial disease (Pad): A review on oxidative disorders. Int. J. Mol. Sci. 2020, 21, 4393. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Ibe, S.; Saita, E.; Sasaki, K.; Niki, H.; Miura, K.; Ikegami, Y.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma Heme Oxygenase-1 Levels in Patients with Coronary and Peripheral Artery Diseases. Dis. Markers 2018, 2018. [Google Scholar] [CrossRef]
- Walker, M.A.; Hoier, B.; Walker, P.J.; Schulze, K.; Bangsbo, J.; Hellsten, Y.; Askew, C.D. Vasoactive enzymes and blood flow responses to passive and active exercise in peripheral arterial disease. Atherosclerosis 2016, 246, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2106–2119. [Google Scholar] [CrossRef]
- Tanaka, L.Y.; Laurindo, F.R.M. Vascular remodeling: A redox-modulated mechanism of vessel caliber regulation. Free Radic. Biol. Med. 2017, 109, 11–21. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Nielsen, S.H.; Bonache, C.; Fernández-Arroyo, S.; Martín-Paredero, V.; Fibla, M.; Karsdal, M.A.; Genovese, F.; Menendez, J.A.; Camps, J.; et al. Assessment of extracellular matrix-related biomarkers in patients with lower extremity artery disease. J. Vasc. Surg. 2018, 68, 1135–1142.e6. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N.; Merrilees, M.J. Proteoglycans in atherosclerosis and restenosis: Key roles for versican. Circ. Res. 2004, 94, 1158–1167. [Google Scholar] [CrossRef]
- Vassiliadis, E.; Barascuk, N.; Karsdal, M.A. Atherofibrosis—A unique and common process of the disease pathogenesis of atherosclerosis and fibrosis—Lessons for biomarker development. Am. J. Transl. Res. 2013, 5, 1–14. [Google Scholar] [PubMed]
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation 2003, 107, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Chang, Z.; Liu, Z. The risk factors for calcification vary among the different sections of the lower extremity artery in patients with symptomatic peripheral arterial disease. BMC Cardiovasc. Disord. 2020, 20. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Calcifying Matrix Vesicles and Atherosclerosis. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, J.; Zhang, Y.; Yu, W.; Zhang, C.; Gong, L.; Shao, L.; Lu, J.; Gao, Y.; Chen, X.; et al. Common genetic variants of MGP are associated with calcification on the arterial wall but not with calcification present in the atherosclerotic plaques. Circ. Cardiovasc. Genet. 2013, 6, 271–278. [Google Scholar] [CrossRef]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Kullo, I.J.; Leeper, N.J. The Genetic Basis of Peripheral Arterial Disease: Current Knowledge, Challenges, and Future Directions. Circ. Res. 2015, 116, 1551–1560. [Google Scholar] [CrossRef]
- Flex, A.; Gaetani, E.; Pola, R.; Santoliquido, A.; Aloi, F.; Papaleo, P.; Dal Lago, A.; Pola, E.; Serricchio, M.; Tondi, P.; et al. The -174 G/C polymorphism of the interleukin-6 gene promoter is associated with peripheral artery occlusive disease. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 264–268. [Google Scholar] [CrossRef]
- Li, R.; Folsom, A.R.; Sharrett, A.R.; Couper, D.; Bray, M.; Tyroler, H.A. Interaction of the glutathione S-transferase genes and cigarette smoking on risk of lower extremity arterial disease: The Atherosclerosis Risk in Communities (ARIC) study. Atherosclerosis 2001, 154, 729–738. [Google Scholar] [CrossRef]
- Arellano Mendoza, M.; Robles, H.; Romo, E.; Rios, A.; Escalante, B. Nitric Oxide-Dependent Neovascularization Role in the Lower Extremity Disease. Curr. Pharm. Des. 2007, 13, 3591–3596. [Google Scholar] [CrossRef]
- Vogel, J.; Niederer, D.; Jung, G.; Troidl, K. Exercise-Induced Vascular Adaptations under Artificially Versus Pathologically Reduced Blood Flow: A Focus Review with Special Emphasis on Arteriogenesis. Cells 2020, 9, 333. [Google Scholar] [CrossRef]
- Chalothorn, D.; Faber, J.E. Formation and maturation of the native cerebral collateral circulation. J. Mol. Cell. Cardiol. 2010, 49, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A.; Nico, B.; Roncali, L.; Dammacco, F. Postnatal vasculogenesis. Mech. Dev. 2001, 100, 157–163. [Google Scholar] [CrossRef]
- Simons, M.; Eichmann, A. Molecular controls of arterial morphogenesis. Circ. Res. 2015, 116, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; Van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef]
- Lasch, M.; Caballero Martinez, A.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Meister, S.; Deindl, E. Contribution of the Potassium Channels KV1.3 and KCa3.1 to Smooth Muscle Cell Proliferation in Growing Collateral Arteries. Cells 2020, 9, 913. [Google Scholar] [CrossRef]
- Ricard, N.; Zhang, J.; Zhuang, Z.W.; Simons, M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells 2019, 9, 38. [Google Scholar] [CrossRef]
- Sealock, R.; Zhang, H.; Lucitti, J.L.; Moore, S.M.; Faber, J.E. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ. Res. 2014, 114, 660–671. [Google Scholar] [CrossRef]
- Peirce, S.M.; Skalak, T.C. Microvascular remodeling: A complex continuum spanning angiogenesis to arteriogenesis. Microcirculation 2003, 10, 99–111. [Google Scholar] [CrossRef]
- Troidl, K.; Tribulova, S.; Cai, W.J.; Rüding, I.; Apfelbeck, H.; Schierling, W.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W. Effects of endogenous nitric oxide and of deta nonoate in arteriogenesis. J. Cardiovasc. Pharmacol. 2010, 55, 153–160. [Google Scholar] [CrossRef]
- Buschmann, I.; Pries, A.; Styp-Rekowska, B.; Hillmeister, P.; Loufrani, L.; Henrion, D.; Shi, Y.; Duelsner, A.; Hoefer, I.; Gatzke, N.; et al. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development 2010, 137, 2187–2196. [Google Scholar] [CrossRef]
- Tronc, F.; Mallat, Z.; Lehoux, S.; Wassef, M.; Esposito, B.; Tedgui, A. Role of Matrix Metalloproteinases in Blood Flow–Induced Arterial Enlargement. Arterioscler. Thromb. Vasc. Biol. 2000, 20. [Google Scholar] [CrossRef]
- Shi, Z.D.; Tarbell, J.M. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann. Biomed. Eng. 2011, 39, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, K.A.; Schwartz, M.A. Ion channels in endothelial responses to fluid shear stress. Physiology 2016, 31, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.; Schierling, W.; Troidl, K.; Rüding, I.; Nelson, K.; Apfelbeck, H.; Benli, I.; Schaper, W.; Schmitz-Rixen, T. Exercise linked to transient increase in expression and activity of cation channels in newly formed hind-limb collaterals. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 81–87. [Google Scholar] [CrossRef][Green Version]
- Troidl, C.; Troidl, K.; Schierling, W.; Cai, W.J.; Nef, H.; Möllmann, H.; Kostin, S.; Schimanski, S.; Hammer, L.; Elsässer, A.; et al. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J. Cell. Mol. Med. 2009, 13, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Wong, C.; Sutter, E.; Cuhlmann, S.; Dunoyer-Geindre, S.; Mach, F.; Horrevoets, A.J.; Evans, P.C.; Krams, R.; Kwak, B.R. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Rubin, J.; Tzima, E. Role of PECAM-1 in arteriogenesis and specification of preexisting collaterals. Circ. Res. 2010, 107, 1355–1363. [Google Scholar] [CrossRef]
- Takeda, Y.; Costa, S.; Delamarre, E.; Roncal, C.; Leite De Oliveira, R.; Squadrito, M.L.; Finisguerra, V.; Deschoemaeker, S.; Bruyère, F.; Wenes, M.; et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 2011, 479, 122–126. [Google Scholar] [CrossRef] [PubMed]
- de Paula, E.V.; Flores-Nascimento, M.C.; Arruda, V.R.; Garcia, R.A.; Ramos, C.D.; Guillaumon, A.T.; Annichino-Bizzacchi, J.M. Dual gene transfer of fibroblast growth factor-2 and platelet derived growth factor-BB using plasmid deoxyribonucleic acid promotes effective angiogenesis and arteriogenesis in a rodent model of hindlimb ischemia. Transl. Res. 2009, 153, 232–239. [Google Scholar] [CrossRef]
- Troidl, K.; Schubert, C.; Vlacil, A.K.; Chennupati, R.; Koch, S.; Schütt, J.; Oberoi, R.; Schaper, W.; Schmitz-Rixen, T.; Schieffer, B.; et al. The Lipopeptide MALP-2 Promotes Collateral Growth. Cells 2020, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; Van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Moraes, F.; Paye, J.; Gabhann, F.M.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909. [Google Scholar] [CrossRef]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The Neuropilin 1 Cytoplasmic Domain Is Required for VEGF-A-Dependent Arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef]
- Van Royen, N.; Piek, J.J.; Buschmann, I.; Hoefer, I.; Voskuil, M.; Schaper, W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553. [Google Scholar] [CrossRef]
- Shireman, P.K. The chemokine system in arteriogenesis and hind limb ischemia. J. Vasc. Surg. 2007, 45, A48–A56. [Google Scholar] [CrossRef]
- Stabile, E.; Kinnaird, T.; La Sala, A.; Hanson, S.K.; Watkins, C.; Campia, U.; Shou, M.; Zbinden, S.; Fuchs, S.; Kornfeld, H.; et al. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin-16. Circulation 2006, 113, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T.; Gamrekelashvili, J.; Beger, C.; Häger, C.; Lozanovski, V.J.; Falk, C.S.; Napp, L.C.; Bauersachs, J.; et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Ribatti, D. A new role of mast cells in arteriogenesis. Microvasc. Res. 2018, 118, 57–60. [Google Scholar] [CrossRef]
- Heil, M.; Ziegelhoeffer, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Hear. Circ. Physiol. 2002, 283. [Google Scholar] [CrossRef]
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled β2 integrin-induced macrophage Rac2-myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Bot, I.; van der Velden, D.; Bouwman, M.; Kröner, M.J.; Kuiper, J.; Quax, P.H.A.; de Vries, M.R. Local Mast Cell Activation Promotes Neovascularization. Cells 2020, 9, 701. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Rubrech, J.; Trumpp, A.; Geissler, P.; Zeller, G.C.; Schnorbus, B.; Schmidt, F.; Gori, T.; Münzel, T.; Espinola-Klein, C. Supervised exercise training in peripheral arterial disease increases vascular shear stress and profunda femoral artery diameter. Eur. J. Prev. Cardiol. 2017, 24, 178–191. [Google Scholar] [CrossRef]
- Pope, Z.K.; Willardson, J.M.; Schoenfeld, B.J. Exercise and blood flow restriction. J. Strength Cond. Res. 2013, 27, 2914–2926. [Google Scholar] [CrossRef] [PubMed]
- Loenneke, J.P.; Wilson, G.J.; Wilson, J.M. A mechanistic approach to blood flow occlusion. Int. J. Sports Med. 2010, 31, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.P.; Madery, B.D.; Curry, T.B.; Eisenach, J.H.; Wilkins, B.W.; Joyner, M.J. Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J. Physiol. 2010, 588, 373–385. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.; Veszeleiova, K.; Steingold, J.; Sethuraman, J. Hypoxia and Cancer Metastasis. Adv. Exp. Med. Biol. 2019, 1136, 71–85. [Google Scholar] [CrossRef]
- De Muinck, E.D.; Simons, M. Re-evaluating therapeutic neovascularization. J. Mol. Cell. Cardiol. 2004, 36, 25–32. [Google Scholar] [CrossRef]
- Ribatti, D. Inflammation and angiogenesis. Inflamm. Angiogenes. 2017, 1–111. [Google Scholar] [CrossRef]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef]
- Cholan, P.M.; Cartland, S.P.; Kavurma, M.M. NADPH oxidases, angiogenesis, and peripheral artery disease. Antioxidants 2017, 6, 56. [Google Scholar] [CrossRef]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef]
- Ebrahimian, T.G.; Heymes, C.; You, D.; Blanc-Brude, O.; Mees, B.; Waeckel, L.; Duriez, M.; Vilar, J.; Brandes, R.P.; Levy, B.I.; et al. NADPH oxidase-derived overproduction of reactive oxygen species impairs postischemic neovascularization in mice with type 1 diabetes. Am. J. Pathol. 2006, 169, 719–728. [Google Scholar] [CrossRef]
- Zhang, J.; Rao, G.; Qiu, J.; He, R.; Wang, Q. MicroRNA-210 improves perfusion recovery following hindlimb ischemia via suppressing reactive oxygen species. Exp. Ther. Med. 2020, 20, 236. [Google Scholar] [CrossRef]
- Ambrose, C.T. Pro-Angiogenesis Therapy and Aging: A Mini-Review. Gerontology 2017, 63, 393–400. [Google Scholar] [CrossRef]
- Pitulescu, M.E.; Schmidt, I.; Giaimo, B.D.; Antoine, T.; Berkenfeld, F.; Ferrante, F.; Park, H.; Ehling, M.; Biljes, D.; Rocha, S.F.; et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat. Cell Biol. 2017, 19, 915–927. [Google Scholar] [CrossRef]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol. Cell Physiol. 2002, 282. [Google Scholar] [CrossRef]
- Kolesnikov, S.S.; Bystrova, M.F. Molecular and cellular mechanisms of taste. Biol. Membr. 2018, 35, 409–423. [Google Scholar] [CrossRef]
- Bleda, S.; De Haro, J.; Varela, C.; Esparza, L.; De Maturana, I.L.; Acin, F. Impact of VEGF polymorphisms on the severity of peripheral artery disease in diabetic patients. Growth Factors 2012, 30, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.D.; Annex, B.H.; McKendall, G.R.; Azrin, M.A.; Lopez, J.J.; Giordano, F.J.; Shah, P.K.; Willerson, J.T.; Benza, R.L.; Berman, D.S.; et al. The VIVA trial: Vascular endothelial growth factor in ischemia for vascular angiogenesis. Circulation 2003, 107, 1359–1365. [Google Scholar] [CrossRef]
- Anderson, E.M.; Silva, E.A.; Hao, Y.; Martinick, K.D.; Vermillion, S.A.; Stafford, A.G.; Doherty, E.G.; Wang, L.; Doherty, E.J.; Grossman, P.M.; et al. VEGF and IGF Delivered from Alginate Hydrogels Promote Stable Perfusion Recovery in Ischemic Hind Limbs of Aged Mice and Young Rabbits. J. Vasc. Res. 2017, 54, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Pontes-Quero, S.; Fernández-Chacón, M.; Luo, W.; Lunella, F.F.; Casquero-Garcia, V.; Garcia-Gonzalez, I.; Hermoso, A.; Rocha, S.F.; Bansal, M.; Benedito, R. High mitogenic stimulation arrests angiogenesis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef]
- Nakamichi, M.; Akishima-Fukasawa, Y.; Fujisawa, C.; Mikami, T.; Onishi, K.; Akasaka, Y. Basic Fibroblast Growth Factor Induces Angiogenic Properties of Fibrocytes to Stimulate Vascular Formation during Wound Healing. Am. J. Pathol. 2016, 186, 3203–3216. [Google Scholar] [CrossRef]
- Itoh, N. Hormone-like (endocrine) Fgfs: Their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res. 2010, 342, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.Y.; Ma, H.X. Significant roles of anti-aging protein klotho and fibroblast growth factor23 in cardiovascular disease. J. Geriatr. Cardiol. 2015, 12, 439–447. [Google Scholar] [PubMed]
- Rhee, E.J.; Oh, K.W.; Yun, E.J.; Jung, C.H.; Lee, W.Y.; Kim, S.W.; Baek, K.H.; Kang, M.I.; Park, S.W. Relationship between polymorphisms G395A in promoter and C1818T in exon 4 of the KLOTHO gene with glucose metabolism and cardiovascular risk factors in Korean women. J. Endocrinol. Investig. 2006, 29, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.H.; Kim, S.G.; Choi, Y.J.; Joo, N.R.; Cho, G.Y.; Choi, S.R.; Kim, E.J.; Kim, H.S.; Kim, H.J.; Rhim, C.Y. KLOTHO gene polymorphism is associated with coronary artery stenosis but not with coronary calcification in a Korean population. Int. Heart J. 2009, 50, 23–32. [Google Scholar] [CrossRef][Green Version]
- Simons, M.; Annex, B.H.; Laham, R.J.; Kleiman, N.; Henry, T.; Dauerman, H.; Udelson, J.E.; Gervino, E.V.; Pike, M.; Whitehouse, M.J.; et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: Double-blind, randomized, controlled clinical trial. Circulation 2002, 105, 788–793. [Google Scholar] [CrossRef]
- Johnson, T.; Zhao, L.; Manuel, G.; Taylor, H.; Liu, D. Approaches to therapeutic angiogenesis for ischemic heart disease. J. Mol. Med. 2019, 97, 141–151. [Google Scholar] [CrossRef]
- Belch, J.; Hiatt, W.R.; Baumgartner, I.; Driver, I.V.; Nikol, S.; Norgren, L.; Van Belle, E. Effect of fibroblast growth factor NV1FGF on amputation and death: A randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet 2011, 377, 1929–1937. [Google Scholar] [CrossRef]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Gene-Therapeutic Strategies Targeting Angiogenesis in Peripheral Artery Disease. Medicines 2018, 5, 31. [Google Scholar] [CrossRef]
- Barrientos, S.; Brem, H.; Stojadinovic, O.; Medicine, R.; Surgery, C. Clinical Application of Growth Factors and Cytokines in Wound Healing. Wound Repair Regen. 2016, 22, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Gaonac’h-Lovejoy, V.; Boscher, C.; Delisle, C.; Gratton, J.-P. Rap1 Is Involved in Angiopoietin-1-Induced Cell-Cell Junction Stabilization and Endothelial Cell Sprouting. Cells 2020, 9, 155. [Google Scholar] [CrossRef]
- Thurston, G.; Suri, C.; Smith, K.; McClain, J.; Sato, T.N.; Yancopoulos, G.D.; McDonald, D.M. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999, 286, 2511–2514. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, C.-H.; Hwang, S.-J.; Choi, H.-H.; Kim, K.-T.; Ahn, S.Y.; Kim, J.-H.; Oh, J.-L.; Lee, G.M.; Koh, G.Y. Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J. 2004, 18, 1200–1208. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006, 108, 3068–3071. [Google Scholar] [CrossRef]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The Endothelial-Specific MicroRNA miR-126 Governs Vascular Integrity and Angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, Z.; Wang, Z.G.; Liu, J. MiRNA-27b Regulates Angiogenesis by Targeting AMPK in Mouse Ischemic Stroke Model. Neuroscience 2019, 398, 12–22. [Google Scholar] [CrossRef]
- McDonald, R.A.; Hata, A.; MacLean, M.R.; Morrell, N.W.; Baker, A.H. MicroRNA and vascular remodelling in acute vascular injury and pulmonary vascular remodelling. Cardiovasc. Res. 2012, 93, 594–604. [Google Scholar] [CrossRef]
- Vogiatzi, G.; Oikonomou, E.; Deftereos, S.; Siasos, G.; Tousoulis, D. Peripheral artery disease: A micro-RNA-related condition? Curr. Opin. Pharmacol. 2018, 39, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Kral-pointner, J.B.; Salzmann, M. Fundamentals of Vascular Biology; Springer Nature Switzerland: Cham, Switzerland, 2019; pp. 145–169. [Google Scholar] [CrossRef]
- Flister, M.J.; Wilber, A.; Hall, K.L.; Iwata, C.; Miyazono, K.; Nisato, R.E.; Pepper, M.S.; Zawieja, D.C.; Ran, S. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-κB and Prox1. Blood 2010, 115, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Wigle, J.T.; Oliver, G. Prox1 function is required for the development of the murine lymphatic system. Cell 1999, 98, 769–778. [Google Scholar] [CrossRef]
- Theos, A.C.; Martina, A.; Hurbain, I.; Peden, A.A.; Sviderskaya, E.V.; Stewart, A.; Robinson, M.S.; Bennett, D.C.; Cutler, D.F.; Bonifacino, J.S.; et al. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol. Biol. Cell 2005, 16, 5356–5372. [Google Scholar] [CrossRef]
- Troidl, K.; Schaper, W. Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes. Metab. Res. Rev. 2012, 32, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.; Lasch, M.; Borgolte, J.; Troidl, K.; Pagel, J.I.; Caballero-Martinez, A.; Kleinert, E.C.; Walzog, B.; Deindl, E. Midkine Controls Arteriogenesis by Regulating the Bioavailability of Vascular Endothelial Growth Factor A and the Expression of Nitric Oxide Synthase 1 and 3. EBioMedicine 2018, 27, 237–246. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Novel biological functions of high-density lipoprotein cholesterol. Circ. Res. 2012, 111, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.L.; Stack, M.S.; Asplin, I.; Enghild, J.J.; Højrup, P.; Everitt, L.; Hubchak, S.; Schnaper, H.W.; Pizzo, S.V. Angiostatin binds ATP synthase on the surface of human endothelial cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Mangiullo, R.; Gnoni, A.; Leone, A.; Gnoni, G.V.; Papa, S.; Zanotti, F. Structural and functional characterization of FoF1-ATP synthase on the extracellular surface of rat hepatocytes. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1326–1335. [Google Scholar] [CrossRef]
- Taurino, F.; Gnoni, A. Systematic review of plasma-membrane ecto-ATP synthase: A new player in health and disease. Exp. Mol. Pathol. 2018, 104, 59–70. [Google Scholar] [CrossRef]
- Norton, K.A.; Popel, A.S. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci. Rep. 2016, 6, 36992. [Google Scholar] [CrossRef]
- Zhao, L.; Johnson, T.; Liu, D. Therapeutic angiogenesis of adipose-derived stem cells for ischemic diseases. Stem Cell Res. Ther. 2017, 8, 125. [Google Scholar] [CrossRef]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal stem cells: Time to change the name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Gangadaran, P.; Rajendran, R.L.; Lee, H.W.; Kalimuthu, S.; Hong, C.M.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J. Control. Release 2017, 264, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.G.; Pouzoulet, F.; Squiban, C.; Buard, V.; André, M.; Cousin, B.; Gourmelon, P.; Benderitter, M.; Casteilla, L.; Tamarat, R. Cell therapy based on adipose tissue-derived stromal cells promotes physiological and pathological wound healing. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Bura, A.; Planat-Benard, V.; Bourin, P.; Silvestre, J.S.; Gross, F.; Grolleau, J.L.; Saint-Lebese, B.; Peyrafitte, J.A.; Fleury, S.; Gadelorge, M.; et al. Phase I trial: The use of autologous cultured adipose-derived stroma/stem cells to treat patients with non-revascularizable critical limb ischemia. Cytotherapy 2014, 16, 245–257. [Google Scholar] [CrossRef]
- Henry, T.D.; Pepine, C.J.; Lambert, C.R.; Traverse, J.H.; Schatz, R.; Costa, M.; Povsic, T.J.; David Anderson, R.; Willerson, J.T.; Kesten, S.; et al. The Athena trials: Autologous adipose-derived regenerative cells for refractory chronic myocardial ischemia with left ventricular dysfunction. Catheter. Cardiovasc. Interv. 2017, 89, 169–177. [Google Scholar] [CrossRef]
- Marędziak, M.; Marycz, K.; Lewandowski, D.; Siudzińska, A.; Śmieszek, A. Static magnetic field enhances synthesis and secretion of membrane-derived microvesicles (MVs) rich in VEGF and BMP-2 in equine adipose-derived stromal cells (EqASCs)—A new approach in veterinary regenerative medicine. Vitr. Cell. Dev. Biol. Anim. 2015, 51, 230–240. [Google Scholar] [CrossRef]
- Kang, T.; Jones, T.M.; Naddell, C.; Bacanamwo, M.; Calvert, J.W.; Thompson, W.E.; Bond, V.C.; Chen, Y.E.; Liu, D. Adipose-Derived Stem Cells Induce Angiogenesis via Microvesicle Transport of miRNA-31. Stem Cells Transl. Med. 2016, 5, 440–450. [Google Scholar] [CrossRef]
- Aung, P.P.; Maxwell, H.G.; Jepson, R.G.; Price, J.F.; Leng, G.C. Lipid-lowering for peripheral arterial disease of the lower limb. Cochrane Database Syst. Rev. 2007, 2007. [Google Scholar] [CrossRef]
- Westin, G.G.; Armstrong, E.J.; Bang, H.; Yeo, K.K.; Anderson, D.; Dawson, D.L.; Pevec, W.C.; Amsterdam, E.A.; Laird, J.R. Association between statin medications and mortality, major adverse cardiovascular event, and amputation-free survival in patients with critical limb ischemia. J. Am. Coll. Cardiol. 2014, 63, 682–690. [Google Scholar] [CrossRef]
- Momsen, A.H.; Jensen, M.B.; Norager, C.B.; Madsen, M.R.; Vestersgaard-Andersen, T.; Lindholt, J.S. Drug Therapy for Improving Walking Distance in Intermittent Claudication: A Systematic Review and Meta-analysis of Robust Randomised Controlled Studies. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 463–474. [Google Scholar] [CrossRef]
- Robertson, L.; Andras, A. Prostanoids for intermittent claudication. Cochrane Database Syst. Rev. 2013, 2013. [Google Scholar] [CrossRef]
- Stevens, J.W.; Simpson, E.; Harnan, S.; Squires, H.; Meng, Y.; Thomas, S.; Michaels, J.; Stansby, G. Systematic review of the efficacy of cilostazol, naftidrofuryl oxalate and pentoxifylline for the treatment of intermittent claudication. Br. J. Surg. 2012, 99, 1630–1638. [Google Scholar] [CrossRef]
- Moazzami, K.; Majdzadeh, R.; Nedjat, S. Local intramuscular transplantation of autologous mononuclear cells for critical lower limb ischaemia. In Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Peeters Weem, S.M.O.; Teraa, M.; De Borst, G.J.; Verhaar, M.C.; Moll, F.L. Bone Marrow derived Cell Therapy in Critical Limb Ischemia: A Meta-analysis of Randomized Placebo Controlled Trials. Eur. J. Vasc. Endovasc. Surg. 2015, 50, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Grossman, P.M.; Mendelsohn, F.; Henry, T.D.; Hermiller, J.B.; Litt, M.; Saucedo, J.F.; Weiss, R.J.; Kandzari, D.E.; Kleiman, N.; Anderson, R.D.; et al. Results from a phase II multicenter, double-blind placebo-controlled study of Del-1 (VLTS-589) for intermittent claudication in subjects with peripheral arterial disease. Am. Heart J. 2007, 153, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Nikol, S.; Baumgartner, I.; Van Belle, E.; Diehm, C.; Visoná, A.; Capogrossi, M.C.; Ferreira-Maldent, N.; Gallino, A.; Graham Wyatt, M.; Dinesh Wijesinghe, L.; et al. Therapeutic Angiogenesis with Intramuscular NV1FGF Improves Amputation-free Survival in Patients With Critical Limb Ischemia. Mol. Ther. 2008, 16, 972–978. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Mohler, E.R.; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional Angiogenesis with Vascular Endothelial Growth Factor in Peripheral Arterial Disease: A Phase II Randomized, Double-Blind, Controlled Study of Adenoviral Delivery of Vascular Endothelial Growth Factor 121 in Patients with Disabling Intermittent Claudication. Circulation 2003, 108, 1933–1938. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutchings, G.; Kruszyna, Ł.; Nawrocki, M.J.; Strauss, E.; Bryl, R.; Spaczyńska, J.; Perek, B.; Jemielity, M.; Mozdziak, P.; Kempisty, B.; et al. Molecular Mechanisms Associated with ROS-Dependent Angiogenesis in Lower Extremity Artery Disease. Antioxidants 2021, 10, 735. https://doi.org/10.3390/antiox10050735
Hutchings G, Kruszyna Ł, Nawrocki MJ, Strauss E, Bryl R, Spaczyńska J, Perek B, Jemielity M, Mozdziak P, Kempisty B, et al. Molecular Mechanisms Associated with ROS-Dependent Angiogenesis in Lower Extremity Artery Disease. Antioxidants. 2021; 10(5):735. https://doi.org/10.3390/antiox10050735
Chicago/Turabian StyleHutchings, Greg, Łukasz Kruszyna, Mariusz J. Nawrocki, Ewa Strauss, Rut Bryl, Julia Spaczyńska, Bartłomiej Perek, Marek Jemielity, Paul Mozdziak, Bartosz Kempisty, and et al. 2021. "Molecular Mechanisms Associated with ROS-Dependent Angiogenesis in Lower Extremity Artery Disease" Antioxidants 10, no. 5: 735. https://doi.org/10.3390/antiox10050735
APA StyleHutchings, G., Kruszyna, Ł., Nawrocki, M. J., Strauss, E., Bryl, R., Spaczyńska, J., Perek, B., Jemielity, M., Mozdziak, P., Kempisty, B., Nowicki, M., & Krasiński, Z. (2021). Molecular Mechanisms Associated with ROS-Dependent Angiogenesis in Lower Extremity Artery Disease. Antioxidants, 10(5), 735. https://doi.org/10.3390/antiox10050735