
Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Cell Irradiation
2.3. Cell Sorting
- B-lymphocytes (Po1): Lin− CD45+ CD34− CD19+;
- nuclear non-specify lineage negative cells (Po2): CD45+ Lin− CD34− CD19−;
- HSPCs (Po3): Lin− CD45+ CD34+ CD19−;
- Pre-Pro B cells (Po4): Lin− CD45+ CD34+ CD19+;
- progenitors (Po5): Lin− CD45+ CD45RA− CD34+ CD38+;
- hematopoietic stem cells/multipotent progenitors (HSCs/MPPs) (Po6): Lin− CD45+ CD45RA− CD34+ CD38−;
- HSCs (Po7): Lin− CD45+ CD45RA− CD34+ CD38− CD90+; and,
- MPPs (Po8): Lin− CD45+ CD45RA− CD34+ CD38− CD90−.
2.4. Cell Expansion
2.5. RNA Isolation and cDNA Synthesis
2.6. Real Time Quantitative PCR
2.7. Validation of PFG Positive RT-qPCR Product by Standard PCR and Sequencing
2.8. Evaluation of DNA DSBs and γH2AX Pan-Staining by Imaging Flow Cytometry
2.9. ROS
2.10. Quantification and Statistical Analysis
3. Results
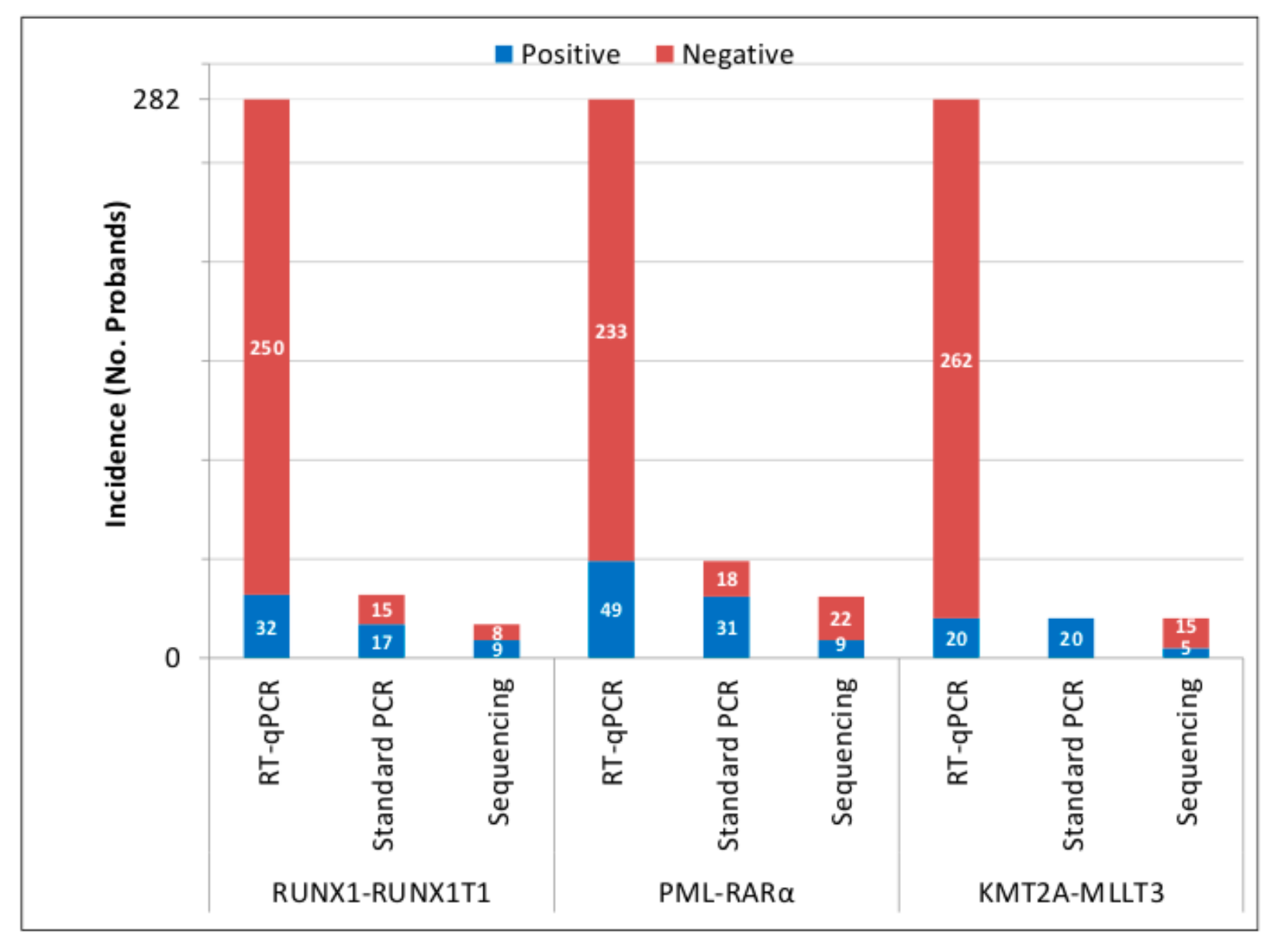
3.1. Presence of AML PFGs in UCB of Newborns
3.2. Inducibility of AML PFGs in Different Subsets of HSPCs by 50 cGy of γ-rays
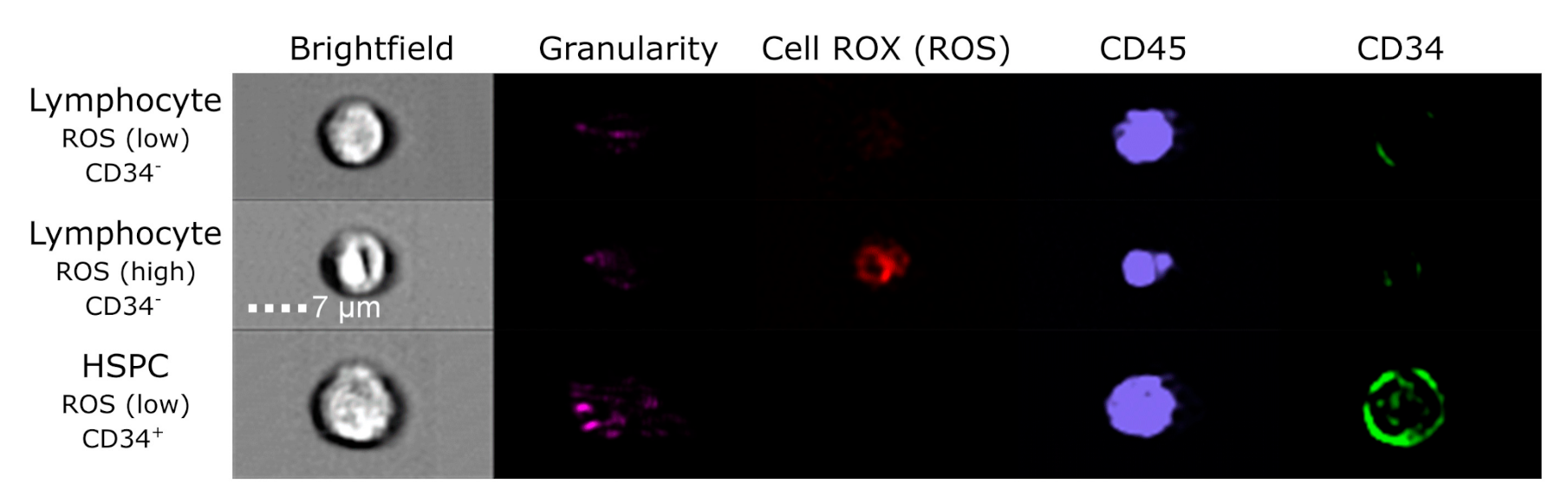
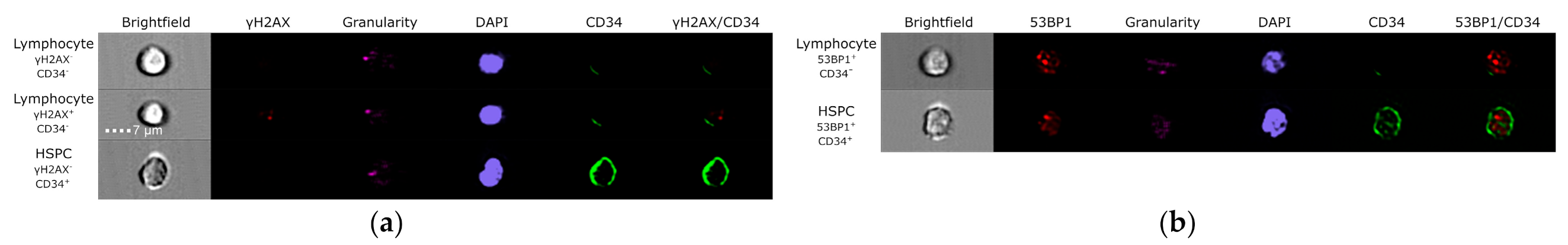
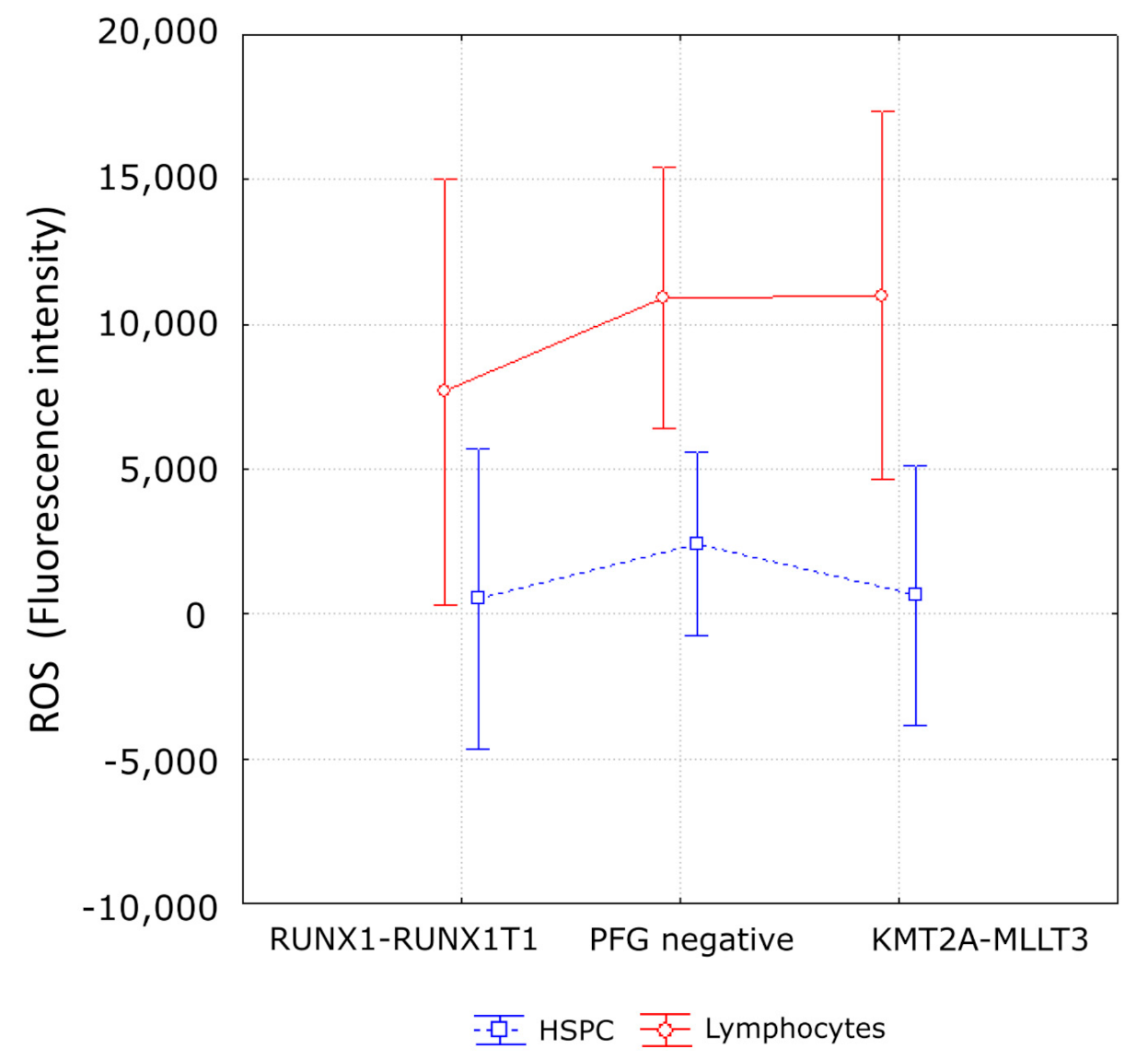
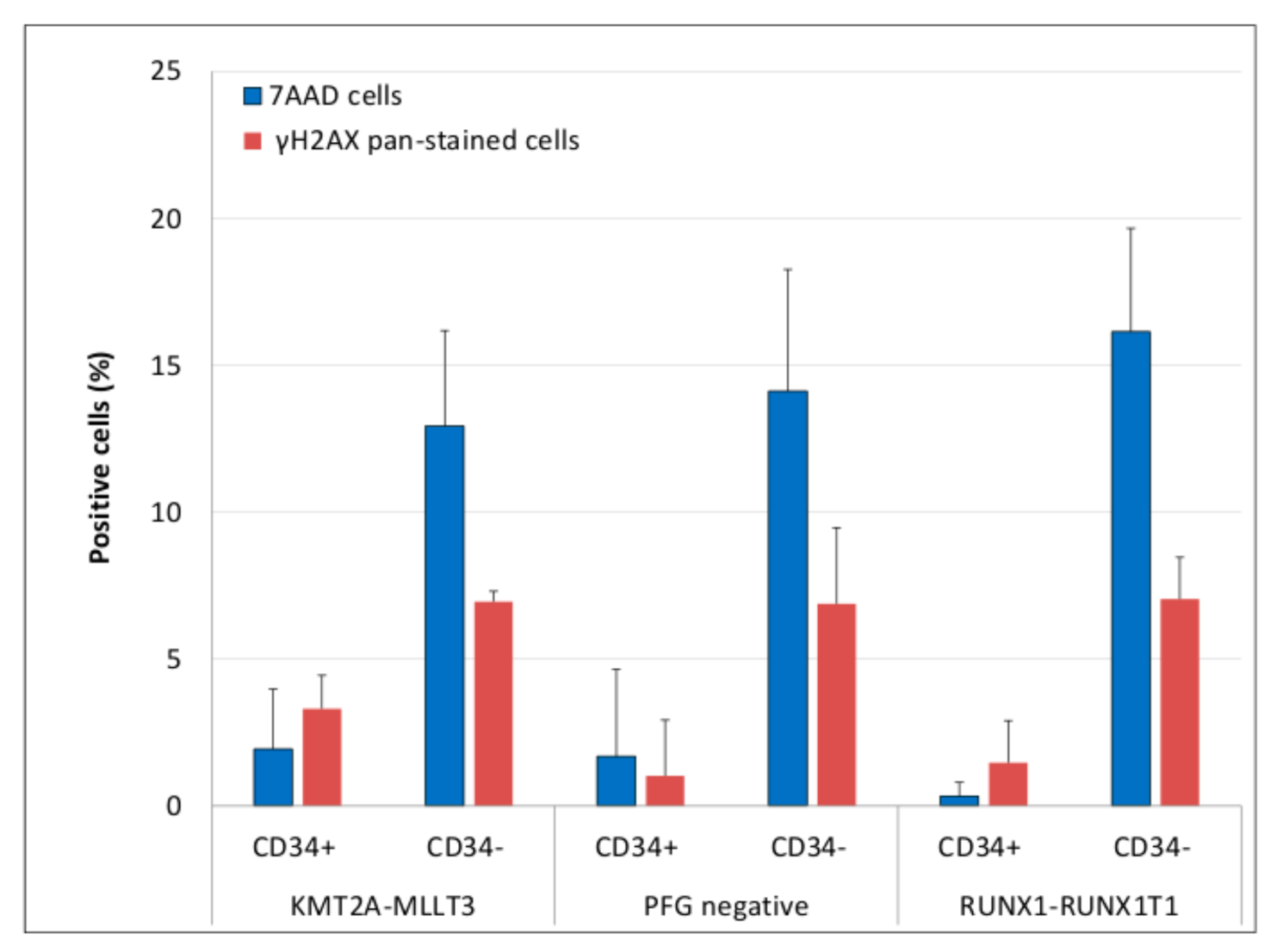
3.3. Evaluation of DNA Damage, Apoptosis and ROS in PFG Positive and Matched Negative Samples by Imaging Flow Cytometry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA A Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Greaves, M. Author Correction: A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 2018, 18, 526. [Google Scholar] [CrossRef]
- Creutzig, U.; Heuvel-Eibrink, M.M.V.D.; Gibson, B.; Dworzak, M.N.; Adachi, S.; De Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: Recommendations from an international expert panel. Blood 2012, 120, 3187–3205. [Google Scholar] [CrossRef]
- Hein, D.; Borkhardt, A.; Fischer, U. Insights into the prenatal origin of childhood acute lymphoblastic leukemia. Cancer Metastasis Rev. 2020, 39, 161–171. [Google Scholar] [CrossRef]
- Kosik, P.; Skorvaga, M.; Belyaev, I. Incidence of preleukemic fusion genes in healthy subjects. Neoplasma 2016, 63, 659–672. [Google Scholar] [CrossRef]
- Kosik, P.; Skorvaga, M.; Durdik, M.; Jakl, L.; Nikitina, E.; Markova, E.; Kozics, K.; Horvathova, E.; Belyaev, I. Low numbers of pre-leukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget 2017, 8, 35824–35834. [Google Scholar] [CrossRef]
- Schäfer, D.; Olsen, M.; Lähnemann, D.; Stanulla, M.; Slany, R.; Schmiegelow, K.; Borkhardt, A.; Fischer, U. Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 2018, 131, 821–826. [Google Scholar] [CrossRef]
- Ornelles, D.A.; Gooding, L.R.; Garnett-Benson, C. Neonatal Infection with Species C Adenoviruses Confirmed in Viable Cord Blood Lymphocytes. PLoS ONE 2015, 10, e0119256. [Google Scholar] [CrossRef]
- Mori, H.; Colman, S.M.; Xiao, Z.; Ford, A.M.; Healy, L.E.; Donaldson, C.; Hows, J.M.; Navarrete, C.; Greaves, M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA 2002, 99, 8242–8247. [Google Scholar] [CrossRef]
- Zuna, J.; Prouzová, Z.; Kalina, T.; Lizcova, L.; Zemanova, Z.; Muzikova, K.; Rahmatová, Š.; Meijerink, J.P.; Trka, J. Backtracking of ALL to cord blood. Leuk. Res. 2009, 33, e107–e108. [Google Scholar] [CrossRef]
- Song, J.; Mercer, D.; Hu, X.; Liu, H.; Li, M.M. Common Leukemia- and lymphoma-associated genetic aberrations in healthy individuals. J. Mol. Diagn. 2011, 13, 213–219. [Google Scholar] [CrossRef]
- Basecke, J.; Cepek, L.; Mannhalter, C.; Krauter, J.; Hildenhagen, S.; Brittinger, G.; Trumper, L.; Griesinger, F. Transcription of AML1/ETO in bone marrow and cord blood of individuals without acute myelogenous leukemia. Blood 2002, 100, 2267. [Google Scholar] [CrossRef]
- National Cancer Institute. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 17 March 2021).
- Škorvaga, M.; Nikitina, E.; Kubeš, M.; Košík, P.; Gajdošechová, B.; Leitnerová, M.; Copáková, L.; Belyaev, I. Incidence of common preleukemic gene fusions in umbilical cord blood in Slovak population. PLoS ONE 2014, 9, e91116. [Google Scholar] [CrossRef] [PubMed]
- Jakl, L.; Skorvaga, M.; Beresova, K.; Kosik, P.; Durdik, M.; Jakubikova, J.; Holop, M.; Kubes, M.; Zastko, L.; Markova, E.; et al. BCR/ABL preleukemic fusion gene in subpopulations of hematopoietic stem and progenitor cells from human UCB. Neoplasma 2020, 67, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Jawdat, D.; Almashaqbeh, W.; Sumaily, A.; Albaloushi, N.; Jammah, S.; Alsultan, A. Screening for pre-leukemia TEL-AML1 chromosomal translocation in banked cord blood units: Cord blood bank perspective. Cell Tissue Bank. 2020, 21, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, C.; Head, D.R.; Mirro, J., Jr.; Behm, F.G.; Ayers, G.D.; Raimondi, S.C. Translocation t(9;11)(p21;q23) in pediatric de novo and secondary acute myeloblastic leukemia. Leukemia 1992, 6, 513–519. [Google Scholar] [PubMed]
- Dang, D.N.; Morris, H.D.; Feusner, J.H.; Koduru, P.; Wilson, K.; Timmons, C.F.; Cavalier, M.; Luu, H.S. Therapy-induced Secondary Acute Myeloid Leukemia With t(11;19)(q23;p13.1) in a pediatric patient with relapsed acute promyelocytic leukemia. J. Pediatr. Hematol. 2014, 36, e546–e548. [Google Scholar] [CrossRef] [PubMed]
- Sulkes, A.; Ruchlemer, R.; Ben Neria, S.; Leizerowitz, R.; Abeliovich, D.; Polliack, A.; Neria, S.B. Acute Monoblastic Leukemia with t(9;11) in a patient receiving chemotherapy for ovarian cancer: Secondary leukemia or fortuitous association of two neoplasias? Leuk. Lymphoma 1991, 4, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kühn, M.W.; Bullinger, L.; Gröschel, S.; Krönke, J.; Edelmann, J.; Rücker, F.G.; Eiwen, K.; Paschka, P.; Gaidzik, V.I.; Holzmann, K.; et al. Genome-wide genotyping of acute myeloid leukemia with translocation t(9;11)(p22;q23) reveals novel recurrent genomic alterations. Hematologica 2014, 99, e133–e135. [Google Scholar] [CrossRef]
- Lesesve, J.F.; Schneider, P.; Dolgopolov, I.; Bastard, C.; Lenormand, B.; Cambon-Michot, E.; Callat, M.P.; Cavelier, B.; Tron, P.H.; Vannier, J.P. Therapy-related acute myeloid leukemia with t(8;21) in a child with previous Ewing’s sarcoma. Med. Pediatr. Oncol. 1997, 29, 132–134. [Google Scholar] [CrossRef]
- Hasan, S.K.; Mays, A.N.; Ottone, T.; Ledda, A.; La Nasa, G.; Cattaneo, C.; Borlenghi, E.; Melillo, L.; Montefusco, E.; Cervera, J.; et al. Molecular analysis of t(15;17) genomic breakpoints in secondary acute promyelocytic leukemia arising after treatment of multiple sclerosis. Blood 2008, 112, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.; Bose, S.; Gora-Tybor, J.; Yan, X.H.; Goldman, J.M.; Melo, J.V. Selective induction of leukemia-associated fusion genes by high-dose ionizing radiation. Cancer Res. 1998, 58, 421–425. [Google Scholar]
- Ito, T.; Seyama, T.; Mizuno, T.; Hayashi, T.; Iwamoto, K.S.; Dohi, K.; Nakamura, N.; Akiyama, M. Induction of BCR-ABL Fusion Genes byin vitroX-irradiation. Jpn. J. Cancer Res. 1993, 84, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Fazel, R.; Krumholz, H.M.; Wang, Y.; Ross, J.S.; Chen, J.; Ting, H.H.; Shah, N.D.; Nasir, K.; Einstein, A.J.; Nallamothu, B.K. Exposure to low-dose ionizing radiation from medical imaging procedures. N. Engl. J. Med. 2009, 361, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-L.; Preston, D.L.; Soda, M.; Sugiyama, H.; Funamoto, S.; Kodama, K.; Kimura, A.; Kamada, N.; Dohy, H.; Tomonaga, M.; et al. The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950–2001. Radiat. Res. 2013, 179, 361–382. [Google Scholar] [CrossRef]
- Bailey, H.D.; Armstrong, B.K.; De Klerk, N.H.; Fritschi, L.; Attia, J.R.; Lockwood, L.; Milne, E. Exposure to diagnostic radiological procedures and the risk of childhood acute lymphoblastic leukemia. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2897–2909. [Google Scholar] [CrossRef]
- Pearce, M.S.; Salotti, J.A.; Little, M.P.; McHugh, K.; Lee, C.; Kim, K.P.; Howe, N.L.; Ronckers, C.M.; Rajaraman, P.; Craft, A.W.; et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet 2012, 380, 499–505. [Google Scholar] [CrossRef]
- Cohen, S.; Liu, A.; Gurvitz, M.; Guo, L.; Therrien, J.; Laprise, C.; Kaufman, J.S.; Abrahamowicz, M.; Marelli, A.J. Exposure to low-dose ionizing radiation from cardiac procedures and malignancy risk in adults with congenital heart disease. Circulation 2018, 137, 1334–1345. [Google Scholar] [CrossRef]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive Oxygen Species (ROS)––Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Kantner, H.-P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B Cells. Neoplasia 2013, 15, 1292-IN28. [Google Scholar] [CrossRef]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species–dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.A.; Carroll, M. BCR-ABL: A multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia 2010, 24, 1105–1112. [Google Scholar] [CrossRef]
- Alcalay, M.; Meani, N.; Gelmetti, V.; Fantozzi, A.; Fagioli, M.; Orleth, A.; Riganelli, D.; Sebastiani, C.; Cappelli, E.; Casciari, C.; et al. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J. Clin. Investig. 2003, 112, 1751–1761. [Google Scholar] [CrossRef]
- Krejci, O.; Wunderlich, M.; Geiger, H.; Chou, F.-S.; Schleimer, D.; Jansen, M.; Andreassen, P.R.; Mulloy, J.C. p53 signaling in response to increased DNA damage sensitizes AML1-ETO cells to stress-induced death. Blood 2008, 111, 2190–2199. [Google Scholar] [CrossRef]
- Somsedíková, A.; Marková, E.; Kolenova, A.; Puskacova, J.; Kubeš, M.; Belyaev, I. Constitutive 53BP1/γH2AX foci are increased in cells of ALL patients dependent on BCR-ABL and TEL-AML1 preleukemic gene fusions. Neoplasma 2014, 62, 617–625. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vasilyev, S.A.; Kubes, M.; Markova, E.; Belyaev, I. DNA damage response in CD133 + stem/progenitor cells from umbilical cord blood: Low level of endogenous foci and high recruitment of 53BP1. Int. J. Radiat. Biol. 2013, 89, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gabert, J.; Beillard, E.; Van Der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cave, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe against cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [PubMed]
- Durdik, M.; Kosik, P.; Gursky, J.; Vokalova, L.; Markova, E.; Belyaev, I. Imaging flow cytometry as a sensitive tool to detect low-dose-induced DNA damage by analyzing 53BP1 and γH2AX foci in human lymphocytes. Cytom. Part A 2015, 87, 1070–1078. [Google Scholar] [CrossRef]
- Durdik, M.; Kosik, P.; Kruzliakova, J.; Jakl, L.; Markova, E.; Belyaev, I. Hematopoietic stem/progenitor cells are less prone to undergo apoptosis than lymphocytes despite similar DNA damage response. Oncotarget 2017, 8, 48846–48853. [Google Scholar] [CrossRef][Green Version]
- Ding, D.; Zhang, Y.; Wang, J.; Zhang, X.; Gao, Y.; Yin, L.; Li, Q.; Li, J.; Chen, H. Induction and inhibition of the pan-nuclear gamma-H2AX response in resting human peripheral blood lymphocytes after X-ray irradiation. Cell Death Discov. 2016, 2, 16011. [Google Scholar] [CrossRef]
- Anglada, T.; Terradas, M.; Hernández, L.; Genescà, A.; Martín, M. Analysis of Residual DSBs in Ataxia-Telangiectasia Lymphoblast Cells Initiating Apoptosis. BioMed Res. Int. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B, and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. MDC1 Cleavage by Caspase-3: A Novel Mechanism for Inactivating the DNA Damage Response during Apoptosis. Cancer Res. 2011, 71, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Durdik, M.; Kosik, P.; Markova, E.; Somsedikova, A.; Gajdosechova, B.; Nikitina, E.; Horvathova, E.; Kozics, K.; Davis, D.; Belyaev, I. Microwaves from mobile phone induce reactive oxygen species but not DNA damage, preleukemic fusion genes and apoptosis in hematopoietic stem/progenitor cells. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Reedijk, A.M.J.; Klein, K.; Coebergh, J.W.W.; Kremer, L.C.; Dinmohamed, A.G.; De Haas, V.; Versluijs, A.B.; Ossenkoppele, G.J.; Beverloo, H.B.; Pieters, R.; et al. Improved survival for children and young adolescents with acute myeloid leukemia: A Dutch study on incidence, survival and mortality. Leukemia 2018, 33, 1349–1359. [Google Scholar] [CrossRef]
- Quina, A.S.; Gameiro, P.; Telhada, M.; Parreira, L. PML-RARA fusion transcripts in irradiated and normal hematopoietic cells. Genes Chromosom. Cancer 2000, 29, 266–275. [Google Scholar] [CrossRef]
- Joannides, M.; Mays, A.N.; Mistry, A.R.; Hasan, S.K.; Reiter, A.; Wiemels, J.L.; Felix, C.A.; Lo-Coco, F.; Osheroff, N.; Solomon, E.; et al. Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011045. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nat. Cell Biol. 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Takatsuki, H.; Sadamura, S.; Umemura, T.; Abe, Y.; Goto, T.; Yufu, Y.; Katsuno, M.; Hirata, J.; Nishimura, J.; Nawata, H. PML/RARα fusion gene is expressed in both granuloid/macrophage and erythroid colonies in acute promyelocytic leukaemia. Br. J. Haematol. 2008, 85, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Radivoyevitch, T.; Kozubek, S.; Sachs, R.K. Biologically based risk estimation for radiation-induced CML. Radiat. Environ. Biophys. 2001, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kozubek, S.; Lukášová, E.; Marečková, A.; Skalníková, M.; Kozubek, M.; Bártová, E.; Kroha, V.; Krahulcová, E.; Šlotová, J. The topological organization of chromosomes 9 and 22 in cell nuclei has a determinative role in the induction of t(9,22) translocations and in the pathogenesis of t(9,22) leukemias. Chromosoma 1999, 108, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Neves, H.; Ramos, C.; da Silva, M.G.; Parreira, A.; Parreira, L. The nuclear topography of ABL, BCR, PML, and RA-Ralpha genes: Evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood 1999, 93, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Nilles, N.; Fahrenkrog, B. Taking a Bad Turn: Compromised DNA Damage Response in Leukemia. Cells 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. TEL-AML1 in cord blood: 1% or 0.01%? Blood 2011, 117, 2–4. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, G.; Schäfer, D.; Gavilán, A.; Vicente-Dueñas, C.; Hauer, J.; Borkhardt, A.; Sánchez-García, I. Modeling the process of childhood ETV6-RUNX1 B-cell leukemias. Oncotarget 2017, 8, 102674–102680. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RUNX1-RUNX1T1 Positive Probands | No. of Positive Reactions/Triplicate | Ct (∆R) | No. of Copies/1 × 105 Cells |
|---|---|---|---|---|
| 1 | P169 | 1/3 | 38.87 | 1 |
| 2 | P174 | 1/3 | 38.29 | 1 |
| 3 | P258 | 1/3 | 39.81 | 0.3 |
| 4 | P267 | 1/3 | 39.64 | 0.3 |
| 5 | P485 | 1/3 | 37.95 | 7.1 |
| 6 | P525 | 1/3 | 40.06 | 1.75 |
| 7 | P412 | 1/3 | 35.62 | 4.3 |
| 8 | P278 | 1/3 | 43.3 | 0.4 |
| 9 | P536 | 1/3 | 40 | 2 |
| No. | PML-RARα Positive Probands | No. of Positive Reactions/Triplicates | Ct (∆R) | No. of Copies/1 × 105 Cells |
|---|---|---|---|---|
| 1 | P417 | 1/3 | 36.11 | 5.7 |
| 2 | P437 | 3/3 | 38.10 | 2.3 |
| 37.75 | 2.9 | |||
| 38.09 | 2.3 | |||
| 3 | P461 | 1/3 + 2/3 | 37.01 | 4.76 |
| 38.49 | 5.97 | |||
| 36.57 | 21.4 | |||
| 4 | P465 | 1/3 + 1/3 | 37.35 | 3.79 |
| 38.14 | 7.5 | |||
| 5 | P469 | 1/3 + 3/3 | 38.44 | 1.83 |
| 38.96 | 4.3 | |||
| 38.44 | 6.17 | |||
| 37.63 | 10.6 | |||
| 6 | P509 | 2/3 | 39.83 | 0.7 |
| 36.99 | 4.8 | |||
| 7 | P510 | 2/3 + 1/3 | 39.63 | 0.8 |
| 38.12 | 2.6 | |||
| 37.35 | 12.7 | |||
| 8 | P511 | 2/3 + 2/3 | 39.27 | 1 |
| 36.87 | 5.3 | |||
| 36.91 | 17 | |||
| 36.43 | 23.5 | |||
| 9 | P525 | 1/3 | 36.25 | 5.2 |
| No. | KMT2A-MLLT3 Positive Probands | No. of Positive Reactions/Triplicates | Ct (∆R) | No. of Copies/1 × 105 Cells |
|---|---|---|---|---|
| 1 | P293 | 1/3 | 38.59 | 17.5 |
| 2 | P369 | 1/3 | 44.22 | 0.4 |
| 3 | P545 | 1/3 | 39.92 | 3 |
| 4 | P549 | 2/3 | 36.81 | 21 |
| 38.05 | 10 | |||
| 5 | P550 | 2/3 | 38.39 | 8 |
| 35.41 | 52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosik, P.; Durdik, M.; Skorvaga, M.; Klimova, D.; Kochanova, D.; Cerna, Z.; Kubes, M.; Holop, M.; Belyaev, I. Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples. Antioxidants 2021, 10, 481. https://doi.org/10.3390/antiox10030481
Kosik P, Durdik M, Skorvaga M, Klimova D, Kochanova D, Cerna Z, Kubes M, Holop M, Belyaev I. Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples. Antioxidants. 2021; 10(3):481. https://doi.org/10.3390/antiox10030481
Chicago/Turabian StyleKosik, Pavol, Matus Durdik, Milan Skorvaga, Daniela Klimova, Dominika Kochanova, Zlatica Cerna, Miroslav Kubes, Marek Holop, and Igor Belyaev. 2021. "Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples" Antioxidants 10, no. 3: 481. https://doi.org/10.3390/antiox10030481
APA StyleKosik, P., Durdik, M., Skorvaga, M., Klimova, D., Kochanova, D., Cerna, Z., Kubes, M., Holop, M., & Belyaev, I. (2021). Induction of AML Preleukemic Fusion Genes in HSPCs and DNA Damage Response in Preleukemic Fusion Gene Positive Samples. Antioxidants, 10(3), 481. https://doi.org/10.3390/antiox10030481