Abstract
RNA binding proteins fulfil a wide number of roles in gene expression. Multiple mechanisms of RNA binding protein dysregulation have been implicated in the pathomechanisms of several neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). Oxidative stress and mitochondrial dysfunction also play important roles in these diseases. In this review, we highlight the mechanistic interplay between RNA binding protein dysregulation, oxidative stress and mitochondrial dysfunction in ALS. We also discuss different potential therapeutic strategies targeting these pathways.
1. Introduction
RNA binding proteins (RBPs) represent a family of >1300 proteins that control all aspects of a RNA life cycle, including regulating transcription, processing, localisation, function and finally decay [1,2]. This carefully controlled interaction allows a diligent regulation of gene expression. Dysfunction of RBPs has been heavily implicated in the pathogenesis of many neurodegenerative diseases, in particular amyotrophic lateral sclerosis (ALS). ALS is characterized by upper and lower motor neuron death [3]. Loss of motor neurons results in progressive muscle weakness, paralysis and eventual death, typically due to denervation of respiratory muscles [4]. Up to half of patients also suffer some form of cognitive decline or behavioural impairment, ~15% of which fulfil diagnostic criteria for frontotemporal dementia (FTD) suggesting that ALS and FTD are part of a disease spectrum [5,6]. The study of familial ALS (fALS) cases, which account for ~5–10% of cases overall, has identified ~30 genes involved in pathogenesis [7]. A genetic basis for disease is not restricted to fALS, as mutations in genes found in fALS cases have been identified in up to 25% of apparently sALS cases, which share many neuropathological features [8]. Mutations in genes implicated in ALS encode proteins with diverse cellular functions with some convergence, however, on RBPs including: TAR DNA-binding protein (TARDBP), fused in sarcoma (FUS), TATA-Box Binding Protein Associated Factor 15 (TAF15), EWS RNA Binding Protein 1 (EWSR1), heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), matrin 3 (MATR3), T-cell-restricted intracellular antigen-1 (TIA1) and senataxin (SETX) (Table 1).

Table 1.
Genes encoding RBPs involved in ALS.
RBP nuclear to cytoplasmic mislocalisation and aggregation is considered a key hallmark of disease, evident across both fALS and sALS cases [9,10,11,12,13,14,15]. Mutations in RBPs can also cause their mislocalisation to the cytoplasm [16,17,18]. Multiple, possibly overlapping, pathomechanisms have been identified in ALS. In addition to RBP dysfunction, increased oxidative stress, mitochondrial dysfunction, impaired nucleocytoplasmic transport, protein dyshomeostasis, deficits in axonal transport, excitotoxicity and non cell autonomous mechanisms of disease arising from glia are thought to contribute to motor neuron death in ALS. Here, we review the dysfunction of RBPs and their interplay with oxidative stress and mitochondrial dysfunction in ALS, as well as highlight potential therapeutic strategies targeting these pathways.
2. Mechanisms of RNA Binding Protein Dysfunction in ALS
RBP dysregulation has been proposed to cause ALS pathogenesis through two main mechanisms: an alteration in RNA metabolism and loss of protein homeostasis. Many RBPs autoregulate their own expression [12,13,14,15] and mouse models either knocking down or overexpressing wildtype forms of certain RBPs have been able to partially recapitulate ALS pathological features [29], highlighting the importance of their tightly regulated physiological expression. Due to the diverse roles of RBPs, dysregulation results in a range of molecular phenotypes such as perturbed gene expression, splicing patterns and splicing machinery, mRNA nuclear export, transport and local translation. This could be the result of aberrant protein localisation, aberrant interactions or post-translational modifications, protein misfolding, aggregate formation and changes in granule dynamics. The following sections will discuss how RBP deregulation can in turn disrupt cellular homeostasis.
2.1. Localisation
Although ALS is highly genetically heterogeneous and most cases have no clear genetic cause, TDP-43 mislocalisation from the nucleus to the cytoplasm of motor neurons has been found to occur in the vast majority (>95%) of ALS cases [9,30]. Furthermore, nuclear to cytoplasmic FUS [10], TAF15 [22] and SFPQ [14] mislocalisation has been identified in sALS motor neurons. Mislocalisation of TDP-43 [31], SFPQ [14] and FUS [32] has also been reported in mutant VCP-ALS iPSC derived motor neurons. These studies and others provide evidence of perturbed protein subcellular localisation in the absence of overt aggregation. It is still unresolved whether disease is caused by a nuclear loss of function or a cytoplasmic gain in function, and if these events occur simultaneously or sequentially, with the possibility remaining that both events have an equal involvement in disease. Mislocalisation of RBPs may be due to defects in the nucleocytoplasmic machinery [33], modification of the nuclear localisation by mutations or post-translational mechanisms discussed below.
2.2. Aggregation
Many proteins implicated in ALS (TDP-43, hnRNPA1, TAF15, EWS, FUS, TIA1) display point mutations in their intrinsically disordered regions (IDRs), which are also called low complexity domains (LCDs). These domains have been shown to be responsible for weak protein-protein interactions, driven by liquid-liquid phase separation (LLPS), allowing the formation of reversible molecular condensates that can assemble/disassemble in response to changes in the cellular environment. Remarkably, over 40 ALS-linked mutations have been found in TDP-43, with only 3 of these not located within the C-terminal LCD [34]. Mutations in the LCD of TDP-43, hnRNPA1, FUS, TIA1, TAF15 and EWS result in increased fibrillar formation of the mutated protein [22,24,27,35,36,37]. This increased fibrillar formation is able to seed prion-like aggregation from the remaining soluble pool of protein, encouraging cytoplasmic aggregation and subsequent nuclear loss. Such seeded aggregation and prion-like behaviour has recently been demonstrated in human iPSC-derived motor neurons and astrocytes, which also exhibited differential vulnerability to recombinant TDP-43 oligomers [38]. In addition, formation of abnormal RNA foci may lead to abnormal RBP mislocalisation. G4C2 hexanucleotide repeat expansion in the first intron of the C9ORF72 gene undergoes bidirectional transcription, which then forms RNA foci [39]. These RNA foci have been shown to sequester RBPs, altering their localisation and thus RNA metabolism, including TDP-43, FUS, SFPQ and hnRNPA1 [40,41,42]. It is still a matter of debate as to whether these aggregates contribute to neurodegeneration [43]. The sequestration of proteins could be both protective and detrimental depending on the disease phase.
2.3. Aberrant Granule Dynamics
The similarity between cytosolic aggregation of RBPs seen in ALS and the formation of membrane-less granules suggests a role for stress granules (SGs) in ALS pathogenesis. SGs are enriched with proteins containing LCDs, including many ALS-linked RBPs, with mutations in these regions shown to alter SG dynamics and increase aggregation [26,44]. Supporting this, ALS-associated RBPs that have been shown to interact with or alter SG dynamics include, TDP-43 [45,46], FUS [47], hnRNPA1 [27], TIA1 [48] and ATXN2 [49]. As many of these ALS causing mutations result in aberrant phase separation, decreased SG dynamics and persistence, it has been suggested these may develop into the cytoplasmic aggregates found at end stage disease. To support this hypothesis, many SG proteins are found in the cytoplasmic protein aggregates, for example TDP-43 and FUS colocalise with SG markers in the cytoplasmic inclusions found in ALS patient samples [45,50]. However, there is no direct evidence to support this phenomenon and it remains unknown whether SGs function as precursors of the inclusions or if SG components are incorporated into inclusions [9,51,52]. Recently, a collection of studies have advanced our insight into this topic, and shown that upon stress TDP-43 also forms droplets that are distinct from SGs, and which can persist into aggregate-like structures [53,54,55]. This suggests aggregation of TDP-43 and the pathogenic cascade of ALS can occur independently of SGs.
Many RBPs can be found in paraspeckles, nuclear ribonucleoprotein membraneless granules that sequester proteins and RNA to regulate gene expression [56]. Paraspeckles have been hypothesised to have a role in ALS pathology, with ALS-linked RBPs, TDP-43, FUS and SFPQ identified as components of paraspeckles. Whilst these proteins have been shown to be mislocalised from the nucleus to the cytoplasm in ALS motor neurons [9,10,14], other core paraspeckle proteins, NONO and PSPC1 have been shown not to be mislocalised in sporadic ALS postmortem tissue [57]. Furthermore, depletion of FUS or TDP-43 have been shown to decrease paraspeckle formation [58,59]. Increased paraspeckle formation has been found in both mutant FUS and TDP-43 ALS post mortem motor neurons [60,61]. Further supporting data come from identification of an increase in paraspeckle formation in early stage sporadic ALS spinal motor neurons [62].
2.4. Post Translational Modifications
Post translational modification (PTM) of RBPs have been shown to tightly regulate their various functions, with aberrant PTMs observed in both fALS and sALS [63]. Phosphorylation of TDP-43 has been linked to aggregation in post mortem ALS tissue [64] but also has been proposed to be a preventive measure to attempt to reduce aggregation [65,66,67,68]. Other modifications to RBPs that affect either RNA binding function or cytoplasmic mislocalisation and/or aggregation include methylation [69,70], sumoylation [71], acetylation [72] and ubiquitination [73]. PTMs of RBPs have been shown to regulate phase separation and SG dynamics with dysregulation in ALS contributing to aggregation and SG dysfunction [70,74,75,76]. Furthermore, RBPs can undergo proteolytic cleavage which generates shorter protein chains, often with modified or new protein activities. Truncated species of TDP-43 can be generated through multiple proteolytic cleavage sites and although the exact function of these proteins remain unclear, they are generally thought to be toxic and are found in aggregates in ALS patients [77,78,79,80,81].
2.5. Deregulated RNA Metabolism
RBPs interact with a diverse range of RNAs. As a single RNA binding protein can bind to many thousands of RNA targets, a disturbance in one or more of these RBPs potentially has a broad and diverse impact on RNA metabolism [82,83,84]. Deregulated RNA metabolism has been described at many levels in ALS, including intron retention [14] and skipping of constitutive exons [85,86]. Loss of TDP-43 function has been shown to result in the downregulation of several transcripts including neuronal growth associated protein stathmin-2 [87]. Specific mutations in TDP-43 have been shown to result in additional splicing dysregulation, including gain of function effects [88].
Similarly to TDP-43, FUS has shown to regulate the stability of hundreds of transcripts [89] and many neuronal function-associated molecules have been identified to be regulated by FUS [83,90,91,92,93,94]. Therefore, loss of function of FUS has the potential to have a large impact in gene expression and/or alternative splicing. Both FUS and SFPQ more avidly bind to retained introns and are hypothesised to be transported out of the nucleus by intron-retaining transcripts [14]. Furthermore, ALS-causing mutant FUS has been shown to result in increased intron retention, with many intron retention events occurring in RBPs including FUS itself [15]. Intron retention and enhanced binding of mutant FUS to growth factor BDNF may impair its function [95]. ALS-causing MATR3 mutations have been shown to result in nuclear global mRNA export defects, including the mRNA of TDP-43 and FUS, demonstrating the interconnected network of RBPs and their involvement in ALS [96]. Furthermore, loss of the interaction between SFPQ and FUS has been identified in ALS-FTD [97].
Dysregulated RNA metabolism extends into the cytoplasm and neuronal processes, as RBPs are essential for mRNA transport and translation. TDP-43 ALS-linked mutants have been shown to have disrupted axonal transport dynamics in vitro and in vivo [98,99]. ALS-linked mutant FUS has been shown to drive toxicity through cytoplasmic gain of function effects such as inhibiting local intra-axonal protein translation and suppressing RNAs encoding proteins essential for synaptic function [100]. This study and others provide evidence for the role of RBPs in synaptic activity [101,102].
3. RNA Binding Proteins and Oxidative Stress in ALS
Oxidative stress is a process where accumulation of reactive oxygen species (ROS) leads to cellular damage and cell death due to an imbalance between free radical production and antioxidant defences. The presence of high levels of ROS causes damage to several different parts of the cellular machinery through lipid peroxidation and oxidation of proteins and/or DNA. Oxidative stress is thought to increase with age, a major risk factor in ALS [103]. Evidence of oxidative stress has been identified in CSF, plasma, serum and urine of sporadic ALS patients [104,105,106,107] and post-mortem ALS spinal cord tissue [108,109,110,111].
Interestingly, links have been made between oxidative stress and RBPs. Oxidative stress has been shown to cause PTMs of RBPs, including cysteine oxidation [112,113] and acetylation [72] of TDP-43, which both led to increased aggregation. Furthermore, oxidative stress induced by multiple experimental methods resulted in TDP-43 phosphorylation, mislocalisation and/or aggregation [114,115,116,117]. TIA1 has also been shown to undergo cysteine oxidation in response to increased ROS production, which subsequently suppressed SG formation and increased apoptosis [118]. Oxidative stress also provokes the recruitment of TDP-43 [119], SFPQ [120], hnRNPA1 [121], TIA1 [48] and ATXN2 [49] to SGs. Thus, oxidative stress driving RBPs to SGs may form a basis for later aggregation. Supporting this, increasing levels of protein oxidation resistance 1 (OXR1) reduced TDP-43 and FUS aggregation [122]. SFPQ has also been implicated in cellular responses to oxidative stress as it has been shown to regulate the transcription of stress response genes and knockdown of SFPQ reduced ROS production in sodium arsenite treated cells [123]. Furthermore paraspeckles, in which SFPQ and FUS are core components, have been shown to form upon recovery from arsenite induced oxidative stress [124]. RBP responses to oxidative stress are summarised in Figure 1.
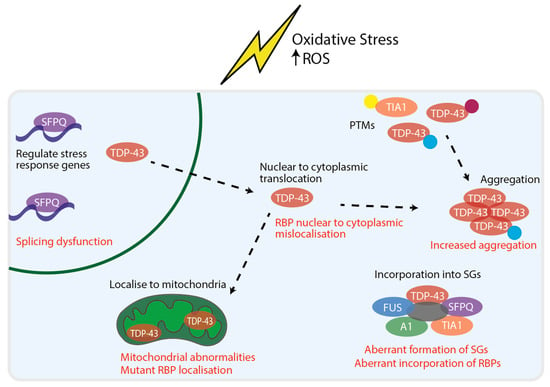
Figure 1.
RBP responses to oxidative stress and dysfunction in ALS. RBPs have been shown to respond to oxidative stress by multiple mechanisms. In the nucleus, SFPQ has been shown to regulate stress response genes. TDP-43 has been shown to undergo nuclear to cytoplasmic translocation. In the cytoplasm, oxidative stress has shown to cause PTMs of RBPs (represented by the yellow, magenta and blue dots). PTMs of RBPs have been associated with RBP aggregation. In addition, upon oxidative stress multiple RBPs are incorporated into SGs and TDP-43 has been shown to localise to mitochondria. The RBP response to oxidative stress overlaps with mechanisms of RBP dysfunction that have been implicated in ALS (displayed in red). This includes nuclear to cytoplasmic mislocalisation, aberrant PTMs, increased aggregation, perturbed stress granule dynamics and damage caused by RBP localisation to mitochondria.
Responses to oxidative stress have been shown to be perturbed in cellular models of ALS. Fibroblasts from ALS patients with TARDBP and C9ORF72 mutations have shown aberrant formation of SG and/or phospho-TDP-43 aggregates in response to chronic oxidative stress [116] and ALS-causing FUS mutations have been shown to result in the aberrant incorporation of FUS into SGs [47,125,126,127]. Mutant FUS expression also increases the presence of RBP, ELAVL4, in SGs formed in response to oxidative stress [128]. Furthermore, mutant TDP-43 expressing motor neurons exposed to oxidative stress displayed reduced formation of SGs, dysfunction of vesicle secretion, an altered protein interactome and reduced survival compared with wild type TDP-43 expressing motor neurons [129].
4. RNA Binding Proteins and Mitochondrial Dysfunction in ALS
Oxidative stress has been heavily associated with mitochondrial dysfunction in ALS. Mitochondria produce ROS as a by-product of energy production, leading to nitration of mitochondrial proteins, mtDNA damage and dysfunction of the mitochondrial respiratory chain. Abnormal morphology of mitochondria has been reported in sALS post-mortem tissue [130,131] and several ALS-linked mutant proteins have been found to localise to mitochondria in ALS models [132,133,134,135].
Several genes directly involved in mitochondrial function are implicated in ALS pathogenesis including SIGMAR1 and CHCHD10 [115,116]. Expression of mutant sigma-1 receptor promoted mitochondrial dysfunction and the mislocalisation of TDP-43 [136]. Interestingly, in this study, supplementation of mutant sigma-1 receptor expressing cells with ATP partially reduced cytoplasmic mislocalisation of TDP-43. Mutations in mitochondrial gene CHCHD10 have also been linked with ALS-FTD [137,138]. CHCHD10 knockin mutations resulted in mitochondrial damage and motor neuron degeneration [139]. Knockdown of CHCHD10 resulted in similar effects on mitochondrial function as expression of mutant CHCHD10, suggesting that mutations in CHCHD10 result in loss of function or dominant negative effects [140]. Interestingly, it was also shown in this study that CHCHD10 interacted with TDP-43 and mutations or depletion of CHCHD10 resulted in TDP-43 cytoplasmic mislocalisation in primary hippocampal neurons.
RBPs have important physiological roles in mitochondrial function. TDP-43 interacts with several mitochondrial proteins [141] and has been proposed to be involved in the stabilisation of mitochondrial transcripts [142]. shRNA knockdown of TDP-43 in primary mouse motor neurons reduced the expression of a number of mitochondrial transcripts, mitochondrial number and mitochondrial membrane potential [143]. Meanwhile, overexpression of wild type TDP-43 causes mitochondrial morphological abnormalities [29], affects mitochondrial dynamics [144] and disrupts mitochondrial-ER contacts [145]. Overexpression of wild type FUS also resulted in mitochondrial abnormalities [146]. Furthermore, hnRNPA1 and TIA1 have been shown to regulate mRNAs of proteins involved in mitochondria fission activity, with down regulation of hnRNPA1 and TIA1 both resulting in mitochondrial elongation or increased abundance and overexpression increasing mitochondrial fragmentation [147,148,149]. EWS has also been shown to have a role in mitochondrial and energy homeostasis, with mitochondrial abundance and activity significantly reduced in EWS-deficient mice [150]. Finally, SFPQ is involved in mitochondrial function, as loss of SFPQ caused decreased abundance of mitochondrial oxidative phosphorylation complexes [151] and SFPQ binds to transcripts of mitochondrially localised LaminB2 and Bcl2 adjacent to mitochondria [152].
ALS-linked mutant RBPs have been shown to localise to mitochondria in ALS and cause damage. Mutant TDP-43 has been associated with multiple mechanisms of mitochondrial dysfunction in neurodegeneration (fully reviewed in [153]). In an important recent study, mutant TDP-43 expression resulted in mitochondrial DNA release, which in turn triggered activation of an immune response through the cGAS-STING pathway [154]. TDP-43 has been found to localise to mitochondria in sALS motor neurons [134] and animal models of ALS [134,155]. Mutant TDP-43 localised to mitochondria at higher levels than expression of wild type TDP-43 and caused downregulation of complex I subunits [134,156]. Blocking TDP-43 import into mitochondria prevented complex I dysfunction and neuronal death in mutant TDP-43 mice [134,157]. However, findings of abnormal mitochondrial bioenergetics in mutant TDP-43 mice and patient fibroblasts could not be confirmed by another group [158]. Truncated forms of TDP-43 have been found to have a higher propensity to accumulate in the intermembrane space of mitochondria [159]. Interestingly, these truncated forms of TDP-43 were less damaging to mitochondria than full length mutant TDP-43, possibly due to the lack of truncated forms in the mitochondrial matrix.
In addition to TDP-43, mutant FUS has also caused damage to mitochondria [133]. Overexpression of mutant FUS led to increased association with mitochondrial transcripts and decreased mitochondrial membrane potential and respiration through a gain in toxic function mechanism [160]. Furthermore, an alternative open reading frame leads to another disease relevant form of FUS, which when overexpressed led to mitochondrial dysfunction [161]. Recent transcriptomic analysis of a FUS mutant mouse has also revealed dysregulation of mitochondrial transcripts [162]. In a more physiological knockin model of humanised mutant FUS, mislocalisation of FUS coincided with downregulated mitochondrial transcripts [163]. Furthermore, transgenic flies with ALS linked mutations in TDP-43, FUS and TAF15 showed mitochondrial fission defects in muscle and motor neurons [164]. Interestingly, this mitochondrial fragmentation could be rescued by the expression of dominant negative mutant form of DRP1, a protein responsible for coordinating mitochondrial dynamics, shown to be regulated by hnRNPA1 [147].
5. Targeting RNA Binding Proteins, Oxidative Stress and Mitochondrial Dysfunction in ALS
Currently there are only two FDA approved drugs for ALS: riluzole and edaravone, which both only have modest effects on disease progression. Riluzole, a glutamatergic neurotransmitter inhibitor which may reduce oxidative stress [165,166], only extends life (or time to tracheostomy) by 3 months [167]. Edaravone (approved in Japan, Korea, US, Canada and Switzerland), is an antioxidant drug in which the exact mechanism of action remains unknown and has been shown to cause a modest reduction in progression in early stage disease [168].
Many small molecules have been shown to reduce the cytoplasmic accumulation and aggregation of TDP-43 and FUS, targeting autophagy and SG pathways [169,170,171]. Small molecules have also been identified to target the RBPs themselves. The RNA recognition motif 1 (RRM1) domain of TDP-43 has shown to be a druggable site, with a small molecule able to reduce RNA binding and improve neuromuscular strength in an ALS drosophila model [172]. Supporting this as a possible therapeutic target, an antibody targeting the RRM1 domain has shown to reduce TDP-43 proteinopathy, cognitive impairment, motor defects and neuroinflammation in a TDP-43 ALS mouse model [173]. Targeting RBPs extends beyond TDP-43, with an ASO targeting ataxin-2 shown to reduce TDP-43 aggregation, improve motor function and improve survival in a TDP-43 transgenic mouse model [174].
Targeting RBP localisation to mitochondria may also be a potentially effective therapeutic target. Peptides designed to block the mitochondrial entry of TDP-43 prevented mitochondrial dysfunction and rescued motor neuron death, neuromuscular junction denervation and motor dysfunction in a mutant TDP-43 overexpressing mouse [134,157].
A number of therapies targeting mitochondria and/or oxidative stress have provided benefits in animal models but have performed disappointingly in clinical trials, such as Vitamin E [175,176], N-acetyl-L-cysteine (NAC) [177,178], Coenzyme Q10 [179,180] and Dexpramipexole (RPPX) [181,182]. This may highlight the lack of translatability and overreliance on animal models, especially the mutant SOD1 mouse model. Despite these failures there are a few ongoing clinical trials that are testing antioxidants. This includes a phase II clinical trial for a compound that targets NAD+, a coenzyme involved in redox reactions in cells, accompanied with the use of over the counter antioxidants (NCT04244630).
6. Conclusions
In this review, we have summarised potential mechanisms of RBP dysfunction in ALS and reviewed the emerging link between RBP dysfunction and both oxidative stress and mitochondrial dysfunction in this context. Although significant overlap has been observed in these pathomechanisms, it is likely that polytherapy targeting multiple pathomechanisms will be required to elicit more efficacious therapeutic benefits. It is important to consider that targeting certain pathways may be more responsive depending on the particular disease phase and thus it is important to resolve the primacy of molecular and cellular pathological events. Further work dissecting these mechanisms may guide the development of new therapies, which are desperately required in this relentlessly progressive and invariably fatal disease.
Funding
This work was supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC010110), the UK Medical Research Council (FC010110), and the Wellcome Trust (FC010110). R.P. holds an MRC Senior Clinical Fellowship [MR/S006591/1].
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The funders had no role in the writing of this manuscript.
References
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Luisier, R.; Taha, D.M.; Neeves, J.; Modic, M.; Mitchell, J.S.; Meyer, I.; Greensmith, L.; Newcombe, J.; Ule, J.; et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 2019, 142, 2572–2580. [Google Scholar] [CrossRef]
- Honda, H.; Hamasaki, H.; Wakamiya, T.; Koyama, S.; Suzuki, S.O.; Fujii, N.; Iwaki, T. Loss of hnRNPA1 in ALS spinal cord motor neurons with TDP-43-positive inclusions. Neuropathology 2015, 35, 37–43. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat. Commun. 2018, 9, 2010. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.; Birsa, N.; Milioto, C.; McLaughlin, M.; Ule, A.M.; Robaldo, D.; Eberle, A.B.; Kräuchi, R.; Bentham, M.; Brown, A.-L.; et al. FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucleic Acids Res. 2020, 48, 6889–6905. [Google Scholar] [CrossRef]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef]
- Bello, M.L.; Di Fini, F.; Notaro, A.; Spataro, R.; Conforti, F.L.; La Bella, V. ALS-related mutant FUS protein is Mislocalized to cytoplasm and is recruited into stress granules of fibroblasts from asymptomatic FUS P525L mutation carriers. Neurodegener. Dis. 2017, 17, 292–303. [Google Scholar] [CrossRef]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 2011, 32, 2323.e27–2323.e40. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 285–290. [Google Scholar] [CrossRef]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.-J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef]
- Harley, J.; Hagemann, C.; Serio, A.; Patani, R. FUS is lost from nuclei and gained in neurites of motor neurons in a human stem cell model of VCP-related ALS. Brain 2020, 143, e103. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Van Damme, P.; Van Den Bosch, L. Inside out: The role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016, 132, 159–173. [Google Scholar] [CrossRef] [PubMed]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; Wei, Y.; Lu, Y.; Song, J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016, 14, e1002338. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Hirsch-Reinshagen, V.; Pottier, C.; Nicholson, A.M.; Baker, M.; Hsiung, G.-Y.R.; Krieger, C.; Sengdy, P.; Boylan, K.B.; Dickson, D.W.; Mesulam, M.; et al. Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol. Commun. 2017, 5, 96. [Google Scholar] [CrossRef]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.-R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef]
- Bajc Česnik, A.; Darovic, S.; Prpar Mihevc, S.; Štalekar, M.; Malnar, M.; Motaln, H.; Lee, Y.-B.; Mazej, J.; Pohleven, J.; Grosch, M.; et al. Nuclear RNA foci from expansion mutation form paraspeckle-like bodies. J. Cell Sci. 2019, 132, jcs224303. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Velde, C.V. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M.; et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, J.; De Santis, R.; de Turris, V.; Morlando, M.; Laneve, P.; Calvo, A.; Caliendo, V.; Chiò, A.; Rosa, A.; Bozzoni, I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell-derived motoneurons. Dis. Model. Mech. 2015, 8, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef]
- Kaehler, C.; Isensee, J.; Nonhoff, U.; Terrey, M.; Hucho, T.; Lehrach, H.; Krobitsch, S. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS ONE 2012, 7, e50134. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.A.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef]
- Fan, A.C.; Leung, A.K.L. RNA Granules and Diseases: A Case Study of Stress Granules in ALS and FTLD. Adv. Exp. Med. Biol. 2016, 907, 263–296. [Google Scholar]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fan, B.; Yang, P.; Temirov, J.; Messing, J.; Kim, H.J.; Taylor, J.P. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. eLife 2019, 8, e39578. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Manferrari, G.; Newcombe, J.; Luscombe, N.M.; Luisier, R.; Patani, R. Paraspeckle components NONO and PSPC1 are not mislocalized from motor neuron nuclei in sporadic ALS. Brain 2020, 143, e66. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.F.; Goshima, N.; Hirose, T. Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 2012, 31, 4020–4034. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2014, 23, 2298–2312. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Kukharsky, M.S.; An, H.; Dimasi, P.; Alexeeva, S.; Shabir, O.; Heath, P.R.; Buchman, V.L. Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol. Neurodegener. 2018, 13, 30. [Google Scholar] [CrossRef]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 2019, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.-I.; Imai, T.; Murayama, S.; et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Yeh, P.-A.; Chiu, H.-C.; Tang, C.-Y.; Tu, B.P.-H. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Rhoads, S.; Monahan, Z.; Yee, D.; Shewmaker, F. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.A.; Neumann, M.; et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.-D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef]
- Maurel, C.; Chami, A.A.; Thépault, R.-A.; Marouillat, S.; Blasco, H.; Corcia, P.; Andres, C.R.; Vourc’h, P. A role for SUMOylation in the Formation and Cellular Localization of TDP-43 Aggregates in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2020, 57, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.-X.; Trojanowski, J.Q.; Lee, V.M.Y. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [PubMed]
- Alexander, E.J.; Ghanbari Niaki, A.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891.e6. [Google Scholar] [CrossRef]
- Wall, M.L.; Lewis, S.M. Methylarginines within the RGG-Motif Region of hnRNP A1 Affect Its IRES Trans-Acting Factor Activity and Are Required for hnRNP A1 Stress Granule Localization and Formation. J. Mol. Biol. 2017, 429, 295–307. [Google Scholar] [CrossRef]
- Kaehler, C.; Guenther, A.; Uhlich, A.; Krobitsch, S. PRMT1-mediated arginine methylation controls ATXN2L localization. Exp. Cell Res. 2015, 334, 114–125. [Google Scholar] [CrossRef]
- Li, Q.; Yokoshi, M.; Okada, H.; Kawahara, Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat. Commun. 2015, 6, 6183. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.-Y. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Lee, K.; Matsuoka, M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J. Biol. Chem. 2011, 286, 13171–13183. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, D.; Campagne, S.; Schmidt, R.; Reber, S.; Mechtersheimer, J.; Gypas, F.; Schweingruber, C.; Colombo, M.; von Schroetter, C.; Loughlin, F.E.; et al. Aberrant interaction of FUS with the U1 snRNA provides a molecular mechanism of FUS induced amyotrophic lateral sclerosis. Nat. Commun. 2020, 11, 6341. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef]
- Humphrey, J.; Emmett, W.; Fratta, P.; Isaacs, A.M.; Plagnol, V. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med. Genom. 2017, 10, 38. [Google Scholar] [CrossRef]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, e98684. [Google Scholar] [CrossRef]
- Kapeli, K.; Pratt, G.A.; Vu, A.Q.; Hutt, K.R.; Martinez, F.J.; Sundararaman, B.; Batra, R.; Freese, P.; Lambert, N.J.; Huelga, S.C.; et al. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun. 2016, 7, 12143. [Google Scholar] [CrossRef]
- Ishigaki, S.; Masuda, A.; Fujioka, Y.; Iguchi, Y.; Katsuno, M.; Shibata, A.; Urano, F.; Sobue, G.; Ohno, K. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2012, 2, 529. [Google Scholar] [CrossRef]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef]
- Honda, D.; Ishigaki, S.; Iguchi, Y.; Fujioka, Y.; Udagawa, T.; Masuda, A.; Ohno, K.; Katsuno, M.; Sobue, G. The ALS/FTLD-related RNA-binding proteins TDP-43 and FUS have common downstream RNA targets in cortical neurons. FEBS Open Bio 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Fujioka, Y.; Ishigaki, S.; Masuda, A.; Iguchi, Y.; Udagawa, T.; Watanabe, H.; Katsuno, M.; Ohno, K.; Sobue, G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci. Rep. 2013, 3, 2388. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Alexiou, P.; Maragkakis, M.; Chang, A.; Mourelatos, Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA 2013, 19, 498–509. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.-Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Boehringer, A.; Garcia-Mansfield, K.; Singh, G.; Bakkar, N.; Pirrotte, P.; Bowser, R. ALS Associated Mutations in Matrin 3 Alter Protein-Protein Interactions and Impede mRNA Nuclear Export. Sci. Rep. 2017, 7, 14529. [Google Scholar] [CrossRef]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L.F. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–E2475. [Google Scholar] [CrossRef]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2020, 106, 354. [Google Scholar] [CrossRef]
- Schoen, M.; Reichel, J.M.; Demestre, M.; Putz, S.; Deshpande, D.; Proepper, C.; Liebau, S.; Schmeisser, M.J.; Ludolph, A.C.; Michaelis, J.; et al. Super-Resolution Microscopy Reveals Presynaptic Localization of the ALS/FTD Related Protein FUS in Hippocampal Neurons. Front. Cell. Neurosci. 2015, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Neubert, T.A.; Jordan, B.A. RNA binding proteins accumulate at the postsynaptic density with synaptic activity. J. Neurosci. 2012, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef]
- Abe, K.; Pan, L.H.; Watanabe, M.; Kato, T.; Itoyama, Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci. Lett. 1995, 199, 152–154. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M.Y. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef]
- Chang, C.-K.; Chiang, M.-H.; Toh, E.K.-W.; Chang, C.-F.; Huang, T.-H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.-I.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.; Patani, R. Stress-Specific Spatiotemporal Responses of RNA-Binding Proteins in Human Stem-Cell-Derived Motor Neurons. Int. J. Mol. Sci. 2020, 21, 8346. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef]
- Meyerowitz, J.; Parker, S.J.; Vella, L.J.; Ng, D.C.; Price, K.A.; Liddell, J.R.; Caragounis, A.; Li, Q.-X.; Masters, C.L.; Nonaka, T.; et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener. 2011, 6, 57. [Google Scholar] [CrossRef]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef]
- Guil, S.; Long, J.C.; Cáceres, J.F. hnRNP A1 Relocalization to the Stress Granules Reflects a Role in the Stress Response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef]
- Finelli, M.J.; Liu, K.X.; Wu, Y.; Oliver, P.L.; Davies, K.E. Oxr1 improves pathogenic cellular features of ALS-associated FUS and TDP-43 mutations. Hum. Mol. Genet. 2015, 24, 3529–3544. [Google Scholar] [CrossRef]
- Guo, P.; Chen, S.; Li, D.; Zhang, J.; Luo, J.; Zhang, A.; Yu, D.; Bloom, M.S.; Chen, L.; Chen, W. SFPQ is involved in regulating arsenic-induced oxidative stress by interacting with the miRNA-induced silencing complexes. Environ. Pollut. 2020, 261, 114160. [Google Scholar] [CrossRef]
- An, H.; Tan, J.T.; Shelkovnikova, T.A. Stress granules regulate stress-induced paraspeckle assembly. J. Cell Biol. 2019, 218, 4127–4140. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Physiol. 2013, 228, 2222–2231. [Google Scholar] [CrossRef]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.K.; Chian, R.-J.; Boggio, K.J.; Quaresma, A.J.C.; Nickerson, J.A.; Bosco, D.A. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef]
- Svetoni, F.; Caporossi, D.; Paronetto, M.P. Oxidative stress affects FET proteins localization and alternative pre-mRNA processing in cellular models of ALS. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S51. [Google Scholar] [CrossRef]
- De Santis, R.; Alfano, V.; de Turris, V.; Colantoni, A.; Santini, L.; Garone, M.G.; Antonacci, G.; Peruzzi, G.; Sudria-Lopez, E.; Wyler, E.; et al. Mutant FUS and ELAVL4 (HuD) Aberrant Crosstalk in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 27, 3818–3831.e5. [Google Scholar] [CrossRef]
- Feneberg, E.; Charles, P.D.; Finelli, M.J.; Scott, C.; Kessler, B.M.; Fischer, R.; Ansorge, O.; Gray, E.; Talbot, K.; Turner, M.R. Detection and quantification of novel C-terminal TDP-43 fragments in ALS-TDP. Brain Pathol. 2020, e12923. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Jaarsma, D.; Rognoni, F.; van Duijn, W.; Verspaget, H.W.; Haasdijk, E.D.; Holstege, J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001, 102, 293–305. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, H.; Shinoda, Y.; Shioda, N.; Fukunaga, K. Methyl pyruvate rescues mitochondrial damage caused by SIGMAR1 mutation related to amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1840, 3320–3334. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chiò, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 2014, 137, e311. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.C.; Madji Hounoum, B.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10 mouse. Acta Neuropathol. 2019, 138, 123–145. [Google Scholar] [CrossRef]
- Woo, J.-A.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef] [PubMed]
- Briese, M.; Saal-Bauernschubert, L.; Lüningschrör, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, H.; Jo, D.S.; Jo, Y.K.; Shin, J.H.; Kim, H.B.; Seo, H.M.; Rubinsztein, D.C.; Koh, J.-Y.; Lee, E.K.; et al. Heterogeneous nuclear ribonucleoprotein A1 post-transcriptionally regulates Drp1 expression in neuroblastoma cells. Biochim. Biophys. Acta 2015, 1849, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Tak, H.; Eun, J.W.; Kim, J.; Park, S.J.; Kim, C.; Ji, E.; Lee, H.; Kang, H.; Cho, D.-H.; Lee, K.; et al. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ. 2017, 24, 49–58. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, C.; Izquierdo, J.M. T-cell intracellular antigen (TIA)-proteins deficiency in murine embryonic fibroblasts alters cell cycle progression and induces autophagy. PLoS ONE 2013, 8, e75127. [Google Scholar] [CrossRef]
- Park, J.H.; Kang, H.-J.; Lee, Y.K.; Kang, H.; Kim, J.; Chung, J.H.; Chang, J.S.; McPherron, A.C.; Lee, S.B. Inactivation of EWS reduces PGC-1α protein stability and mitochondrial homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 6074–6079. [Google Scholar] [CrossRef]
- Hosokawa, M.; Takeuchi, A.; Tanihata, J.; Iida, K.; Takeda, S.; Hagiwara, M. Loss of RNA-Binding Protein Sfpq Causes Long-Gene Transcriptopathy in Skeletal Muscle and Severe Muscle Mass Reduction with Metabolic Myopathy. iScience 2019, 13, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Cosker, K.E.; Fenstermacher, S.J.; Pazyra-Murphy, M.F.; Elliott, H.L.; Segal, R.A. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016, 19, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Yin, H.Z.; Nalbandian, A.; Hsu, C.-I.; Li, S.; Llewellyn, K.J.; Mozaffar, T.; Kimonis, V.E.; Weiss, J.H. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012, 3, e374. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Duan, W.; Guo, Y.; Jiang, H.; Li, Z.; Huang, J.; Hong, K.; Li, C. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res. Bull. 2012, 89, 185–190. [Google Scholar] [CrossRef]
- Wang, P.; Wander, C.M.; Yuan, C.-X.; Bereman, M.S.; Cohen, T.J. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef]
- Kawamata, H.; Peixoto, P.; Konrad, C.; Palomo, G.; Bredvik, K.; Gerges, M.; Valsecchi, F.; Petrucelli, L.; Ravits, J.M.; Starkov, A.; et al. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol. Neurodegener. 2017, 12, 37. [Google Scholar] [CrossRef]
- Salvatori, I.; Ferri, A.; Scaricamazza, S.; Giovannelli, I.; Serrano, A.; Rossi, S.; D’Ambrosi, N.; Cozzolino, M.; Giulio, A.D.; Moreno, S.; et al. Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 2018, 146, 585–597. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.A.; Jacques, J.-F.; Nassari, S.; Tyzack, G.E.; McGoldrick, P.; Zinman, L.; Jean, S.; Robertson, J.; Patani, R.; Roucou, X. The FUS gene is dual-coding with both proteins contributing to FUS-mediated toxicity. EMBO Rep. 2020, 22, e50640. [Google Scholar]
- Ho, W.Y.; Agrawal, I.; Tyan, S.-H.; Sanford, E.; Chang, W.-T.; Lim, K.; Ong, J.; Tan, B.S.Y.; Moe, A.A.K.; Yu, R.; et al. Dysfunction in nonsense-mediated decay, protein homeostasis, mitochondrial function, and brain connectivity in ALS-FUS mice with cognitive deficits. Acta Neuropathol. Commun. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Devoy, A.; Kalmar, B.; Stewart, M.; Park, H.; Burke, B.; Noy, S.J.; Redhead, Y.; Humphrey, J.; Lo, K.; Jaeger, J.; et al. Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in “FUSDelta14” knockin mice. Brain 2017, 140, 2797–2805. [Google Scholar] [CrossRef] [PubMed]
- Altanbyek, V.; Cha, S.-J.; Kang, G.-U.; Im, D.S.; Lee, S.; Kim, H.-J.; Kim, K. Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2016, 481, 259–264. [Google Scholar] [CrossRef]
- Koh, J.Y.; Kim, D.K.; Hwang, J.Y.; Kim, Y.H.; Seo, J.H. Antioxidative and proapoptotic effects of riluzole on cultured cortical neurons. J. Neurochem. 1999, 72, 716–723. [Google Scholar] [CrossRef]
- Deng, Y.; Xu, Z.-F.; Liu, W.; Xu, B.; Yang, H.-B.; Wei, Y.-G. Riluzole-triggered GSH synthesis via activation of glutamate transporters to antagonize methylmercury-induced oxidative stress in rat cerebral cortex. Oxid. Med. Cell. Longev. 2012, 2012, 534705. [Google Scholar] [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 61–70. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717.e9. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef]
- François-Moutal, L.; Felemban, R.; Scott, D.D.; Sayegh, M.R.; Miranda, V.G.; Perez-Miller, S.; Khanna, R.; Gokhale, V.; Zarnescu, D.C.; Khanna, M. Small Molecule Targeting TDP-43′s RNA Recognition Motifs Reduces Locomotor Defects in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS). ACS Chem. Biol. 2019, 14, 2006–2013. [Google Scholar] [CrossRef]
- Pozzi, S.; Thammisetty, S.S.; Codron, P.; Rahimian, R.; Plourde, K.V.; Soucy, G.; Bareil, C.; Phaneuf, D.; Kriz, J.; Gravel, C.; et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J. Clin. Investig. 2019, 129, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, D.F.; Outeiro, T.F.; Bellucci, A.; Brundin, P. Protein Misfolding and Spreading Pathology in Neurodegenerative Diseases; Frontiers Media SA: Lausanne, Switzerland, 2020; ISBN 9782889635078. [Google Scholar]
- Gurney, M.E.; Cutting, F.B.; Zhai, P.; Doble, A.; Taylor, C.P.; Andrus, P.K.; Hall, E.D. Benefit of vitamin E, riluzole, and gababapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 147–157. [Google Scholar] [CrossRef]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A double-blind, placebo-controlled randomized clinical trial of α-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 9–18. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Flint Beal, M.; Bush, A.I. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. NeuroReport 2000, 11, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.E.; de Jong, J.M. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Kaufmann, P.; Thompson, J.L.P.; Levy, G.; Buchsbaum, R.; Shefner, J.; Krivickas, L.S.; Katz, J.; Rollins, Y.; Barohn, R.J.; Jackson, C.E.; et al. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 2009, 66, 235–244. [Google Scholar] [CrossRef]
- Danzeisen, R.; Schwalenstoecker, B.; Gillardon, F.; Buerger, E.; Krzykalla, V.; Klinder, K.; Schild, L.; Hengerer, B.; Ludolph, A.C.; Dorner-Ciossek, C.; et al. Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride]. J. Pharmacol. Exp. Ther. 2006, 316, 189–199. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; van den Berg, L.H.; Shefner, J.M.; Mitsumoto, H.; Mora, J.S.; Ludolph, A.; Hardiman, O.; Bozik, M.E.; Ingersoll, E.W.; Archibald, D.; et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): A randomised, double-blind, phase 3 trial. Lancet Neurol. 2013, 12, 1059–1067. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).