
Generation of Reactive Oxygen Species by Mitochondria

Abstract

1. Introduction
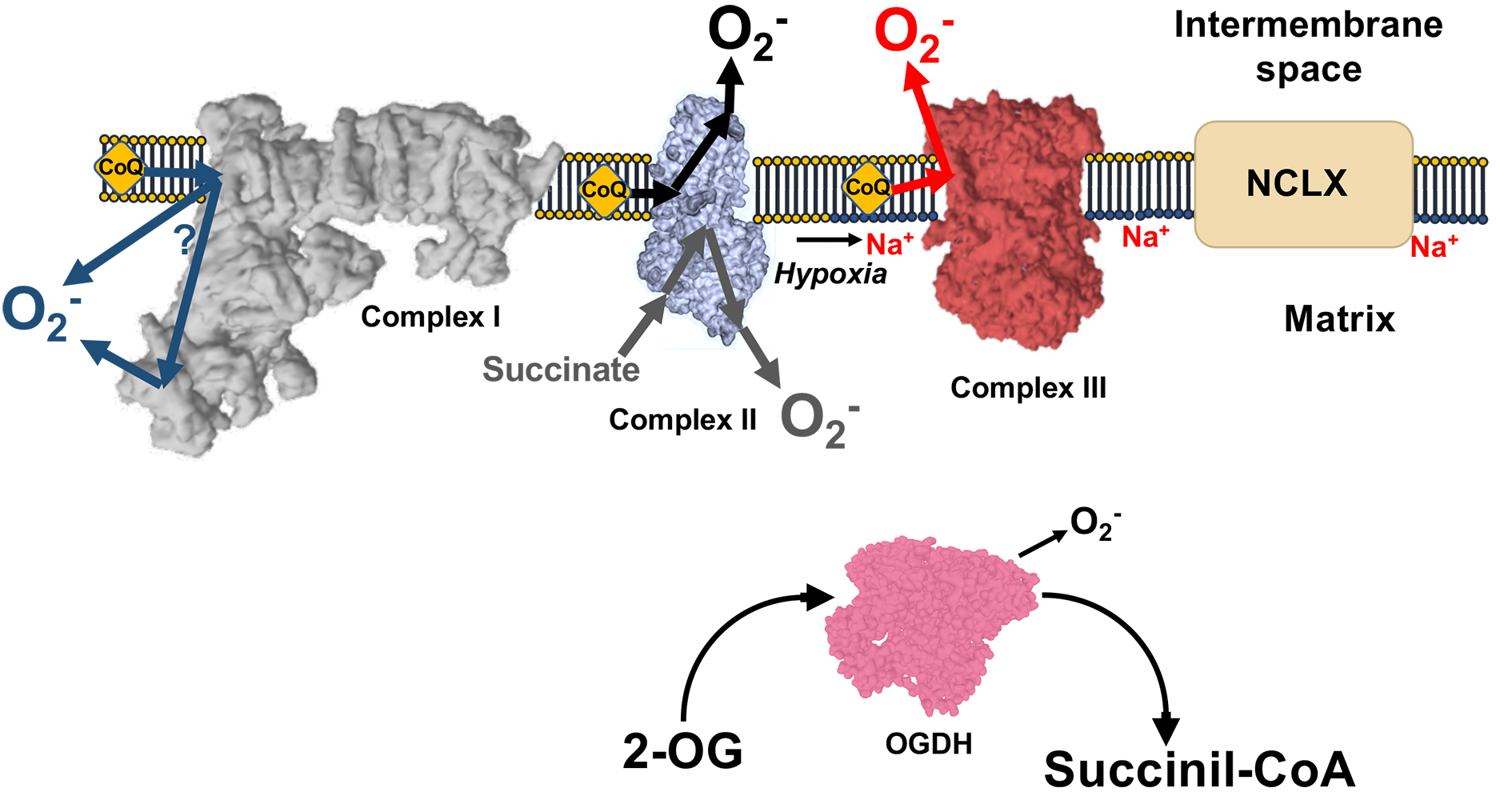
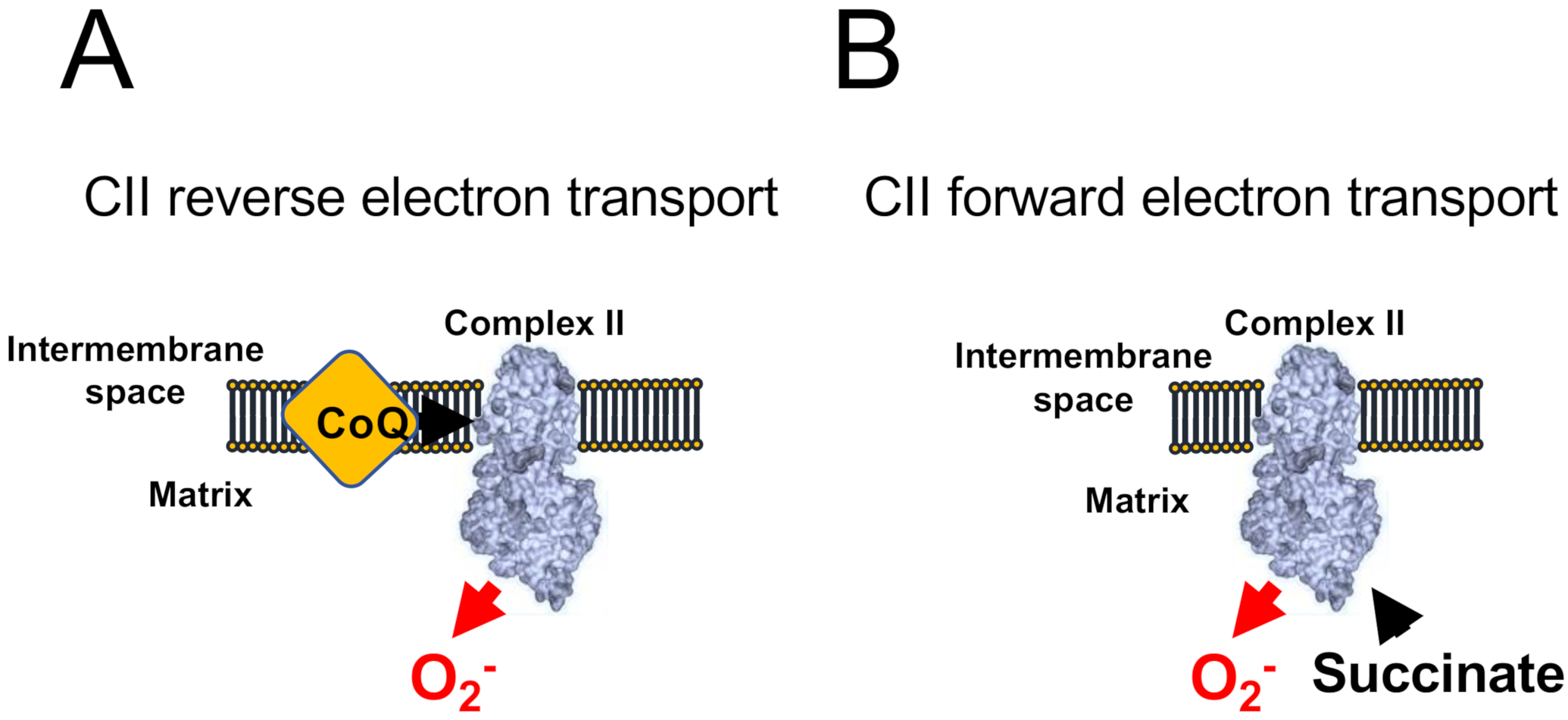
2. Modes of ROS Production by Mitochondria
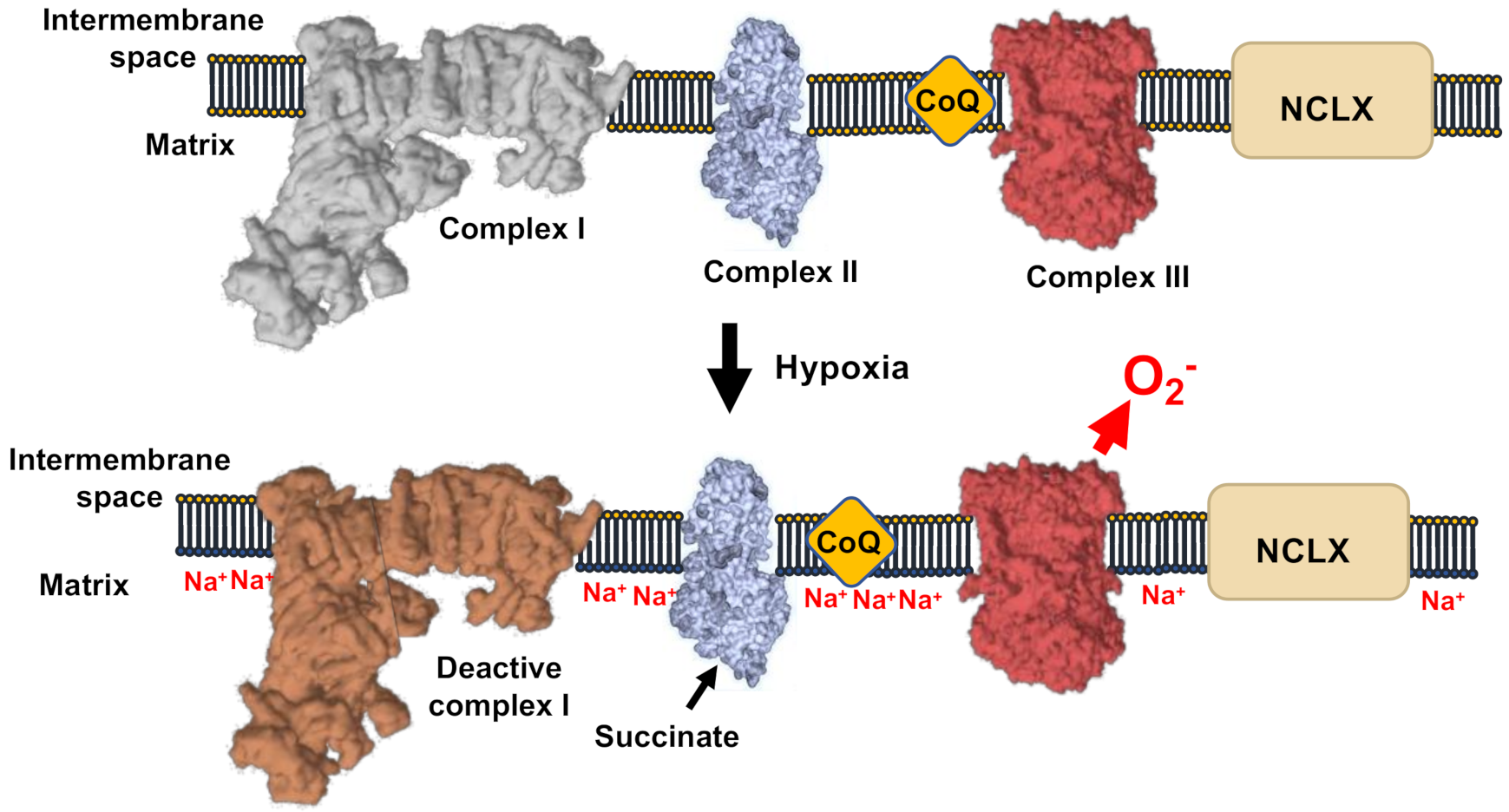
3. ROS in Acute Hypoxia
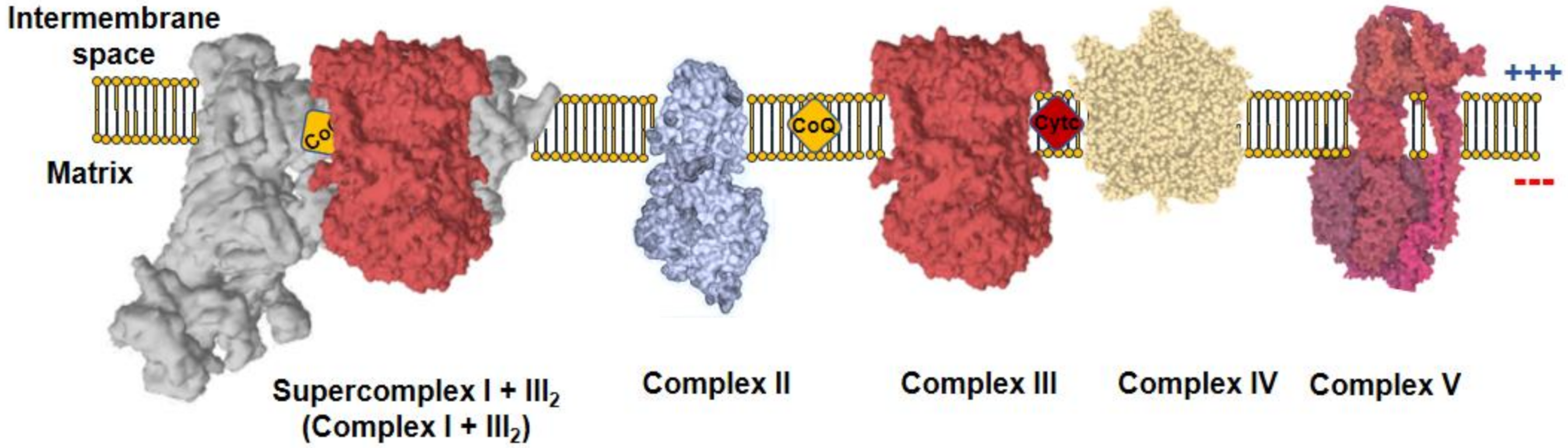
4. The Production of ROS and Supercomplexes
5. ROS, Acute Oxygen Sensing, and Calcium Homeostasis
6. Mitochondrial ROS Production in (Patho)Physiology
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Enriquez, J.A. Supramolecular Organization of Respiratory Complexes. Annu. Rev. Physiol. 2016, 78, 533–561. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. Structure and mechanism of mitochondrial proton-translocating transhydrogenase. Nature 2019, 573, 291–295. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Jung, D.W.; Apel, L.M.; Brierley, G.P. Transmembrane gradients of free Na+ in isolated heart mitochondria estimated using a fluorescent probe. Am. J. Physiol. 1992, 262, C1047–C1055. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Iborra, S.; Marti-Mateos, Y.; Cook, E.C.L.; Conde-Garrosa, R.; Petcherski, A.; Munoz, M.D.M.; Martinez de Mena, R.; Krishnan, K.C.; Jimenez, C.; et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988. [Google Scholar] [CrossRef]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sanchez-Cabo, F.; Torroja, C.; Acin-Perez, R.; Calvo, E.; Aix, E.; Gonzalez-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acin-Perez, R.; Fernandez-Silva, P.; Movilla, N.; Perez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.E.; Enriquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Galkin, A.; Brandt, U. Chapter 26 Measurement of superoxide formation by mitochondrial complex I of Yarrowia lipolytica. Methods Enzymol. 2009, 456, 475–490. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Izyumov, D.S.; Lyamzaev, K.G.; Pashkovskaya, A.A.; Pletjushkina, O.Y.; Antonenko, Y.N.; Sakharov, D.V.; Wirtz, K.W.; Skulachev, V.P. Production of reactive oxygen species in mitochondria of HeLa cells under oxidative stress. Biochim. Biophys. Acta 2006, 1757, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Guaras, A.; Perales-Clemente, E.; Calvo, E.; Acin-Perez, R.; Loureiro-Lopez, M.; Pujol, C.; Martinez-Carrascoso, I.; Nunez, E.; Garcia-Marques, F.; Rodriguez-Hernandez, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Ramos, E.; Navarro, E.; Parada, E.; Sanchez-Lopez, N.; Pelaez-Aguado, L.; Cabrera-Garcia, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef]
- Korge, P.; Calmettes, G.; Weiss, J.N. Reactive oxygen species production in cardiac mitochondria after complex I inhibition: Modulation by substrate-dependent regulation of the NADH/NAD(+) ratio. Free Radic. Biol. Med. 2016, 96, 22–33. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Martinez-Ruiz, A. Measurement of Superoxide Production in Acute Hypoxia by Fixed-Cell Microscopy. Methods Mol. Biol. 2021, 2202, 43–50. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Izquierdo-Alvarez, A.; Sanchez-Gomez, F.J.; Ramos, E.; Villa-Pina, T.; Lamas, S.; Bogdanova, A.; Martinez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Fernandez-Bueno, G.A.; Wohlgemuth, S.; Fernandez, J.; Chen, J.; Mathews, C.E. Respiration and substrate transport rates as well as reactive oxygen species production distinguish mitochondria from brain and liver. BMC Biochem. 2015, 16, 22. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Stepanova, A.; Kahl, A.; Konrad, C.; Ten, V.; Starkov, A.S.; Galkin, A. Reverse electron transfer results in a loss of flavin from mitochondrial complex I: Potential mechanism for brain ischemia reperfusion injury. J. Cereb. Blood Flow Metab. 2017, 37, 3649–3658. [Google Scholar] [CrossRef]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Borch Jensen, M.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide-H2O2 Production at Site IQ of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Ohnishi, S.T.; Ohnishi, T.; Muranaka, S.; Fujita, H.; Kimura, H.; Uemura, K.; Yoshida, K.; Utsumi, K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J. Bioenerg. Biomembr. 2005, 37, 1–15. [Google Scholar] [CrossRef]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef]
- Komlodi, T.; Geibl, F.F.; Sassani, M.; Ambrus, A.; Tretter, L. Membrane potential and delta pH dependency of reverse electron transport-associated hydrogen peroxide production in brain and heart mitochondria. J. Bioenerg. Biomembr. 2018, 50, 355–365. [Google Scholar] [CrossRef]
- Albracht, S.P.; Meijer, A.J.; Rydstrom, J. Mammalian NADH:ubiquinone oxidoreductase (Complex I) and nicotinamide nucleotide transhydrogenase (Nnt) together regulate the mitochondrial production of H(2)O(2)--implications for their role in disease, especially cancer. J. Bioenerg. Biomembr. 2011, 43, 541–564. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, M.S.; Stevens, D.; Kamunde, C. Mitochondrial transition ROS spike (mTRS) results from coordinated activities of complex I and nicotinamide nucleotide transhydrogenase. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 955–965. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Burger, N.; Kula-Alwar, D.; Aksentijevic, D.; Bridges, H.R.; Prag, H.A.; Grba, D.N.; Viscomi, C.; James, A.M.; Mottahedin, A.; et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 2021, 12, 707. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef]
- Drose, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef]
- Hernansanz-Agustin, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Pina, T.; Moreno, L.; Izquierdo-Alvarez, A.; Cabrera-Garcia, J.D.; Cortes, A.; et al. Na(+) controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Barneo, J.; Simon, M.C. Cellular adaptation to oxygen deficiency beyond the Nobel award. Nat. Commun. 2020, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Huang, J.; Henry, T.; Peterson, D.; Weir, E.K. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 1993, 73, 1100–1112. [Google Scholar] [CrossRef]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mayenco, I.; Gonzalez-Rodriguez, P.; Torres-Torrelo, H.; Gao, L.; Fernandez-Aguera, M.C.; Bonilla-Henao, V.; Ortega-Saenz, P.; Lopez-Barneo, J. Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab. 2018, 28, 145–158.e144. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Aguera, M.C.; Gao, L.; Gonzalez-Rodriguez, P.; Pintado, C.O.; Arias-Mayenco, I.; Garcia-Flores, P.; Garcia-Perganeda, A.; Pascual, A.; Ortega-Saenz, P.; Lopez-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [CrossRef]
- Smith, K.A.; Schumacker, P.T. Sensors and signals: The role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J. Physiol. 2019, 597, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Aspects Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef]
- Agip, A.A.; Blaza, J.N.; Bridges, H.R.; Viscomi, C.; Rawson, S.; Muench, S.P.; Hirst, J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018, 25, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Blaza, J.N.; Vinothkumar, K.R.; Hirst, J. Structure of the Deactive State of Mammalian Respiratory Complex I. Structure 2018, 26, 312–319.e313. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.; Meyer, B.; Wittig, I.; Karas, M.; Schagger, H.; Vinogradov, A.; Brandt, U. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J. Biol. Chem. 2008, 283, 20907–20913. [Google Scholar] [CrossRef]
- Kampjut, D.; Sazanov, L.A. The coupling mechanism of mammalian respiratory complex I. Science 2020, 370, eabc4209. [Google Scholar] [CrossRef]
- Drose, S.; Stepanova, A.; Galkin, A. Ischemic A/D transition of mitochondrial complex I and its role in ROS generation. Biochim. Biophys. Acta 2016, 1857, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Cogliati, S.; Hernansanz-Agustin, P.; Loureiro-Lopez, M.; Guaras, A.; Casuso, R.A.; Garcia-Marques, F.; Acin-Perez, R.; Marti-Mateos, Y.; Silla-Castro, J.C.; et al. Functional role of respiratory supercomplexes in mice: SCAF1 relevance and segmentation of the Qpool. Sci. Adv. 2020, 6, eaba7509. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Aksentijevic, D.; Karlstaedt, A.; Basalay, M.V.; O’Brien, B.A.; Sanchez-Tatay, D.; Eminaga, S.; Thakker, A.; Tennant, D.A.; Fuller, W.; Eykyn, T.R.; et al. Intracellular sodium elevation reprograms cardiac metabolism. Nat. Commun. 2020, 11, 4337. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Hoffman, D.L.; Brookes, P.S. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.L.; Salter, J.D.; Brookes, P.S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: Implications for hypoxic cell signaling. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H101–H108. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goldberger, O.; Marutani, E.; Wojtkiewicz, G.R.; Ast, T.; Wang, H.; Schleifer, G.; Stepanova, A.; Brepoels, K.; et al. Leigh Syndrome Mouse Model Can Be Rescued by Interventions that Normalize Brain Hyperoxia, but Not HIF Activation. Cell Metab. 2019, 30, 824–832.e823. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Genova, M.L.; Parenti Castelli, G.; Lenaz, G. The mitochondrial respiratory chain is partially organized in a supercomplex assembly: Kinetic evidence using flux control analysis. J. Biol. Chem. 2004, 279, 36562–36569. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890.e876. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; De Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed]
- Blaza, J.N.; Serreli, R.; Jones, A.J.; Mohammed, K.; Hirst, J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. USA 2014, 111, 15735–15740. [Google Scholar] [CrossRef]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531.e524. [Google Scholar] [CrossRef]
- Lobo-Jarne, T.; Nyvltova, E.; Perez-Perez, R.; Timon-Gomez, A.; Molinie, T.; Choi, A.; Mourier, A.; Fontanesi, F.; Ugalde, C.; Barrientos, A. Human COX7A2L Regulates Complex III Biogenesis and Promotes Supercomplex Organization Remodeling without Affecting Mitochondrial Bioenergetics. Cell Rep. 2018, 25, 1786–1799.e1784. [Google Scholar] [CrossRef]
- Trouillard, M.; Meunier, B.; Rappaport, F. Questioning the functional relevance of mitochondrial supercomplexes by time-resolved analysis of the respiratory chain. Proc. Natl. Acad. Sci. USA 2011, 108, E1027–E1034. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Poyatos, C.; Cogliati, S.; Calvo, E.; Hernansanz-Agustin, P.; Lagarrigue, S.; Magni, R.; Botos, M.; Langa, X.; Amati, F.; Vazquez, J.; et al. Scaf1 promotes respiratory supercomplexes and metabolic efficiency in zebrafish. EMBO Rep. 2020, 21, e50287. [Google Scholar] [CrossRef]
- Letts, J.A.; Fiedorczuk, K.; Degliesposti, G.; Skehel, M.; Sazanov, L.A. Structures of Respiratory Supercomplex I+III2 Reveal Functional and Conformational Crosstalk. Mol. Cell 2019, 75, 1131–1146.e1136. [Google Scholar] [CrossRef]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148137. [Google Scholar] [CrossRef] [PubMed]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolanos, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Lechuga-Vieco, A.V.; Latorre-Pellicer, A.; Johnston, I.G.; Prota, G.; Gileadi, U.; Justo-Mendez, R.; Acin-Perez, R.; Martinez-de-Mena, R.; Fernandez-Toro, J.M.; Jimenez-Blasco, D.; et al. Cell identity and nucleo-mitochondrial genetic context modulate OXPHOS performance and determine somatic heteroplasmy dynamics. Sci. Adv. 2020, 6, eaba5345. [Google Scholar] [CrossRef]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, N.; Vazquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nature 2016, 539, 579–582. [Google Scholar] [CrossRef]
- Moreno-Dominguez, A.; Ortega-Saenz, P.; Gao, L.; Colinas, O.; Garcia-Flores, P.; Bonilla-Henao, V.; Aragones, J.; Huttemann, M.; Grossman, L.I.; Weissmann, N.; et al. Acute O2 sensing through HIF2alpha-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelkovic, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Pak, O.; Schorner, S.; Derfuss, T.; Krug, A.; Gnaiger, E.; Ghofrani, H.A.; Schermuly, R.T.; Huckstorf, C.; Seeger, W.; et al. Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur. Respir. J. 2010, 36, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Ebert, N.; Ahrens, M.; Ghofrani, H.A.; Schermuly, R.T.; Hanze, J.; Fink, L.; Rose, F.; Conzen, J.; Seeger, W.; et al. Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 721–732. [Google Scholar] [CrossRef]
- Gupte, R.S.; Rawat, D.K.; Chettimada, S.; Cioffi, D.L.; Wolin, M.S.; Gerthoffer, W.T.; McMurtry, I.F.; Gupte, S.A. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J. Biol. Chem. 2010, 285, 19561–19571. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, F.; Tse, F.W.; Tse, A. ATP inhibits the hypoxia response in type I cells of rat carotid bodies. J. Neurochem. 2005, 92, 1419–1430. [Google Scholar] [CrossRef]
- Douglas, M.G.; Cockrell, R.S. Mitochondrial cation-hydrogen ion exchange. Sodium selective transport by mitochondria and submitochondrial particles. J. Biol. Chem. 1974, 249, 5464–5471. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Sauer, S.W.; Klemke, C.D.; Suss, D.; Okun, J.G.; Krammer, P.H.; Gulow, K. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: Mechanism of ciprofloxacin-mediated immunosuppression. J. Immunol. 2010, 184, 4827–4841. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef]
- Goh, J.; Enns, L.; Fatemie, S.; Hopkins, H.; Morton, J.; Pettan-Brewer, C.; Ladiges, W. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011, 11, 191. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Bohm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Katoshevski, T.; Sekler, I. Allosteric Regulation of NCLX by Mitochondrial Membrane Potential Links the Metabolic State and Ca(2+) Signaling in Mitochondria. Cell Rep. 2018, 25, 3465–3475.e3464. [Google Scholar] [CrossRef] [PubMed]
- Katoshevski, T.; Ben-Kasus Nissim, T.; Sekler, I. Recent studies on NCLX in health and diseases. Cell Calcium 2021, 94, 102345. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Kostic, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca(2+) efflux inhibition can be rescued by Na(+)/Ca(2+)/Li(+) exchanger upregulation. Cell Death Dis. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolanos, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Sherer, T.B.; Betarbet, R.; Panov, A.V. Complex I and Parkinson’s disease. IUBMB Life 2001, 52, 135–141. [Google Scholar] [CrossRef]
- Stojakovic, A.; Trushin, S.; Sheu, A.; Khalili, L.; Chang, S.Y.; Li, X.; Christensen, T.; Salisbury, J.L.; Geroux, R.E.; Gateno, B.; et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Commun. Biol. 2021, 4, 61. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2015, 2, 294–305. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Carrascoso, I.; Baixauli, F.; Roche-Molina, M.; Latorre-Pellicer, A.; Fernandez-Silva, P.; Mittelbrunn, M.; Sanchez-Madrid, F.; Perez-Martos, A.; Lowell, C.A.; et al. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab. 2014, 19, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Acin-Perez, R.; Martinez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villan, E.; Hervas-Stubbs, S.; Pelegrin, P.; Sander, L.E.; Enriquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef]
- Bleier, L.; Wittig, I.; Heide, H.; Steger, M.; Brandt, U.; Drose, S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radic. Biol. Med. 2015, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
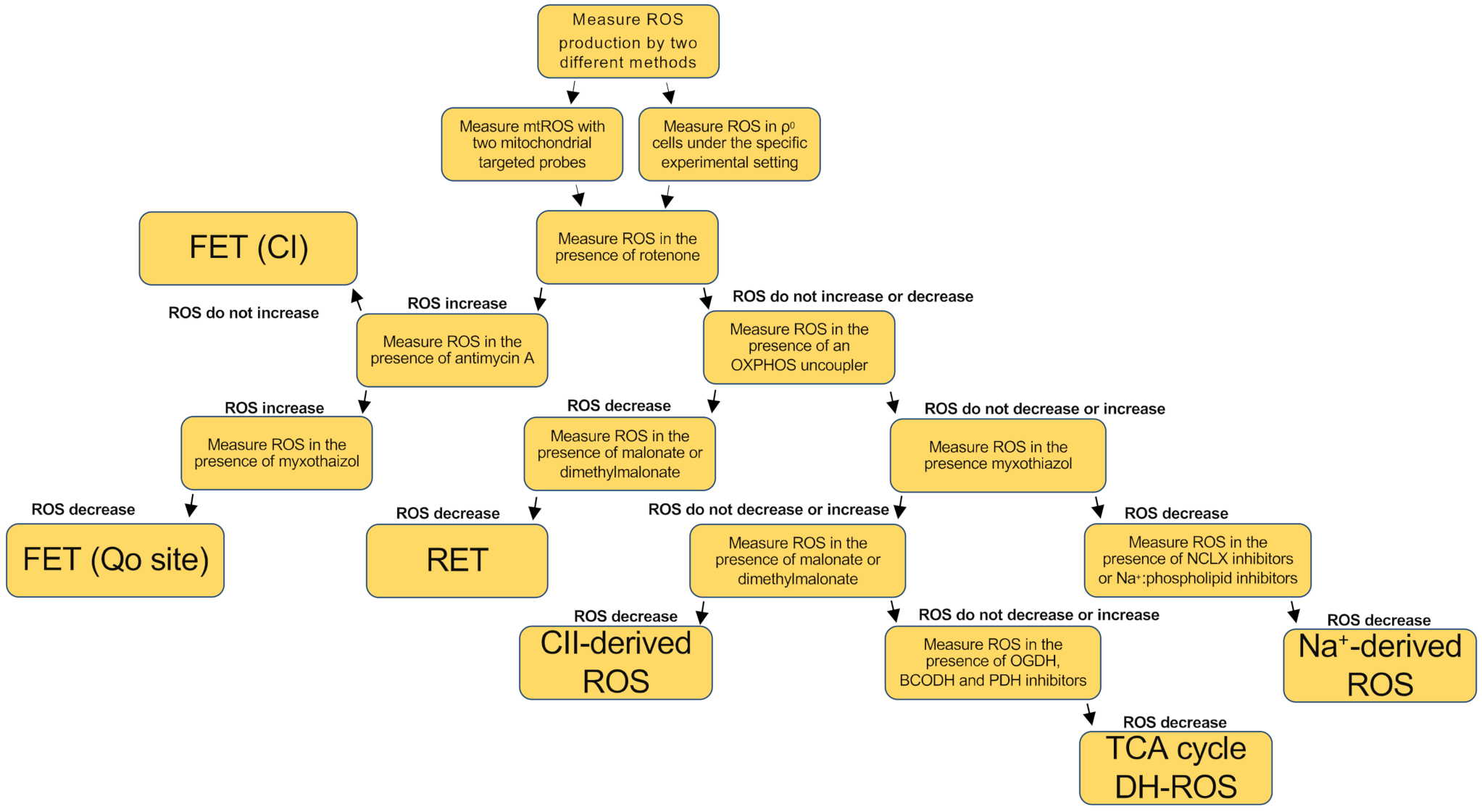{kind=link}
| Name and Source | System | Substrate(s)/Conditions | Potentiator(s) | Inhibitor(s) | References |
|---|---|---|---|---|---|
| FET (CI and CIIIo) | Tissues, cells, and isolated mitochondria | Cells and Tissues: standard culture media Isolated Mitochondria: pyruvate, malate and glutamate | Rotenone, Piericidin A or Antimycin A | DPI, myxothiazol, stigmatellin or mucidin | [25,26,28,29,30,31,32,33,34] |
| RET (CIN) | Cells and isolated mitochondria | Succinate or G3P | CV inhibitors or ATP | CI and CII inhibitors and OXPHOS uncouplers (FCCP) | [13,16,20,22,36,37,38,39,40,41,42,44] |
| CIIIo | Cells and isolated mitochondria | NADH or succinate | Antimycin A | Myxothiazol or stigmatellin | [29,30,51] |
| CII-derived forward ROS production | Isolated mitochondria | Low succinate concentration, CI and CIII inhibited | - | Malonate | [49] |
| CII-derived reverse ROS production | Isolated mitochondria | Ubiquinol concentration, CI and CIII inhibited | - | Atpenin A5 and malonate | [49] |
| Hypoxic ROS | Tissues, cells and isolated mitochondria | Cells and Tissues: normal culture media Isolated Mitochondria: Malate, glutamate, CaCl2 and NaCl | Monensin, Nigericin, FCCP (in normoxic cells) | Rotenone, piericidin A, myxothiazol, malonate, NCLX inhibitors (preincubated) | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernansanz-Agustín, P.; Enríquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. https://doi.org/10.3390/antiox10030415
Hernansanz-Agustín P, Enríquez JA. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants. 2021; 10(3):415. https://doi.org/10.3390/antiox10030415
Chicago/Turabian StyleHernansanz-Agustín, Pablo, and José Antonio Enríquez. 2021. "Generation of Reactive Oxygen Species by Mitochondria" Antioxidants 10, no. 3: 415. https://doi.org/10.3390/antiox10030415
APA StyleHernansanz-Agustín, P., & Enríquez, J. A. (2021). Generation of Reactive Oxygen Species by Mitochondria. Antioxidants, 10(3), 415. https://doi.org/10.3390/antiox10030415