
Oxidative Stress in Cancer Cell Metabolism


 ,
,  ,
,  ,
, 
 ,
,  and
and 
Abstract
1. Introduction
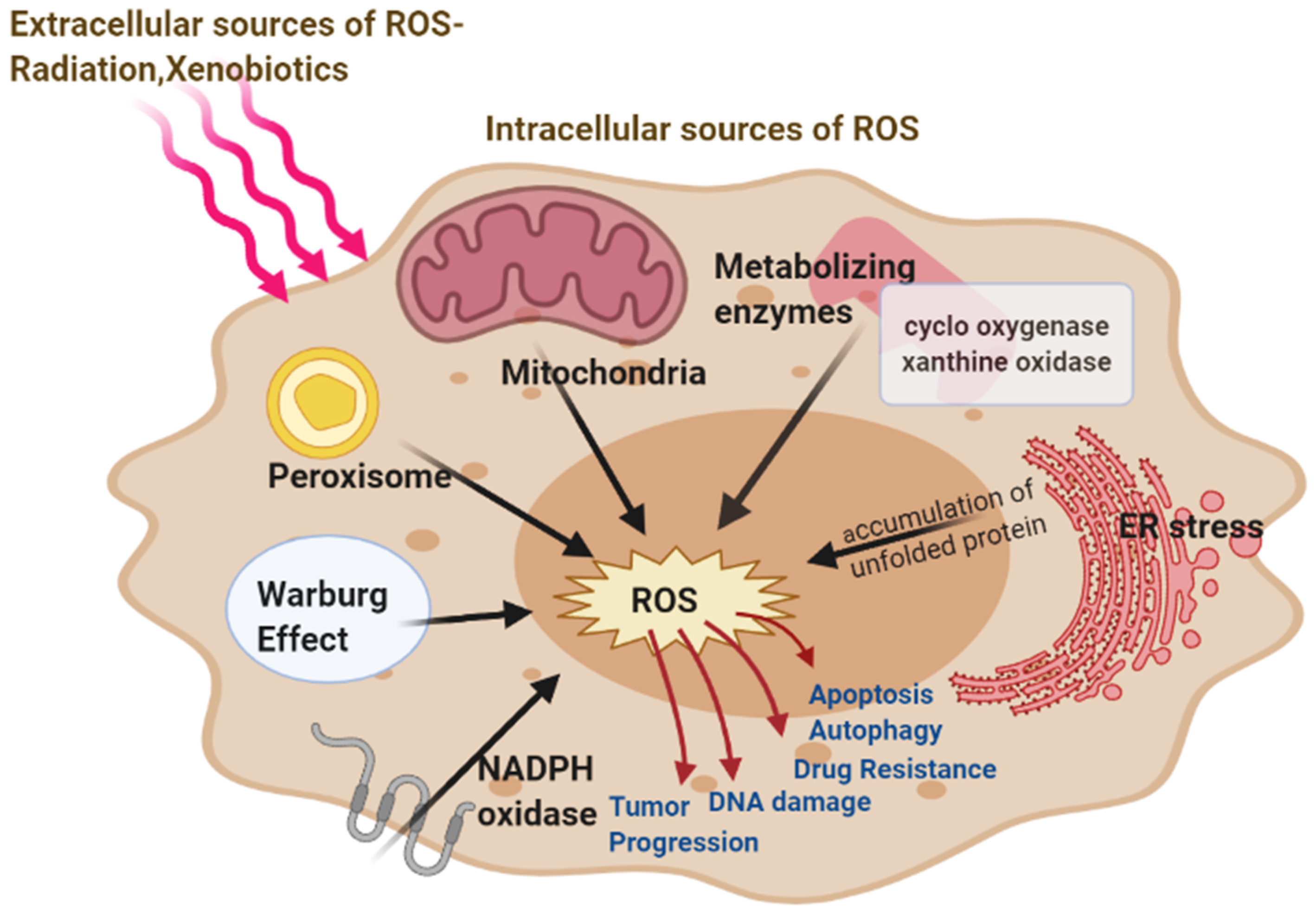
2. Source of Reactive Oxygen Species in Cancer Cells
2.1. Mitochondrial ROS
2.2. Role of Warburg Effect in ROS
2.3. NADPH Oxidase, Cox, and Xanthine Oxidase Produce ROS
2.4. ER Stress Leads to ROS
3. Mechanism of Oxidative Stress-Related Carcinogenesis
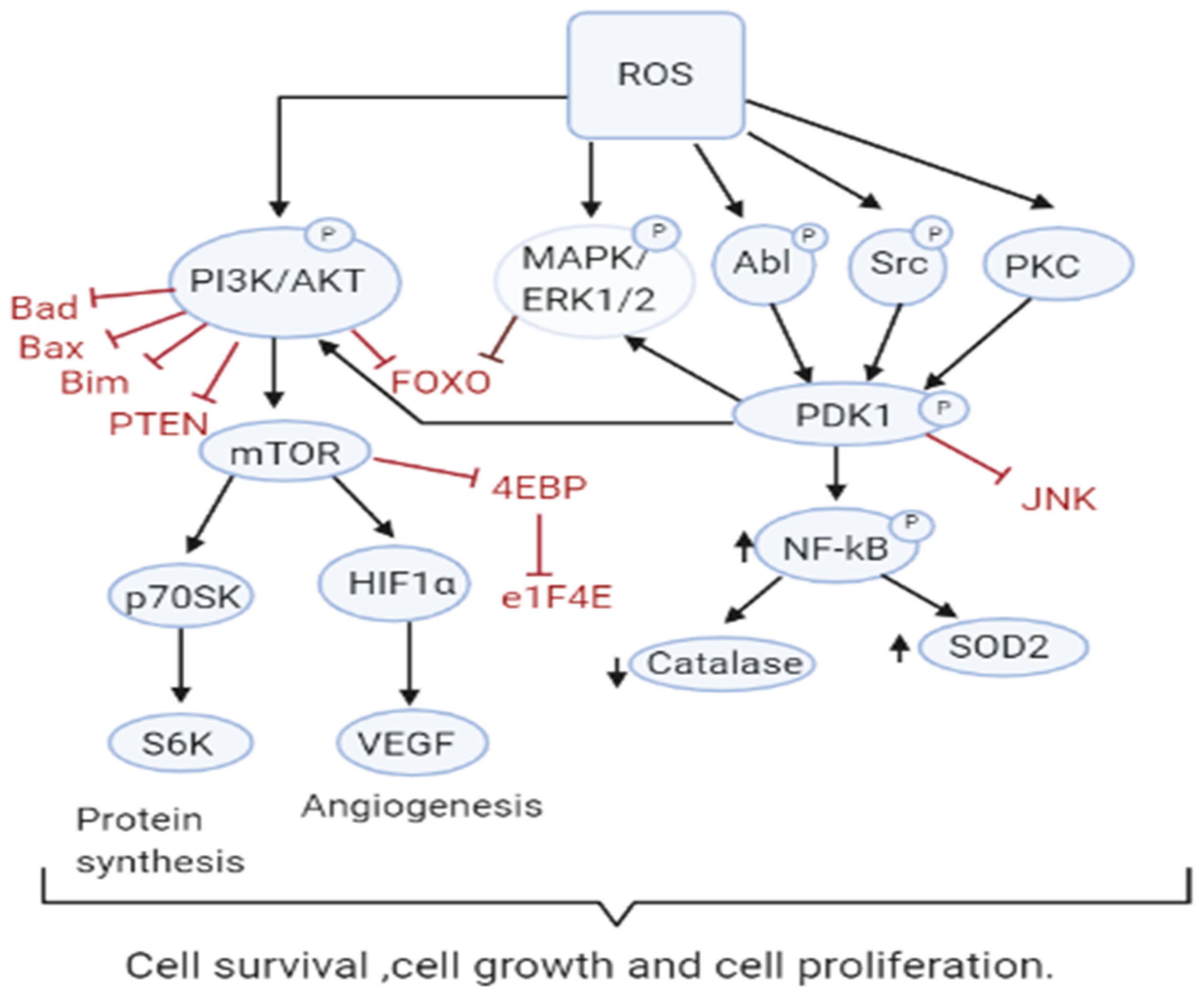
3.1. Role of ROS in Tumor Cell Proliferation, Survival and Tumor Progression
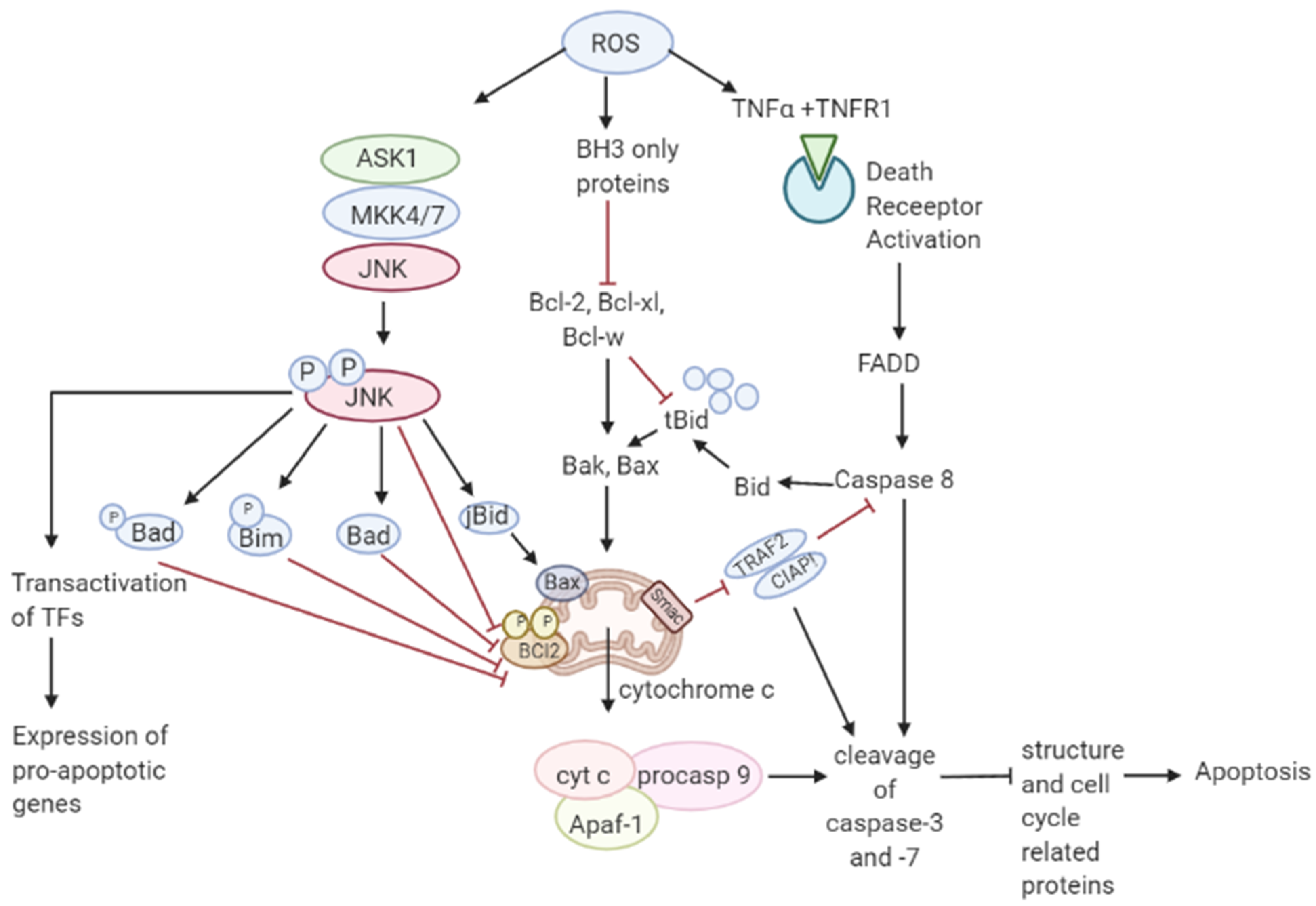
3.2. Role of ROS in Apoptosis-Tumor Suppressive Role
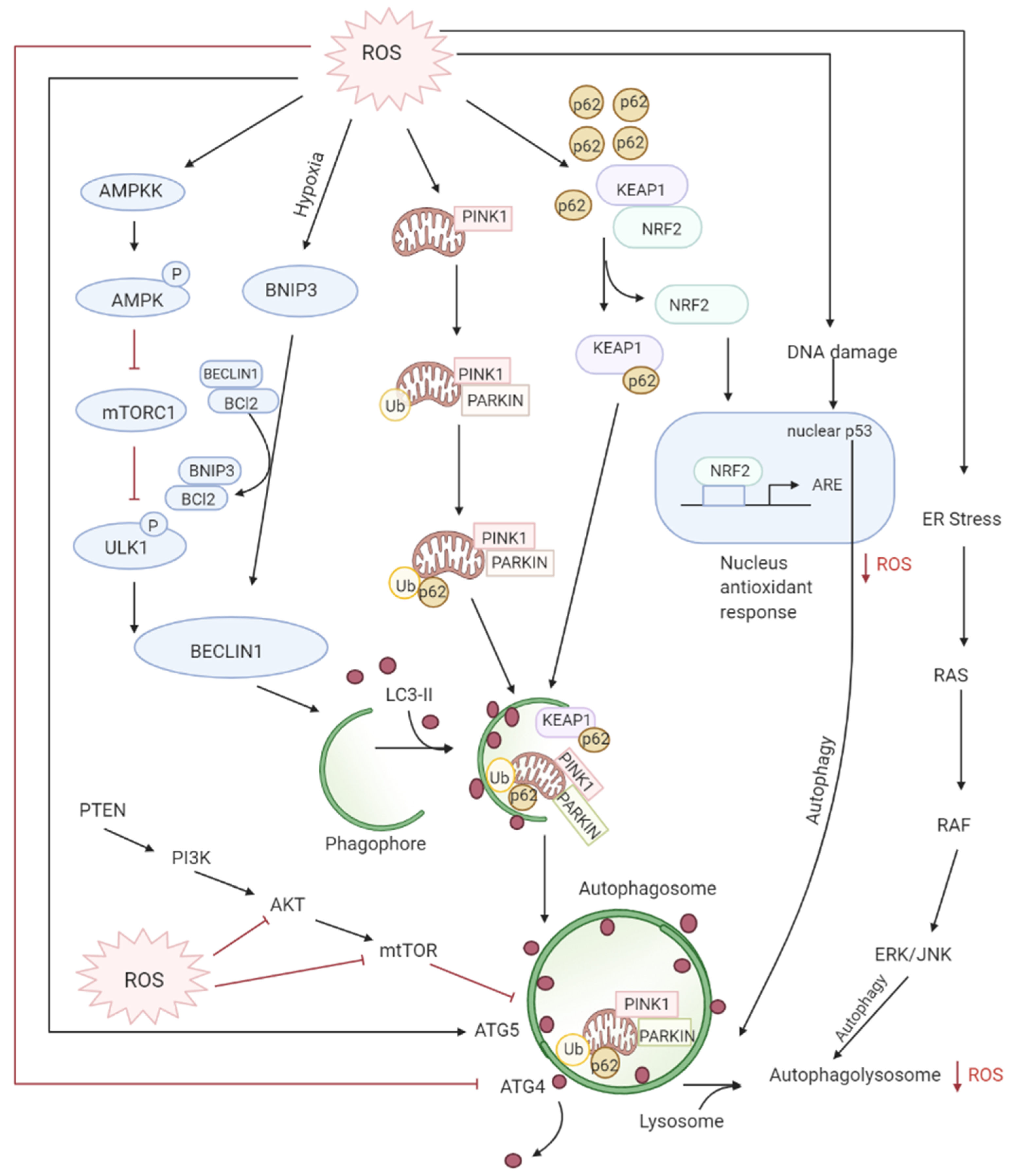
3.3. Role of ROS in Autophagy-Both Tumor Suppressive and Tumor Promoting Roles
3.4. ROS and Inflammation
3.5. ROS and DNA Damage
3.6. ROS-Mediated Alterations in Protein Stability and Lipid Peroxidation
3.7. Adaptation of Cancer Cells to ROS
3.8. Role of ROS in Drug Resistance
3.9. Role of ROS in Angiogenesis
3.10. Role of ROS in Metastasis
4. Targeting ROS
4.1. Targeting Tumor Death by Upregulation of ROS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target Cancer Type | Primary Action | Secondary Action | Reference |
|---|---|---|---|---|
| Arsenic trioxide | Ovarian cancer | Induces beclin-1-independent autophagic pathway, modulating SnoN/SkiL expression | Alters TGFβ signaling via ROS generation | [194] |
| Artemisinin | Cancer cells | Weakens the levels of glutathione, Supply extra ferrous ion to elevate ROS levels | Self-amplification of oxidative stress | [195] |
| Buthionine-sulfoximine | Cancer cells | Deplete intracellular GSH, may affect STAT3 pathway | Induce oxidative stress | [195,196] |
| Chloroquine | MCF-7, HT29, U373 cancer cells | Sensitizes cells to hypoxia, due to increased ROS, incapacity to reduce mitochondrial content | Inhibition of autophagy, increases cell death | [197] |
| Cisplatin | Head and neck cancer | Enhances ROS levels | Induce DNA damage | [198,199] |
| Curcumin | Colon cancer cells | Induces ROS production, activation of ERK1/2 and p38 MAPK | Autophagic cell death | [200] |
| Daunorubicin | Breast cancer | Induce ROS, activates the JNK pathway | Lead to apoptosis | [114,201] |
| Doxorubicin | Breast, esophageal carcinomas, endometrial carcinomas, bile duct, pancreatic, gastric, liver cancer | NO synthase inhibition, Generates ROS, activates p53 | Induces tumor cell death | [126,127,128,202] |
| Diphenylene iodonium | pancreatic cancer | Jak/STAT pathway inhibited, dephosphorylation of AKT/ASK1 pathway | Decrease ROS, lead to apoptosis | [203,204] |
| Fullerene C60 (Nano-C60) | Normal and drug-resistant cancer cells | Activation of Atg5 | Causes autophagy in a ROS-dependent fashion | [205] |
| Gemcitabine | Head and neck cancer, pancreatic cancer | Activate antioxidant agents, suppress Nox4, block ROS-related signaling pathways, inactivate stromal cells | scavenge ROS | [198,206] |
| Idarubicin (IDR) | Breast cancer | Induce oxidative DNA damage | [207] | |
| Itraconazole | Liver cancer | Increase ROS | Upregulate expression of death receptor protein FAS, pro-apoptotic protein Bax, decreased expression of anti-apoptotic protein Bcl-2, activating apoptosis | [208] |
| Medroxyprogesterone | Head and neck cancer | Induction of 15d-PGJ2-ligand of PPARγ, increased ROS | Induce apoptosis | [198,209] |
| Metformin | Pancreatic cancer | Increase MnSOD/SOD2 expression, decrease NOX2 and NOX4 protein expression | Pro-apoptotic effects | [210] |
| OSU-03012 | Hepatocellular carcinoma | Inhibit PDK/AKT signaling pathway inducing apoptotic cell death | ROS accumulation and subsequent autophagic cell death | [211] |
| Panitumumab (EGFR antibody) | EGFR-expressing metastatic colorectal carcinoma | Increase in GSH levels, reduced stability of proteins | Redox imbalance induced autophagy | [212,213,214] |
| Proton pump inhibitor, Esomeprazole | Melanoma | Mitochondrial dysfunctions, involvement of NADPH oxidase | Accumulation of reactive oxygen species (ROS) | [215] |
| Proscillaridin A (PSD-A) | Breast cancer colorectal cancer | ROS generation, Ca2+ oscillation | inhibits STAT3 activation, induces apoptosis and autophagy | [216] |
| Recombinant human HMGB1 | Glioblastoma cells | Bind to TLR2 and TLR4, induce NADPH oxidase to produce ROS | activate MAPK and NFκB, release Cytokines | [217] |
| Resveratrol | Colon cancer cells | Induce ROS and subsequent cytotoxic autophagy | Caspase-8/Caspase-3-dependent apoptosis | [218] |
| Ruthenium (II) complexes | Cancer cells | DNA damage, Induce ROS | subsequent protective autophagy along with apoptosis | [219] |
| Suberoylanilide hydroxamic acid (Zolinza, Vorinostat) | Cutaneous T-cell lymphoma, leukemia | Regulate gene expression, Induce ROS | autophagy, prosurvival | [220,221] |
| Sulforaphane | Therapy-resistant pancreatic carcinoma cell | Promote mitochondria-derived ROS | initiate protective autophagy | [222,223] |
| Sulindac | colon and lung cancer | mitochondrial damage | elevate ROS production | [129] |
| Tamoxifen | MCF-7 breast cancer cells | Induced ROS, increased expression of Beclin-1 | protective autophagy | [224] |
| Temozolomide | Malignant gliomas | Suppress ROS/ERK-mediated autophagy | Induce apoptosis | [225] |
| Valproic acid | Glioma cells | Mitochondrial ROS activates the ERK1/2 pathway | Autophagic cell death | [226] |
| Vitamin A | Testis tumor Leydig cell lines | Modulate antioxidant enzyme activities | Induce protective autophagy or apoptosis at different doses | [227] |
| 2 deoxy glucose (2DG) | pancreatic and prostate cancer | Disrupt hydroperoxide metabolism, increased glutathione disulfide accumulation, NADP (+)/NADPH ratios | Elevated ROS production leading to cell death | [132,133] |
| 7-formyl-10-methyisoellipticine | AML | Increase mitochondrial ROS production | Induces apoptosis | [134,228] |
4.2. Targeting Tumor Proliferation by Downregulation of ROS
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2DG | 2-deoxyglucose |
| 8-OHdG | 8-hydroxydeoxy guanosine |
| ALL | acute lymphoblastic leukemia |
| APL | acute promyelocytic leukemia |
| Apaf-1 | apoptotic protein-activating factor 1 |
| Ask1 | apoptosis signal-regulating kinase-1 |
| AREs | antioxidant response elements (AREs) |
| ATM | ataxia telangiectasia mutated |
| Cdk1 | cyclin-dependent kinase 1 |
| COX2 | cyclooxygenase 2 |
| CSC | cancer stem cells |
| DR | death receptor |
| EGF | epidermal growth factor |
| EGF-R | epidermal growth factor-receptor |
| Erk1/2 | extracellular-regulated kinases 1/2 |
| FAD | flavin adenine dinucleotide |
| FLIP | FLICE inhibitory protein |
| FOXO | forkhead homeobox type O |
| GF | growth factor |
| GF-R | growth factor receptor |
| GSH | glutathione |
| GSSG | glutathione disulphide |
| GPX | glutathione peroxidase |
| GST | Glutathione S-transferase |
| GLUT1 | Glucose transporter 1 |
| H2O2 | hydrogen peroxide |
| HIF-1 | hypoxia-inducible factor-1 |
| HK2 | hexokinase 2 |
| JNK | c-Jun N-terminal Kinase |
| KPNA6 | Karyopherin-6 |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LDHA | lactate dehydrogenase A |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| MOMP | mitochondrial outer-membrane permeabilization |
| mTORC1 | mammalian target of rapamycin complex1 |
| mROS | mitochondrial ROS |
| Nox | NADPH oxidases |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NFκB | nuclear factor κ-B |
| NOX | NADPH oxidase |
| OXPHOS | Oxidative Phosphorylation |
| PARP | poly ADP ribose polymerase |
| PDK1 | phosphoinositide-dependent kinase 1 |
| PDGF | platelet-derived growth factor |
| PDGF-R | platelet-derived growth factor-receptor |
| PEP | Phospho enol pyruvate |
| PERK | PKR-like endoplasmic reticulum kinase |
| PKM2 | Pyruvate kinase isozyme M2 |
| PTPs | protein tyrosine phosphatases |
| PTP1B | protein tyrosine phosphatase 1B |
| PINK1 | putative kinase 1 |
| PI3-K | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PKD | protein kinase D |
| PPP | Pentose phosphate pathways |
| Prdx | peroxiredoxin |
| PTEN | phosphatase and tensin homolog |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| SOCS | suppressors of cytokine signaling |
| TGFβ | transforming growth factor β |
| TIMP | tissue inhibitor of metalloproteinases |
| TNFα | tumor necrosis factor α |
| TP1 | triosephosphate isomerase |
| TNTs | intercellular tunneling nanotubes |
| TrxR | Thioredoxin reductase |
| VEGF | vascular epithelial growth factor |
References
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Ramirez-Tortosa, M.; Perez-Lopez, P.; Granados-Principal, S.; Battino, M.; Quiles, J.L. Long-term effects of systemic cancer treatment on DNA oxidative damage: The potential for targeted therapies. Cancer Lett. 2012, 327, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Robert, F.F. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Dada, L.A.; Sznajder, J.I. Mitochondrial Ca2+ and ROS Take Center Stage to Orchestrate TNF-α-Mediated Inflammatory Responses. J. Clin. Investig. 2011, 121, 1683–1685. [Google Scholar] [CrossRef]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Baudry, N.; Laemmel, E.; Vicaut, E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: Involvement of xanthine oxidase and mitochondria. Am. J. Physiol. Circ. Physiol. 2008, 294, H821–H828. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ceylan-Isik, A.F.; Guo, K.K.; Carlson, E.C.; Privratsky, J.R.; Liao, S.-J.; Cai, L.; Chen, A.F.; Ren, J. Metallothionein Abrogates GTP Cyclohydrolase I Inhibition–Induced Cardiac Contractile and Morphological Defects. Hypertension 2009, 53, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nat. Cell Biol. 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical Role for Mitochondrial Oxidative Phosphorylation in the Activation of Tumor Suppressors Bax and Bak. J. Natl. Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle 2010, 9, 3276–3296. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Venkataraman, S.; Kalen, A.L.; Oberley, L.W.; Goswami, P.C. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell 2008, 7, 405–417. [Google Scholar] [CrossRef]
- Wang, M.; Kirk, J.S.; Venkataraman, S.; Domann, F.E.; Zhang, H.J.; Schafer, F.Q.; Flanagan, S.W.; Weydert, C.J.; Spitz, D.R.; Buettner, G.R.; et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1α and vascular endothelial growth factor. Oncogene 2005, 24, 8154–8166. [Google Scholar] [CrossRef]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. Tumor cell redox state and mitochondria at the center of the non-canonical activity of telomerase reverse transcriptase. Mol. Asp. Med. 2010, 31, 21–28. [Google Scholar] [CrossRef]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef]
- Ruckenstuhl, C.; Büttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Fröhlich, K.-U.; Madeo, F. The Warburg Effect Suppresses Oxidative Stress Induced Apoptosis in a Yeast Model for Cancer. PLoS ONE 2009, 4, e4592. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Warburg Effect and Redox Balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef]
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b inhibits growth of human gastric cancer cells by silencing pyruvate kinase muscle-splicer polypyrimidine tract-binding protein 1. Cancer Sci. 2016, 107, 1767–1775. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Liu, J.; Levens, D. Making Myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.-W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W. Element Partitioning: The Role of Melt Structure and Composition. Science 2006, 312, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Won, K.Y.; Lim, S.-J.; Kim, G.Y.; Kim, Y.W.; Han, S.-A.; Song, J.Y.; Lee, D.-K. Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer. Hum. Pathol. 2012, 43, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Kotsinas, A.; Aggarwal, V.; Tan, E.-J.; Levy, B.; Gorgoulis, V.G. PIG3: A novel link between oxidative stress and DNA damage response in cancer. Cancer Lett. 2012, 327, 97–102. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Bulleid, N.J. Glutathione Is Required to Regulate the Formation of Native Disulfide Bonds within Proteins Entering the Secretory Pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.Z.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Mazzeo, L.E.M.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Weijzen, S.; De Vries-Smits, A.M.M.; Saarloos, I.; De Ruiter, N.D.; Bos, J.L.; Burgering, B.M.T. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic Augmentation of Rapamycin-Induced Autophagy in Malignant Glioma Cells by Phosphatidylinositol 3-Kinase/Protein Kinase B Inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.-C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nat. Cell Biol. 2003, 423, 769–773. [Google Scholar] [CrossRef]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Casciaro, M.; Musolino, C.; Gangemi, S. Synergic Crosstalk between Inflammation, Oxidative Stress, and Genomic Alterations in BCR–ABL-Negative Myeloproliferative Neoplasm. Antioxidants 2020, 9, 1037. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic Signaling Mediated by Oxidants in Ras-Transformed Fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Park, S.-A.; Na, H.-K.; Kim, E.-H.; Cha, Y.-N.; Surh, Y.-J. 4-Hydroxyestradiol Induces Anchorage-Independent Growth of Human Mammary Epithelial Cells via Activation of IκB Kinase: Potential Role of Reactive Oxygen Species. Cancer Res. 2009, 69, 2416–2424. [Google Scholar] [CrossRef]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Limaye, V.; Li, X.; Hahn, C.; Xia, P.; Berndt, M.C.; Vadas, M.A.; Gamble, J.R. Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1–dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. Blood 2005, 105, 3169–3177. [Google Scholar] [CrossRef]
- Liu, L.-Z.; Hu, X.-W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.-H. Reactive oxygen species regulate epidermal growth factor-induced vascular endothelial growth factor and hypoxia-inducible factor-1α expression through activation of AKT and P70S6K1 in human ovarian cancer cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Döppler, H.; Toker, A. Protein Kinase D Mediates Mitochondrion-to-Nucleus Signaling and Detoxification from Mitochondrial Reactive Oxygen Species. Mol. Cell. Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Toker, A. NF-κB Signaling: An ALternate Pathway for Oxidate Stress Responses. Cell Cycle 2003, 2, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, J.; Lulla, A.; Evers, B.M.; Chung, D.H. Protein kinase D protects against oxidative stress-induced intestinal epithelial cell injury via Rho/ROK/PKC-delta pathway activation. Am. J. Physiol. Physiol. 2006, 290, C1469–C1476. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Schattenberg, J.M.; Rigoli, R.M.; Storz, P.; Czaja, M.J. Hepatocyte Resistance to Oxidative Stress Is Dependent on Protein Kinase C-mediated Down-regulation of c-Jun/AP-1. J. Biol. Chem. 2004, 279, 31089–31097. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Nishitoh, H.; Tobiume, K.; Takeda, K.; Ichijo, H. Physiological Roles of ASK1-Mediated Signal Transduction in Oxidative Stress- and Endoplasmic Reticulum Stress-Induced Apoptosis: Advanced Findings from ASK1 Knockout Mice. Antioxid. Redox Signal. 2002, 4, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O.; Kim, M.-O.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kim, G.-Y. Butein induces G2/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010, 288, 204–213. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.-H.; Rojanasakul, Y. The Fas Death Signaling Pathway Connecting Reactive Oxygen Species Generation and FLICE Inhibitory Protein Down-Regulation1. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Terauchi, Y.; Solomon, G.G.; Aizawa, S.; Rangarajan, P.N.; Yazaki, Y.; Kadowaki, T.; Barrett, J.C. Involvement of p85 in p53-dependent apoptotic response to oxidative stress. Nat. Cell Biol. 1998, 391, 707–710. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.-I.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of p53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardó, F.V.; Viña, J. Mitochondrial Oxidative Stress Plays a Key Role in Aging and Apoptosis. IUBMB Life 2000, 49, 427–435. [Google Scholar] [CrossRef]
- Carmody, R.; Cotter, T. Signalling apoptosis: A radical approach. Redox Rep. 2001, 6, 77–90. [Google Scholar] [CrossRef]
- Role of JNK in Tumor Development—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12734425/ (accessed on 13 January 2021).
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen Peroxide as a Cell-Survival Signaling Molecule. Antioxid. Redox Signal. 2009, 11, 2655–2671. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed]
- Yodkeeree, S.; Sung, B.; Limtrakul, P.; Aggarwal, B.B. Zerumbone Enhances TRAIL-Induced Apoptosis through the Induction of Death Receptors in Human Colon Cancer Cells: Evidence for an Essential Role of Reactive Oxygen Species. Cancer Res. 2009, 69, 6581–6589. [Google Scholar] [CrossRef]
- Kelekar, A.; Thompson, C.B. Bcl-2-family proteins: The role of the BH3 domain in apoptosis. Trends Cell Biol. 1998, 8, 324–330. [Google Scholar] [CrossRef]
- Chen, Q.; Lesnefsky, E.J. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic. Biol. Med. 2006, 40, 976–982. [Google Scholar] [CrossRef]
- Lu, C.; Armstrong, J. Role of calcium and cyclophilin D in the regulation of mitochondrial permeabilization induced by glutathione depletion. Biochem. Biophys. Res. Commun. 2007, 363, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic. Biol. Med. 2012, 53, 1399–1410. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Vousden, K.H. Metabolic regulation by p53. J. Mol. Med. 2011, 89, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.-M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta (BBA)—Bioenerg. 2009, 1793, 1524–1532. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Mariño, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive Functions of Autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2010, 12, 9–14. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nat. Cell Biol. 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nat. Cell Biol. 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and Lung Cancer: Roles of Reactive Oxygen/Nitrogen Species. J. Toxicol. Environ. Health Part B 2008, 11, 1–15. [Google Scholar] [CrossRef]
- Aggarwal, C. Targeted therapy for lung cancer: Present and future. Ann. Palliat. Med. 2014, 3, 229–22935. [Google Scholar] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernández-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef]
- Chung, C.; Seo, W.; Silwal, P.; Jo, E.-K. Crosstalks between inflammasome and autophagy in cancer. J. Hematol. Oncol. 2020, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Rowlands, D.J.; Islam, M.N.; Das, S.R.; Huertas, A.; Quadri, S.K.; Horiuchi, K.; Inamdar, N.; Emin, M.T.; Lindert, J.; Ten, V.S.; et al. Activation of TNFR1 ectodomain shedding by mitochondrial Ca2+ determines the severity of inflammation in mouse lung microvessels. J. Clin. Investig. 2011, 121, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef]
- Fujisawa, T.; Takeda, K.; Ichijo, H. ASK Family Proteins in Stress Response and Disease. Mol. Biotechnol. 2007, 37, 13–18. [Google Scholar] [CrossRef]
- Yao, Z.; Liu, N.; Zhu, X.; Wang, L.; Zhao, Y.; Liu, Q.; Gao, C.; Li, J. Subanesthetic isoflurane abates ROS-activated MAPK/NF-κB signaling to repress ischemia-induced microglia inflammation and brain injury. Aging 2020, 12, 26121–26139. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Cao, N.; Li, J.J. HER-2 and NFκB as the Targets for Therapy-Resistant Breast Cancer. Anticancer Res. 2006, 26, 4235–4243. [Google Scholar]
- Bourguignon, L.Y.W.; Xia, W.; Wong, G. Hyaluronan-mediated CD44 Interaction with p300 and SIRT1 Regulates β-Catenin Signaling and NFκB-specific Transcription Activity Leading to MDR1 and Bcl-xL Gene Expression and Chemoresistance in Breast Tumor Cells. J. Biol. Chem. 2009, 284, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Li, N.-L.; Li, J.-T.; Yu, F.; Zhao, Y.-L.; Wang, L.; Yi, J.; Bian, J.-F.; Chen, J.-H.; et al. Subanesthetic Isoflurane Reduces Zymosan-Induced Inflammation in Murine Kupffer Cells by Inhibiting ROS-Activated p38 MAPK/NF-κB Signaling. Oxidative Med. Cell. Longev. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- No, J.H.; Kim, Y.-B.; Song, Y.S. Targeting Nrf2 Signaling to Combat Chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef]
- Nakano, H.; Nakajima, A.; Sakon-Komazawa, S.; Piao, J.-H.; Xue, X.; Okumura, K. Reactive oxygen species mediate crosstalk between NF-κB and JNK. Cell Death Differ. 2005, 13, 730–737. [Google Scholar] [CrossRef]
- Chung, F.-L.; Xu, Y. Increased 8-oxodeoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with the tobacco-specific nitrosamine 4-(methyhiitrosamine)-l-(3-pyridyl)-1-butanone. Carcinogenesis 1992, 13, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2009, 220, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1–NRF2 Interaction in Non-Small-Cell Lung Cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 Induces Antioxidant Gene Expression and Resistance to Oxidative Stress. 2004. Available online: http://www.aecom.yu.edu/cancer/new/cores/microarray (accessed on 28 January 2021).
- Rosen, E.M.; Fan, S.; Pestell, R.G.; Goldberg, I.D. BRCA1 gene in breast cancer. J. Cell. Physiol. 2003, 196, 19–41. [Google Scholar] [CrossRef]
- Seo, Y.; Kinsella, T.J. Essential Role of DNA Base Excision Repair on Survival in an Acidic Tumor Microenvironment. Cancer Res. 2009, 69, 7285–7293. [Google Scholar] [CrossRef]
- Bos, J.L. Review Ras Oncogenes in Human Cancer: A Review1. 1989. Available online: https://cancerres.aacrjournals.org/content/49/17/4682.short (accessed on 28 January 2021).
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Young, J.J.; Burton, D.G.A.; Giribaldi, M.G.; Onder, T.T.; Weinberg, R.A. Enhanced elimination of oxidized guanine nucleotides inhibits oncogenic RAS-induced DNA damage and premature senescence. Oncogene 2010, 30, 1489–1496. [Google Scholar] [CrossRef]
- Weyemi, U.; Lagentechevallier, O.; Boufraqech, M.; Prenois, F.; Courtin, F.; Caillou, B.; Talbot, M.R.; Dardalhon, M.; Al Ghuzlan, A.; Bidart, J.-M.; et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2011, 31, 1117–1129. [Google Scholar] [CrossRef]
- Kim, H.-S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L.P. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole Blood Transcriptional Profiling Reveals Deregulation of Oxidative and Antioxidative Defence Genes in Myelofibrosis and Related Neoplasms. Potential Implications of Downregulation of Nrf2 for Genomic Instability and Disease Progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Uedono, Y.; Ishikura, H.; Takeyama, N.; Tanaka, T. DNA Damage Induced by Tumour Necrosis Factor-α in L929 Cells Is Mediated by Mitochondrial Oxygen Radical Formation. Immunology 1995, 84, 543–548. Available online: https://pmc/articles/PMC1415148/?report=abstract (accessed on 6 January 2021).
- Barzilai, A.; Rotman, G.; Shiloh, Y. ATM deficiency and oxidative stress: A new dimension of defective response to DNA damage. DNA Repair 2002, 1, 3–25. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The Antioxidant Function of the P53 Tumor Suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Ando, K.; Hirao, S.; Kabe, Y.; Ogura, Y.; Sato, I.; Yamaguchi, Y.; Wada, T.; Handa, H. A new APE1/Ref-1-dependent pathway leading to reduction of NF- B and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008, 36, 4327–4336. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef]
- Cai, F.; Dupertuis, Y.M.; Pichard, C. Role of polyunsaturated fatty acids and lipid peroxidation on colorectal cancer risk and treatments. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 99–106. [Google Scholar] [CrossRef]
- Volinsky, R.; Kinnunen, P.K.J. Oxidized phosphatidylcholines in membrane-level cellular signaling: From biophysics to physiology and molecular pathology. FEBS J. 2013, 280, 2806–2816. [Google Scholar] [CrossRef]
- MohammadAlipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 603292. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.K.; Lin, Z.; Balliet, R.M.; Howell, A.; et al. Understanding the “lethal” drivers of tumor-stroma co-evolution. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef]
- Chai, Y.; Ashraf, S.; Rokutan, K.; Johnston, R.; Thomas, J. S-Thiolation of Individual Human Neutrophil Proteins Including Actin by Stimulation of the Respiratory Burst: Evidence against a Role for Glutathione Disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef]
- Van Muiswinkel, F.L.; Kuiperij, F.L.V.M.A.H.B. The Nrf2-ARE Signalling Pathway: Promising Drug Target to Combat Oxidative Stress in Neurodegenerative Disorders. Curr. Drug Target CNS Neurol. Disord. 2005, 4, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, T.; Zhao, F.; Lau, A.; Birch, C.M.; Zhang, N.D. KPNA6 (Importin 7)-Mediated Nuclear Import of Keap1 Represses the Nrf2-Dependent Antioxidant Response. Mol. Cell. Biol. 2011, 31, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Slocum, S.L.; Kensler, T.W. Nrf2: Control of sensitivity to carcinogens. Arch. Toxicol. 2011, 85, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Skórski, T. BCR/ABL regulates response to DNA damage: The role in resistance to genotoxic treatment and in genomic instability. Oncogene 2002, 21, 8591–8604. [Google Scholar] [CrossRef]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species–dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of Glycolysis in Cancer Cells: A Novel Strategy to Overcome Drug Resistance Associated with Mitochondrial Respiratory Defect and Hypoxia. 2005. Available online: www.aacrjournals.org (accessed on 14 January 2021).
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-Mediated Down-Regulation of Bid and Bax in Tumors Occurs via Hypoxia-Inducible Factor 1-Dependent and -Independent Mechanisms and Contributes to Drug Resistance. Mol. Cell. Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Matsumoto, T.; Claesson-Welsh, L. VEGF Receptor Signal Transduction. Sci. Signal. 2001, 2001, re21. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Davis-Smyth, T. The Biology of Vascular Endothelial Growth Factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Sun, C.; Lambeth, J.D.; Marshall, F.; Amin, M.; Chung, L.; Petros, J.A.; Arnold, R.S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2004, 62, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Corbin, Z.; Kennedy, T.P.; Hemendinger, R.; Thornton, L.; Bommarius, B.; Arnold, R.S.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; et al. NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am. J. Physiol. Physiol. 2003, 285, C353–C369. [Google Scholar] [CrossRef]
- Powis, G.; Kirkpatrick, D.L. Thioredoxin signaling as a target for cancer therapy. Curr. Opin. Pharmacol. 2007, 7, 392–397. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1α and reactive oxygen species. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Belkhiri, A.; Richards, C.; Whaley, M.; McQueen, S.A.; Orr, F.W. Increased Expression of Activated Matrix Metalloproteinase-2 by Human Endothelial Cells after Sublethal H2O2 Exposure—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/9389796/ (accessed on 9 April 2021).
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose Oxidation Modulates Anoikis and Tumor Metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.K.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Gouazé, V.; Mirault, M.-E.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione Peroxidase-1 Overexpression Prevents Ceramide Production and Partially Inhibits Apoptosis in Doxorubicin-Treated Human Breast Carcinoma Cells. Mol. Pharmacol. 2001, 60, 488–496. [Google Scholar]
- Mas, V.M.-D.; Bezombes, C.; Quillet-Mary, A.; Bettaïeb, A.; D’Orgeix, A.D.T.; Laurent, G.; Jaffrézou, J.-P. Implication of Radical Oxygen Species in Ceramide Generation, c-Jun N-Terminal Kinase Activation and Apoptosis Induced by Daunorubicin. Mol. Pharmacol. 1999, 56, 867–874. [Google Scholar] [CrossRef]
- Goodman, J.; Hochstein, P. Generation of free radicals and lipid peroxidation by redox cycling of adriamycin and daunomycin. Biochem. Biophys. Res. Commun. 1977, 77, 797–803. [Google Scholar] [CrossRef]
- Doroshow, J.H. Anthracycline antibiotic-stimulated superoxide, hydrogen peroxide, and hydroxyl radical production by NADH dehydrogenase. Cancer Res. 1983, 43, 4543–4551. [Google Scholar]
- Pan, S.S.; Pedersen, L.; Bachur, N.R. Comparative flavoprotein catalysis of anthracycline antibiotic. Reductive cleavage and oxygen consumption. Mol. Pharmacol. 1981, 19, 184–186. [Google Scholar]
- Bates, D.A.; Winterbourn, C.C. Deoxyribose breakdown by the adriamycin semiquinone and H2O2: Evidence for hydroxyl radical participation. FEBS Lett. 1982, 145, 137–142. [Google Scholar] [CrossRef]
- Bachur, N.R.; Gordon, S.L.; Gee, M.V. Anthracycline antibiotic augmentation of microsomal electron transport and free radical formation. Mol. Pharmacol. 1977, 13, 901–910. [Google Scholar] [PubMed]
- Sinha, B.K. Free radicals in anticancer drug pharmacology. Chem. Interact. 1989, 69, 293–317. [Google Scholar] [CrossRef]
- Bustamante, J.; Galleano, M.; Medrano, E.E.; Boveris, A. Adriamycin effects on hydroperoxide metabolism and growth of human breast tumor cells. Breast Cancer Res. Treat. 1990, 17, 145–153. [Google Scholar] [CrossRef]
- Serrano, J.; Palmeira, C.; Kuehl, D.; Wallace, K. Cardioselective and cumulative oxidation of mitochondrial DNA following subchronic doxorubicin administration. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 201–205. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Marcus, C.; Blumenschein, G.R.; Smith, T.L. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer 1985, 55, 2761–2765. [Google Scholar] [CrossRef]
- Wang, X.; Han, W.; Yu, Y.; Zhu, J.; Zhang, R. Doxorubicin-induced cardiomyopathy. In Doxorubicin: Biosynthesis, Clinical Uses and Health Implications; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2014; pp. 43–87. [Google Scholar]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin Induces Apoptosis in Normal and Tumor Cells via Distinctly Different Mechanisms. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.; Bodis, S.; McClatchey, A.; Remington, L.; Ruley, H.; Fisher, D.; Housman, D.; Jacks, T. p53 status and the efficacy of cancer therapy in vivo. Science 1994, 266, 807–810. [Google Scholar] [CrossRef]
- Lotem, J.; Peled-Kamar, M.; Groner, Y.; Sachs, L. Cellular Oxidative Stress and the Control of Apoptosis by Wild-Type p53, Cytotoxic Compounds, and Cytokines. 1996. Available online: https://www.pnas.org/content/93/17/9166.short (accessed on 14 January 2021).
- Marchetti, M.; Resnick, L.; Gamliel, E.; Kesaraju, S.; Weissbach, H.; Binninger, D. Sulindac Enhances the Killing of Cancer Cells Exposed to Oxidative Stress. PLoS ONE 2009, 4, e5804. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.M.; Abdalla, M.Y.; Aykin-Burns, N.; Simons, A.L.; Oberley, L.W.; Domann, F.E.; Spitz, D.R. 2-Deoxyglucose combined with wild-type p53 overexpression enhances cytotoxicity in human prostate cancer cells via oxidative stress. Free. Radic. Biol. Med. 2008, 44, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.C.; Asbury, C.R.; Daniels, D.; Du, J.; Aykin-Burns, N.; Smith, B.J.; Li, L.; Spitz, D.R.; Cullen, J.J. 2-Deoxy-d-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free. Radic. Biol. Med. 2008, 44, 322–331. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef]
- Russell, E.G.; Guo, J.; O’Sullivan, E.C.; O’Driscoll, C.M.; McCarthy, F.O.; Cotter, T.G. 7-formyl-10-methylisoellipticine, a novel ellipticine derivative, induces mitochondrial reactive oxygen species (ROS) and shows anti-leukaemic activity in mice. Investig. New Drugs 2016, 34, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Raffoul, F.; Campla, C.; Nanjundan, M. SnoN/SkiL, a TGFβ signaling mediator. Autophagy 2010, 6, 955–957. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, X.; Huang, L.; Yan, J.; Yu, B.-Y.; Tian, J. Artemisinin-Based Smart Nanomedicines with Self-Supply of Ferrous Ion to Enhance Oxidative Stress for Specific and Efficient Cancer Treatment. ACS Appl. Mater. Interfaces 2019, 11, 29490–29497. [Google Scholar] [CrossRef]
- Li, Q.; Yin, X.; Wang, W.; Zhan, M.; Zhao, B.; Hou, Z.; Wang, J. The effects of buthionine sulfoximine on the proliferation and apoptosis of biliary tract cancer cells induced by cisplatin and gemcitabine. Oncol. Lett. 2015, 11, 474–480. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Ramaekers, C.H.; Schaaf, M.B.; Keulers, T.G.; Savelkouls, K.G.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416. [Google Scholar] [CrossRef]
- Yen, C.-J.; Hung, C.-H.; Tsai, W.-M.; Cheng, H.-C.; Yang, H.-L.; Lu, Y.-J.; Tsai, K.-L. Effect of Exercise Training on Exercise Tolerance and Level of Oxidative Stress for Head and Neck Cancer Patients Following Chemotherapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Mirzaei, S.; Mohammadi, A.T.; Gholami, M.H.; Hashemi, F.; Zarrabi, A.; Zabolian, A.; Hushmandi, K.; Makvandi, P.; Samec, M.; Liskova, A.; et al. Nrf2 signaling pathway in cisplatin chemotherapy: Potential involvement in organ protection and chemoresistance. Pharmacol. Res. 2021, 167, 105575. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, N.-Y.; Suh, Y.-A.; Lee, C. Involvement of ROS in Curcumin-induced Autophagic Cell Death. Korean J. Physiol. Pharmacol. 2011, 15, 1–7. [Google Scholar] [CrossRef]
- Al-Aamri, H.M.; Ku, H.; Irving, H.R.; Tucci, J.; Meehan-Andrews, T.; Bradley, C. Time dependent response of daunorubicin on cytotoxicity, cell cycle and DNA repair in acute lymphoblastic leukaemia. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Asensio-López, M.C.; Soler, F.; Pascual-Figal, D.; Fernández-Belda, F.; Lax, A. Doxorubicin-induced oxidative stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [Google Scholar] [CrossRef]
- Doroshow, J.H.; Juhasz, A.; Ge, Y.; Holbeck, S.; Lu, J.; Antony, S.; Wu, Y.; Jiang, G.; Roy, K. Antiproliferative mechanisms of action of the flavin dehydrogenase inhibitors diphenylene iodonium and di-2-thienyliodonium based on molecular profiling of the NCI-60 human tumor cell panel. Biochem. Pharmacol. 2012, 83, 1195–1207. [Google Scholar] [CrossRef]
- Fang, B. Genetic Interactions of STAT3 and Anticancer Drug Development. Cancers 2014, 494–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, W.; Man, N.; Zheng, F.; Shen, Y.; Sun, K.; Li, Y.; Wen, L.-P. Autophagy-mediated chemosensitization in cancer cells by fullerene C60 nanocrystal. Autophagy 2009, 5, 1107–1117. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Mizutani, H.; Shiga, C.; Imai, M.; Ikemura, K.; Kitamura, Y.; Ohta, K.; Miyazawa, D.; Sakanashi, M.; Tahira, T.; Maeda, T.; et al. Idarubicin, an Anthracycline, Induces Oxidative DNA Damage in the Presence of Copper (II). Anticancer Res. 2020, 40, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Dong, X.; Liu, Y.; Ni, B.; Sai, N.; You, L.; Sun, M.; Yao, Y.; Qu, C.; Yin, X.; et al. Itraconazole exerts anti-liver cancer potential through the Wnt, PI3K/AKT/mTOR, and ROS pathways. Biomed. Pharmacother. 2020, 131, 110661. [Google Scholar] [CrossRef]
- Khanim, F.L.; Hayden, R.E.; Birtwistle, J.; Lodi, A.; Tiziani, S.; Davies, N.J.; Ride, J.P.; Viant, M.R.; Günther, U.L.; Mountford, J.C.; et al. Combined Bezafibrate and Medroxyprogesterone Acetate: Potential Novel Therapy for Acute Myeloid Leukaemia. PLoS ONE 2009, 4, e8147. [Google Scholar] [CrossRef] [PubMed]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin Transiently Inhibits Colorectal Cancer Cell Proliferation as a Result of Either AMPK Activation or Increased ROS Production. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yeh, P.Y.; Lu, Y.-S.; Hsu, C.-H.; Chen, K.-F.; Lee, W.-C.; Feng, W.-C.; Chen, C.-S.; Kuo, M.-L.; Cheng, A.-L. OSU-03012, a Novel Celecoxib Derivative, Induces Reactive Oxygen Species–Related Autophagy in Hepatocellular Carcinoma. Cancer Res. 2008, 68, 9348–9357. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, E.; Antonacopoulou, A.; Matsouka, P.; Kalofonos, H.P. Autophagy: Novel action of panitumumab in colon cancer. Anticancer. Res. 2009, 29, 5077–5082. [Google Scholar]
- Peeters, M.; Balfourf, J.; Arnold, D. Review Article: Panitumumab A Fully Human Anti-EGFR Monoclonal Antibody for Treatment of Metastatic Colorectal Cancer. Aliment. Pharmacol. Ther. 2008, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Gemmete, J.J.; Mukherji, S.K. Panitumumab (Vectibix). Am. J. Neuroradiol. 2011, 32, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Fais, S.; Djavaherimergny, M.; Villa, A.; Meschini, S.; Lozupone, F.; Venturi, G.M.; Della Mina, P.; Pattingre, S.; Rivoltini, L.; et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010, 1, e87. [Google Scholar] [CrossRef]
- Saleem, M.Z.; Alshwmi, M.; Zhang, H.; Din, S.R.U.; Nisar, M.A.; Khan, M.; Alam, S.; Alam, G.; Jin, L.; Ma, T. Inhibition of JNK-Mediated Autophagy Promotes Proscillaridin A- Induced Apoptosis via ROS Generation, Intracellular Ca+2 Oscillation and Inhibiting STAT3 Signaling in Breast Cancer Cells. Front. Pharmacol. 2020, 11, 1055. [Google Scholar] [CrossRef]
- Gdynia, G.; Keith, M.; Kopitz, J.; Bergmann, M.; Fassl, A.; Weber, A.N.; George, J.; Kees, T.; Zentgraf, H.-W.; Wiestler, O.D.; et al. Danger Signaling Protein HMGB1 Induces a Distinct Form of Cell Death Accompanied by Formation of Giant Mitochondria. Cancer Res. 2010, 70, 8558–8568. [Google Scholar] [CrossRef]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef]
- Tan, C.; Lai, S.; Wu, S.; Hu, S.; Zhou, L.; Chen, Y.; Wang, M.; Zhu, Y.; Lian, W.; Peng, W.; et al. Nuclear Permeable Ruthenium(II) β-Carboline Complexes Induce Autophagy To Antagonize Mitochondrial-Mediated Apoptosis. J. Med. Chem. 2010, 53, 7613–7624. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R. Regulation of apoptosis-associated genes by histone deacetylase inhibitors: Implications in cancer therapy. Anti-Cancer Drugs 2010, 21, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef]
- Xiao, D.; Powolny, A.A.; Antosiewicz, J.; Hahm, E.-R.; Bommareddy, A.; Zeng, Y.; Desai, D.; Amin, S.; Herman-Antosiewicz, A.; Singh, S.V. Cellular Responses to Cancer Chemopreventive Agent D,L-Sulforaphane in Human Prostate Cancer Cells Are Initiated by Mitochondrial Reactive Oxygen Species. Pharm. Res. 2009, 26, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, F.; Naumann, P.; Zentgraf, H.; Büchler, M.W.; Herr, I.; Werner, J. Autophagy and cell death signaling following dietary sulforaphane act independently of each other and require oxidative stress in pancreatic cancer. Int. J. Oncol. 2011, 39, 101–109. [Google Scholar] [CrossRef]
- De Medina, P.; Silvente-Poirot, S.; Poirot, M. Tamoxifen and AEBS ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy 2009, 5, 1066–1067. [Google Scholar] [CrossRef][Green Version]
- Lin, C.-J.; Lee, C.-C.; Shih, Y.-L.; Lin, T.-Y.; Wang, S.-H.; Lin, Y.-F.; Shih, C.-M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Fu, J.; Shao, C.-J.; Chen, F.-R.; Ng, H.-K.; Chen, Z.-P. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro-Oncology 2009, 12, 328–340. [Google Scholar] [CrossRef]
- Perri, M.; Pingitore, A.; Cione, E.; Vilardi, E.; Perrone, V.; Genchi, G. Proliferative and anti-proliferative effects of retinoic acid at doses similar to endogenous levels in Leydig MLTC-1/R2C/TM-3 cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2010, 1800, 993–1001. [Google Scholar] [CrossRef]
- Miller, C.M.; O’sullivan, E.C.; McCarthy, F.O. Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmaceuticals 2019, 12, 90. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Cheng, G.; Lanza-Jacoby, S. Metformin decreases growth of pancreatic cancer cells by decreasing reactive oxygen species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.; et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef]
- Stanicka, J.; Russell, E.G.; Woolley, J.F.; Cotter, T.G. NADPH Oxidase-generated Hydrogen Peroxide Induces DNA Damage in Mutant FLT3-expressing Leukemia Cells. J. Biol. Chem. 2015, 290, 9348–9361. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.P.; Zhong, W.; Askeland, R.W.; Domann, F.E. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene 2014, 33, 358–368. [Google Scholar] [CrossRef]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.R.; Cullen, M.R.; Glass, A.G.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.G.; Williams, J.H.; et al. Effects of a Combination of Beta Carotene and Vitamin A on Lung Cancer and Cardiovascular Disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef]
- Kein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the Risk of Prostate Cancer: The selenium and vitamin e cancer prevention trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. https://doi.org/10.3390/antiox10050642
Arfin S, Jha NK, Jha SK, Kesari KK, Ruokolainen J, Roychoudhury S, Rathi B, Kumar D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants. 2021; 10(5):642. https://doi.org/10.3390/antiox10050642
Chicago/Turabian StyleArfin, Saniya, Niraj Kumar Jha, Saurabh Kumar Jha, Kavindra Kumar Kesari, Janne Ruokolainen, Shubhadeep Roychoudhury, Brijesh Rathi, and Dhruv Kumar. 2021. "Oxidative Stress in Cancer Cell Metabolism" Antioxidants 10, no. 5: 642. https://doi.org/10.3390/antiox10050642
APA StyleArfin, S., Jha, N. K., Jha, S. K., Kesari, K. K., Ruokolainen, J., Roychoudhury, S., Rathi, B., & Kumar, D. (2021). Oxidative Stress in Cancer Cell Metabolism. Antioxidants, 10(5), 642. https://doi.org/10.3390/antiox10050642