
Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Human Atherosclerotic Tissue
2.3. Animals
2.4. Cell Culture
2.5. Western Blot
2.6. Immunohistochemistry
2.7. LEC Tube Formation Assay
2.8. LEC Migration Assay
2.9. LEC Proliferation Assays
2.10. Cell Cycle Analysis
2.11. In Vivo Lymphangiogenesis Assay
2.12. Quantitative Real-Time PCR
2.13. Gene Silencing
2.14. Lipid Internalization Assay
2.15. Reactive Oxygen Species Generation
2.16. Statistical Analysis
3. Results
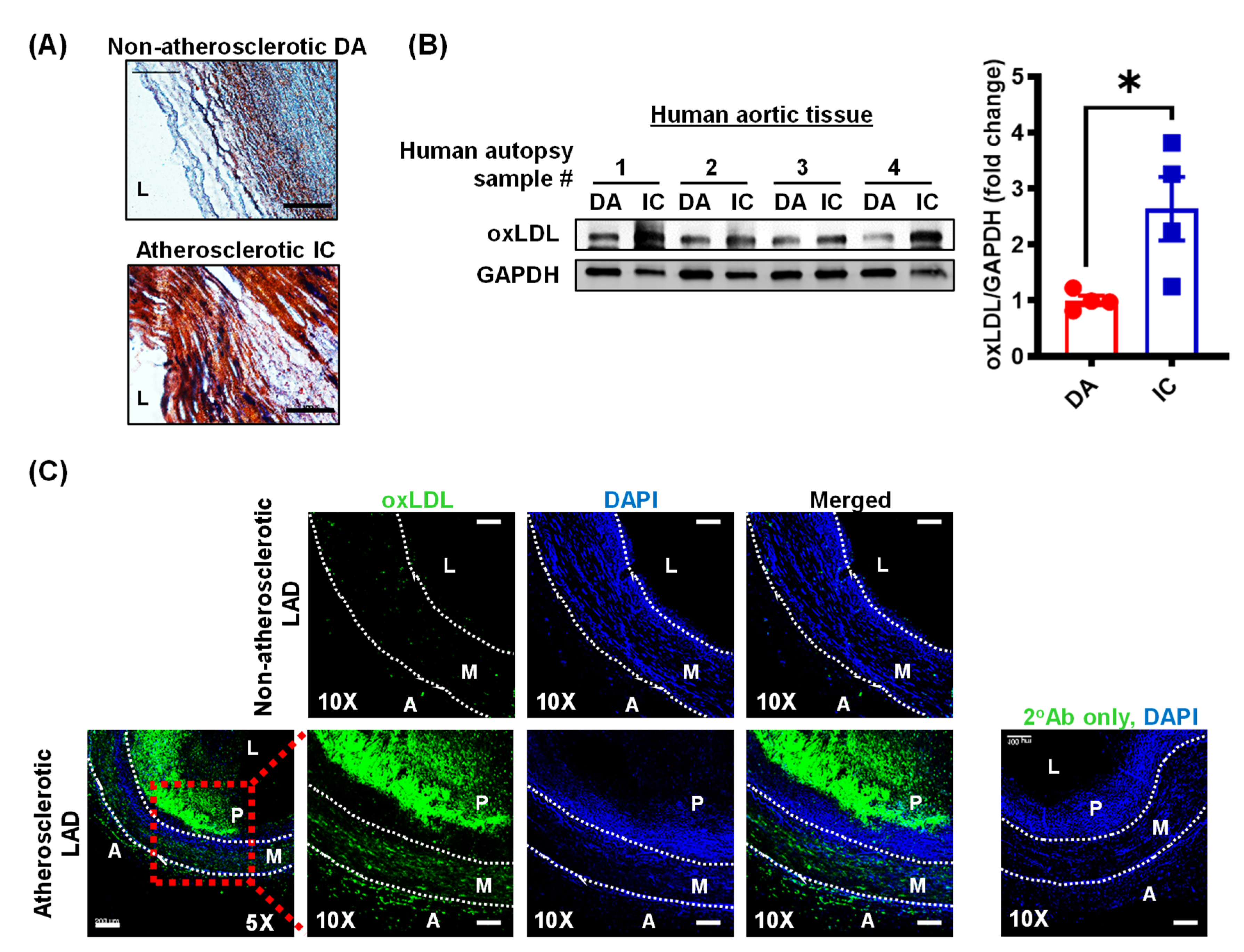
3.1. Oxidized LDL Levels Are Elevated in Human Atherosclerotic Arteries
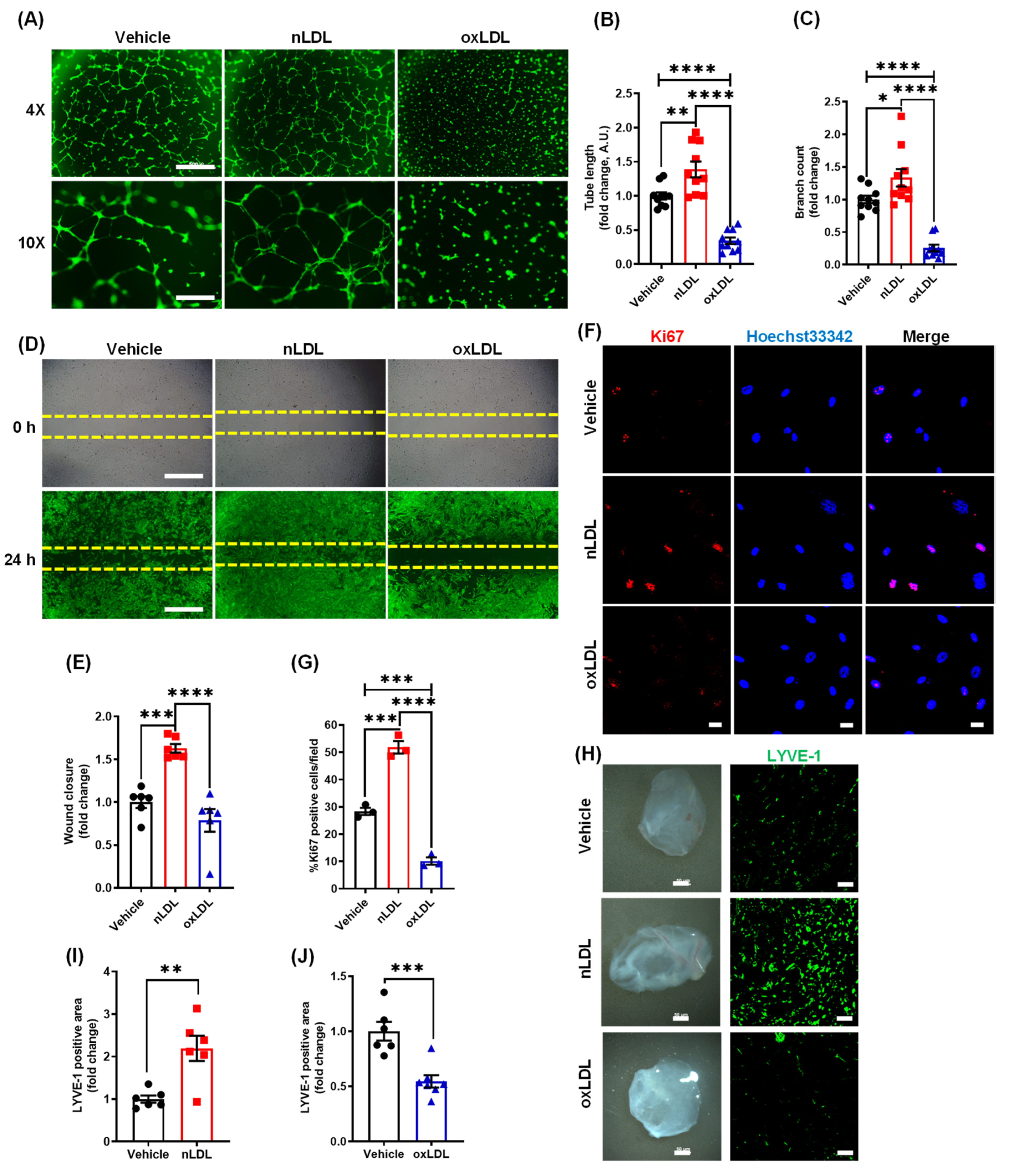
3.2. Oxidized LDL Suppresses Lymphangiogenesis In Vitro and In Vivo
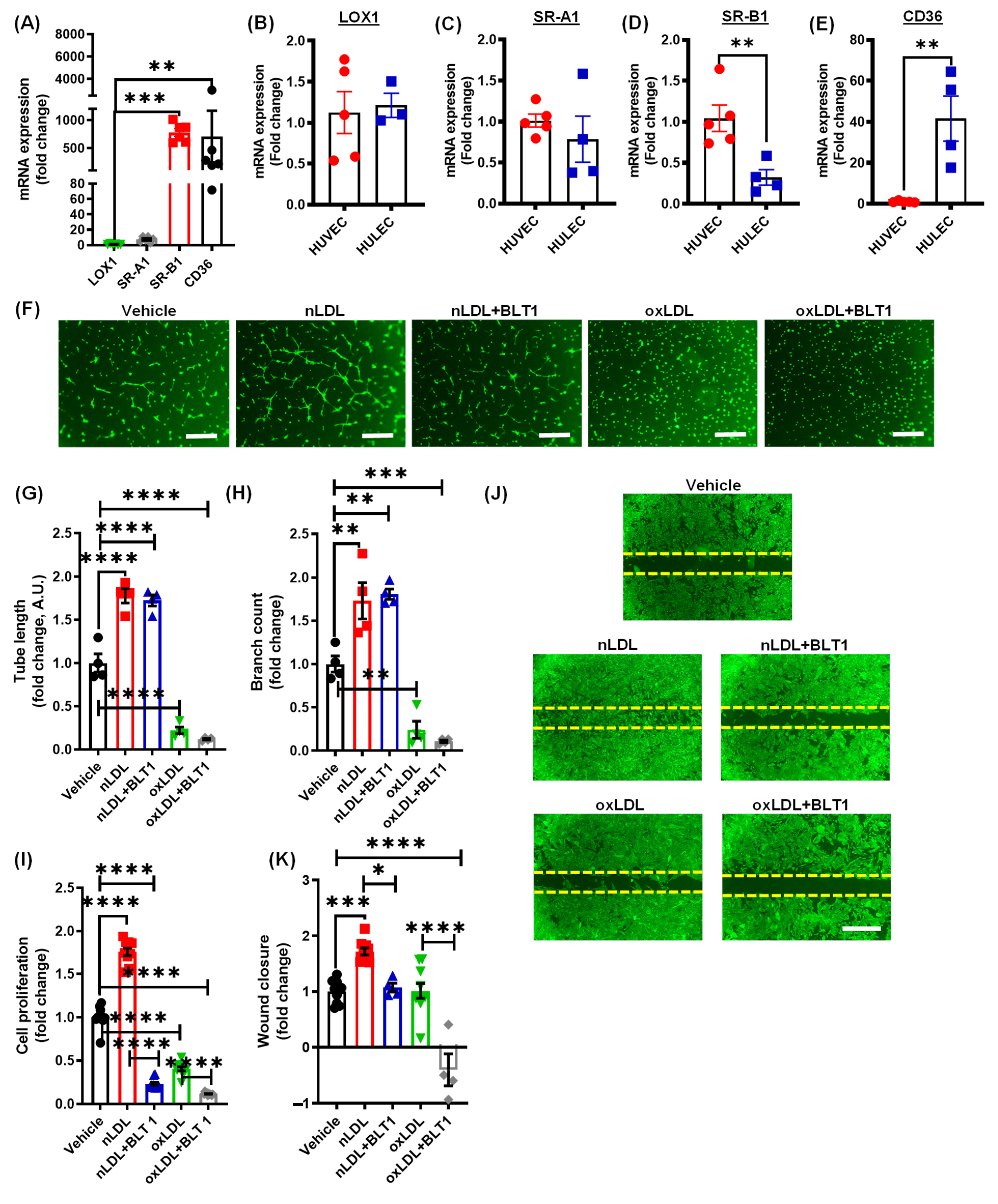
3.3. Pharmacological Blockade of SR-B1 Does Not Rescue OxLDL-Induced Inhibition of Lymphatic Endothelial Cell Proliferation and Tube Formation
3.4. Oxidized LDL Mediates Its Anti-Lymphangiogenic Effect via CD36
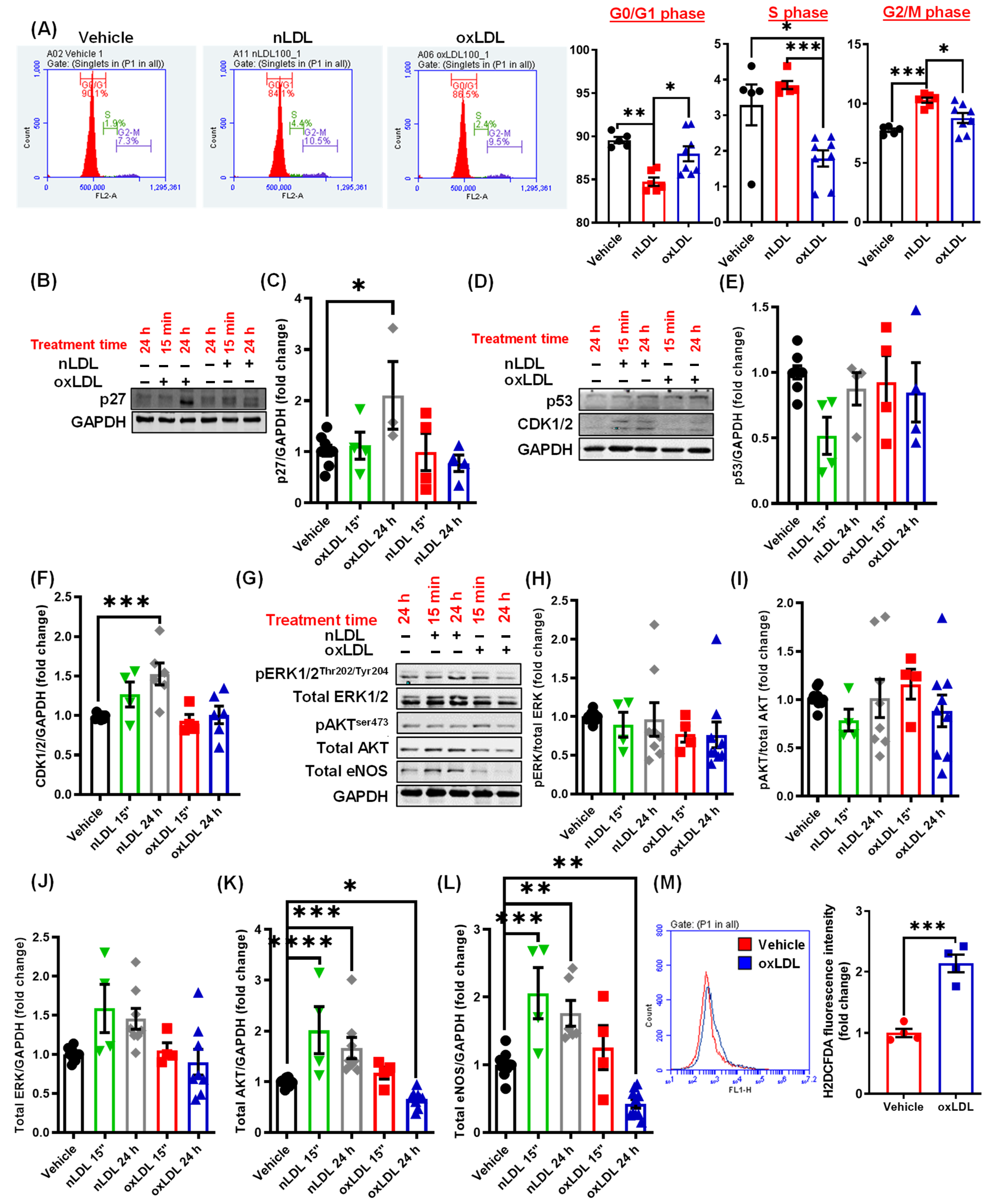
3.5. Oxidized LDL Induces Cell Cycle Arrest and Inhibits AKT and eNOS Expression in LEC
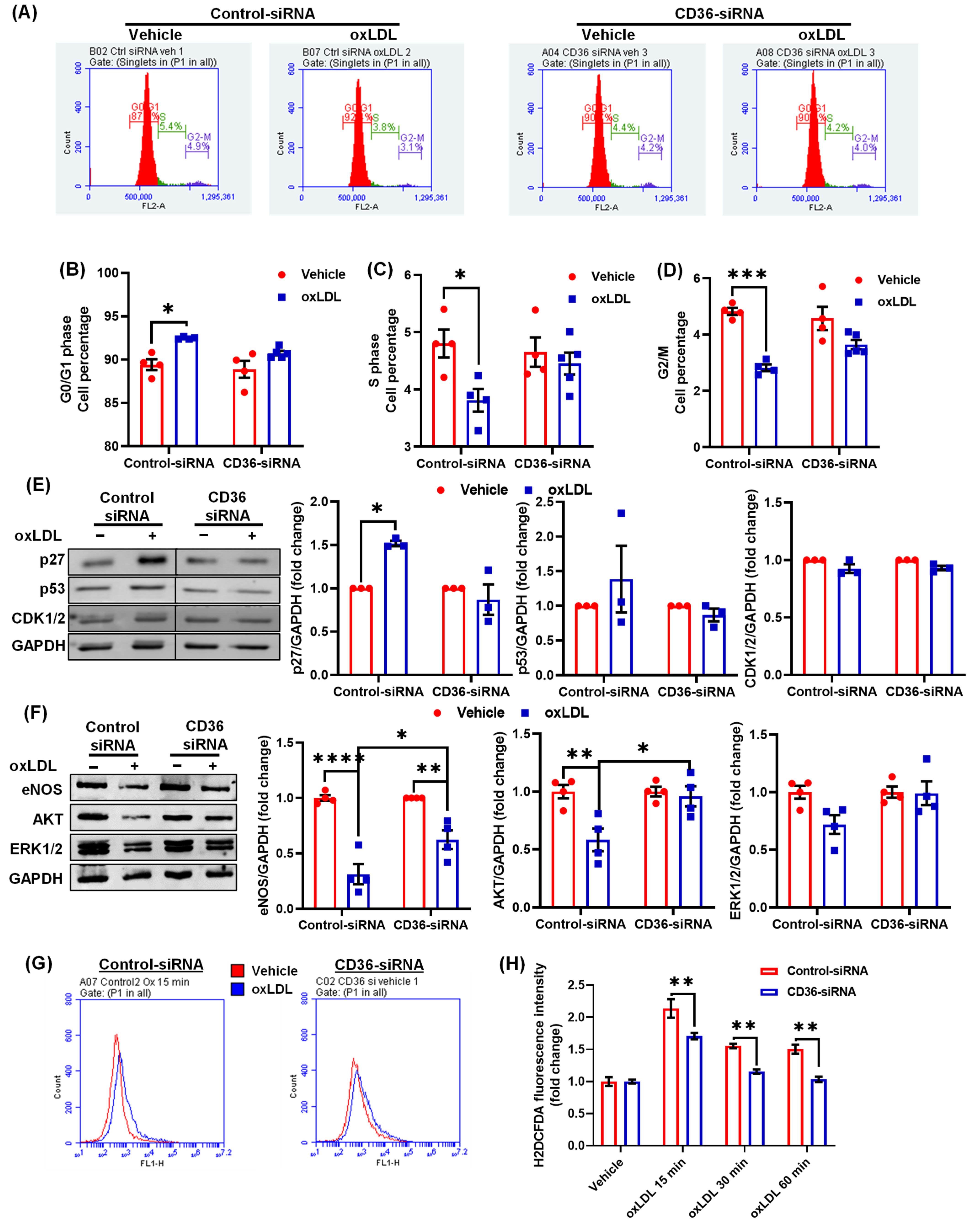
3.6. CD36 Knockdown Attenuates the Inhibitory Effects of oxLDL on Cell Cycle
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Li, W.; Fulp, B.; Platt, A.M.; Gautier, E.L.; Westerterp, M.; Bittman, R.; Tall, A.R.; Chen, S.-H.; Thomas, M.J.; et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Investig. 2013, 123, 1571–1579. [Google Scholar] [CrossRef]
- Yeo, K.P.; Lim, H.Y.; Thiam, C.H.; Azhar, S.H.; Tan, C.; Tang, Y.; See, W.Q.; Koh, X.H.; Zhao, M.H.; Phua, M.L.; et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci. Adv. 2020, 6, eabc2697. [Google Scholar] [CrossRef]
- Rademakers, T.; Van Der Vorst, E.P.C.; Daissormont, I.T.M.N.; Otten, J.J.T.; Theodorou, K.; Theelen, T.L.; Gijbels, M.; Anisimov, A.; Nurmi, H.; Lindeman, J.H.N.; et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci. Rep. 2017, 7, srep45263. [Google Scholar] [CrossRef] [PubMed]
- Vuorio, T.; Nurmi, H.; Moulton, K.; Kurkipuro, J.; Robciuc, M.R.; Öhman, M.; Heinonen, S.E.; Samaranayake, H.; Heikura, T.; Alitalo, K.; et al. Lymphatic Vessel Insufficiency in Hypercholesterolemic Mice Alters Lipoprotein Levels and Promotes Atherogenesis. Arter. Thromb. Vasc. Biol. 2014, 34, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Lewis, A. Conner Memorial Lecture. Circulation 1997, 95, 1062–1071. [Google Scholar] [CrossRef]
- Berliner, J.A.; Navab, M.; Fogelman, A.M.; Frank, J.S.; Demer, L.L.; Edwards, P.A.; Watson, A.D.; Lusis, A.J. Atherosclerosis: Basic Mechanisms. Circulation 1995, 91, 2488–2496. [Google Scholar] [CrossRef]
- Yurdagul, A.; Sulzmaier, F.J.; Chen, X.L.; Pattillo, C.B.; Schlaepfer, D.D.; Orr, A.W. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-κB activation and VCAM-1 expression. J. Cell Sci. 2016, 129, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Itabe, H.; Uno, M.; Kitazato, K.T.; Horiguchi, H.; Shinno, K.; Nagahiro, S. Oxidized LDL in Carotid Plaques and Plasma Associates With Plaque Instability. Arter. Thromb. Vasc. Biol. 2002, 22, 1649–1654. [Google Scholar] [CrossRef]
- Uchida, Y.; Uchida, Y.; Shimoyama, E.; Hiruta, N.; Kishimoto, T.; Watanabe, S. Pericoronary Adipose Tissue as Storage and Supply Site for Oxidized Low-Density Lipoprotein in Human Coronary Plaques. PLoS ONE 2016, 11, e0150862. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Vanhaecke, J.; Janssens, S.; Van De Werf, F.; Collen, D. Oxidized LDL and Malondialdehyde-Modified LDL in Patients With Acute Coronary Syndromes and Stable Coronary Artery Disease. Circulation 1998, 98, 1487–1494. [Google Scholar] [CrossRef]
- Toshima, S.-I.; Hasegawa, A.; Kurabayashi, M.; Itabe, H.; Takano, T.; Sugano, J.; Shimamura, K.; Kimura, J.; Michishita, I.; Suzuki, T.; et al. Circulating Oxidized Low Density Lipoprotein Levels. Arter. Thromb. Vasc. Biol. 2000, 20, 2243–2247. [Google Scholar] [CrossRef]
- Itabe, H.; Takeshima, E.; Iwasaki, H.; Kimura, J.; Yoshida, Y.; Imanaka, T.; Takano, T. A monoclonal antibody against oxidized lipoprotein recognizes foam cells in atherosclerotic lesions. Complex formation of oxidized phosphatidylcholines and polypeptides. J. Biol. Chem. 1994, 269, 15274–15279. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S.; Palinski, W.; Rosenfeld, M.E.; Parthasarathy, S.; Carew, T.E.; Butler, S.; Witztum, J.L.; Steinberg, D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Investig. 1989, 84, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Rutkowski, J.M.; Helft, J.; Reddy, S.T.; Swartz, M.A.; Randolph, G.J.; Angeli, V. Hypercholesterolemic Mice Exhibit Lymphatic Vessel Dysfunction and Degeneration. Am. J. Pathol. 2009, 175, 1328–1337. [Google Scholar] [CrossRef]
- Taher, M.; Nakao, S.; Zandi, S.; Melhorn, M.I.; Hayes, K.C.; Hafezi-Moghadam, A. Phenotypic transformation of intimal and adventitial lymphatics in atherosclerosis: A regulatory role for soluble VEGF receptor 2. FASEB J. 2016, 30, 2490–2499. [Google Scholar] [CrossRef]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A Specific CD36-Dependent Signaling Pathway Is Required for Platelet Activation by Oxidized Low-Density Lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Stiko-Rahm, A.; Hultgårdh-Nilsson, A.; Regnström, J.; Hamsten, A.; Nilsson, J. Native and oxidized LDL enhances production of PDGF AA and the surface expression of PDGF receptors in cultured human smooth muscle cells. Arter. Thromb. Vasc. Biol. 1992, 12, 1099–1109. [Google Scholar] [CrossRef]
- Hansson, G.K.; Robertson, A.-K.L.; Söderberg-Nauclér, C. Inflammation and atherosclerosiS. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Dandapat, A.; Hu, C.; Sun, L.; Mehta, J.L. Small Concentrations of oxLDL Induce Capillary Tube Formation From Endothelial Cells via LOX-1–Dependent Redox-Sensitive Pathway. Arter. Thromb. Vasc. Biol. 2007, 27, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Pasini, A.F.; Anselmi, M.; Garbin, U.; Franchi, E.; Stranieri, C.; Nava, M.; Boccioletti, V.; Vassanelli, C.; Cominacini, L. Enhanced Levels of Oxidized Low-Density Lipoprotein Prime Monocytes to Cytokine Overproduction via Upregulation of CD14 and Toll-Like Receptor 4 in Unstable Angina. Arter. Thromb. Vasc. Biol. 2007, 27, 1991–1997. [Google Scholar] [CrossRef]
- Lin, F.-Y.; Tsao, N.-W.; Shih, C.-M.; Lin, Y.-W.; Yeh, J.-S.; Chen, J.-W.; Nakagami, H.; Morishita, R.; Sawamura, T.; Huang, C.-Y. The Biphasic Effects of Oxidized-Low Density Lipoprotein on the Vasculogenic Function of Endothelial Progenitor Cells. PLoS ONE 2015, 10, e0123971. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, G.; Afonyushkin, T.; Binder, C.J. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J. Thromb. Haemost. 2018, 16, 418–428. [Google Scholar] [CrossRef]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; García-Caballero, M.; Missiaen, R.; Huang, H.; Brüning, U.; et al. The role of fatty acid β-oxidation in lymphangiogenesis. Nat. Cell Biol. 2017, 542, 49–54. [Google Scholar] [CrossRef]
- Singla, B.; Lin, H.-P.; Chen, A.; Ahn, W.; Ghoshal, P.; Cherian-Shaw, M.; White, J.; Stansfield, B.K.; Csányi, G. Role of R-spondin 2 in arterial lymphangiogenesis and atherosclerosis. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Singla, B.; Lin, H.-P.; Ghoshal, P.; Cherian-Shaw, M.; Csányi, G. PKCδ stimulates macropinocytosis via activation of SSH1-cofilin pathway. Cell. Signal. 2019, 53, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Rahman, H.A.; Dong, Y.; Liu, X.; Lee, Y.; Wen, A.; To, K.H.; Xiao, L.; Birsner, A.E.; Bazinet, L.; et al. Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J. Clin. Investig. 2018, 128, 4025–4043. [Google Scholar] [CrossRef]
- Goyal, T.; Mitra, S.; Khaidakov, M.; Wang, X.; Singla, S.; Ding, Z.; Liu, S.; Mehta, J.L. Current Concepts of the Role of Oxidized LDL Receptors in Atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, Y.; Li, M.; Qu, Z.; Ruan, Q. Oxidized low density lipoprotein-induced transdifferentiation of bone marrow-derived smooth muscle-like cells into foam-like cellsin vitro. Int. J. Exp. Pathol. 2010, 91, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Drozdz, K.; Janczak, D.; Dzięgiel, P.; Podhorska, M.; Patrzałek, D.; Ziółkowski, P.; Andrzejak, R.; Szuba, A. Adventitial lymphatics of internal carotid artery in healthy and atherosclerotic vessels. Folia Histochem. Cytobiol. 2009, 46, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nat. Cell Biol. 2019, 569, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Thiam, C.H.; Yeo, K.P.; Bisoendial, R.; Hii, C.S.; McGrath, K.C.; Tan, K.W.; Heather, A.; Alexander, J.S.J.; Angeli, V. Lymphatic Vessels Are Essential for the Removal of Cholesterol from Peripheral Tissues by SR-BI-Mediated Transport of HDL. Cell Metab. 2013, 17, 671–684. [Google Scholar] [CrossRef]
- Csányi, G.; Feck, D.M.; Ghoshal, P.; Singla, B.; Lin, H.; Nagarajan, S.; Meijles, D.N.; Al Ghouleh, I.; Cantu-Medellin, N.; Kelley, E.E.; et al. CD47 and Nox1 Mediate Dynamic Fluid-Phase Macropinocytosis of Native LDL. Antioxid. Redox Signal. 2017, 26, 886–901. [Google Scholar] [CrossRef]
- Chang, S.-F.; Chang, C.A.; Lee, D.-Y.; Lee, P.-L.; Yeh, Y.-M.; Yeh, C.-R.; Cheng, C.-K.; Chien, S.; Chiu, J.-J. Tumor cell cycle arrest induced by shear stress: Roles of integrins and Smad. Proc. Natl. Acad. Sci. USA 2008, 105, 3927–3932. [Google Scholar] [CrossRef]
- Zuo, S.; Liu, C.; Wang, J.; Wang, F.; Xu, W.; Cui, S.; Yuan, L.; Chen, X.; Fan, W.; Cui, M.; et al. IGFBP-rP1 induces p21 expression through a p53-independent pathway, leading to cellular senescence of MCF-7 breast cancer cells. J. Cancer Res. Clin. Oncol. 2012, 138, 1045–1055. [Google Scholar] [CrossRef]
- Abbastabar, M.; Kheyrollah, M.; Azizian, K.; Bagherlou, N.; Tehrani, S.S.; Maniati, M.; Karimian, A. Multiple functions of p27 in cell cycle, apoptosis, epigenetic modification and transcriptional regulation for the control of cell growth: A double-edged sword protein. DNA Repair 2018, 69, 63–72. [Google Scholar] [CrossRef]
- Dotto, G. p21WAF1/Cip1: More than a break to the cell cycle? Biochim. Biophys. Acta (BBA) Bioenergy 2000, 1471, M43–M56. [Google Scholar] [CrossRef]
- Geng, X.; Yanagida, K.; Akwii, R.G.; Choi, D.; Chen, L.; Ho, Y.; Cha, B.; Mahamud, R.; de Ruiz, K.B.; Ichise, H.; et al. S1PR1 regulates the quiescence of lymphatic vessels by inhibiting laminar shear stress–dependent VEGF-C signaling. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Lao, Y.; Zhang, Y.; Gillespie, D.A. Akt: A Double-Edged Sword in Cell Proliferation and Genome Stability. J. Oncol. 2012, 2012, 1–15. [Google Scholar] [CrossRef]
- Scallan, J.P.; Hill, M.A.; Davis, M.J. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc. Res. 2015, 107, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gelston, C.A.L.; Balasubbramanian, D.; Abouelkheir, G.R.; Lopez, A.H.; Hudson, K.R.; Johnson, E.R.; Muthuchamy, M.; Mitchell, B.M.; Rutkowski, J.M. Enhancing Renal Lymphatic Expansion Prevents Hypertension in Mice. Circ. Res. 2018, 122, 1094–1101. [Google Scholar] [CrossRef]
- Chakraborty, A.; Barajas, S.; Lammoglia, G.M.; Reyna, A.J.; Morley, T.S.; Johnson, J.A.; Scherer, P.E.; Rutkowski, J.M. Vascular Endothelial Growth Factor–D (VEGF-D) Overexpression and Lymphatic Expansion in Murine Adipose Tissue Improves Metabolism in Obesity. Am. J. Pathol. 2019, 189, 924–939. [Google Scholar] [CrossRef]
- Hasegawa, S.; Nakano, T.; Torisu, K.; Tsuchimoto, A.; Eriguchi, M.; Haruyama, N.; Masutani, K.; Tsuruya, K.; Kitazono, T. Vascular endothelial growth factor-C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab. Investig. 2017, 97, 1439–1452. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Leitinger, N.; Ley, K. Effects of Native and Modified Low-Density Lipoproteins on Monocyte Recruitment in Atherosclerosis. Hypertension 2007, 50, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Panchal, S.K.; Brown, L. Cholesterol versus Inflammation as Cause of Chronic Diseases. Nutrients 2019, 11, 2332. [Google Scholar] [CrossRef] [PubMed]
- Tsoupras, A.; Lordan, R.; Zabetakis, I. Inflammation, not Cholesterol, Is a Cause of Chronic Disease. Nutrients 2018, 10, 604. [Google Scholar] [CrossRef]
- Zinöcker, M.K.; Svendsen, K.; Dankel, S.N. The homeoviscous adaptation to dietary lipids (HADL) model explains controversies over saturated fat, cholesterol, and cardiovascular disease risk. Am. J. Clin. Nutr. 2021, 113, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Kromhout, D. Serum cholesterol in cross-cultural perspective. The Seven Countries Study. Acta Cardiol. 1999, 54, 155–158. [Google Scholar]
- Libby, P. Inflammation in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Burchill, M.A.; Finlon, J.M.; Goldberg, A.R.; Gillen, A.E.; Dahms, P.A.; Mcmahan, R.H.; Tye, A.; Winter, A.B.; Reisz, J.A.; Bohrnsen, E.; et al. Oxidized Low-Density Lipoprotein Drives Dysfunction of the Liver Lymphatic System. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Love-Gregory, L.; Sherva, R.; Schappe, T.; Qi, J.-S.; McCrea, J.; Klein, S.; Connelly, M.A.; Abumrad, N.A. Common CD36 SNPs reduce protein expression and may contribute to a protective atherogenic profile. Hum. Mol. Genet. 2010, 20, 193–201. [Google Scholar] [CrossRef]
- Ding, Z.; Wang, X.; Schnackenberg, L.K.; Khaidakov, M.; Liu, S.; Singla, S.; Dai, Y.; Mehta, J.L. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7g. Int. J. Cardiol. 2013, 168, 1378–1385. [Google Scholar] [CrossRef]
- Luo, Y.; Meng, X.; Zhou, P.; Lu, S.; Qin, M.; Xu, X.; Sun, G.; Sun, X. Elatoside C protects against ox-LDL-induced HUVECs injury by FoxO1-mediated autophagy induction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1654–1665. [Google Scholar] [CrossRef]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E−/− mice. Sci. Rep. 2015, 4, 5519. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Ding, Y.; Zhang, Y.; Zhou, Y.; Li, M.; Wang, C. Curcumin Suppresses Proliferation and Migration of MDA-MB-231 Breast Cancer Cells through Autophagy-Dependent Akt Degradation. PLoS ONE 2016, 11, e0146553. [Google Scholar] [CrossRef]
- Zamora, A.; Alves, M.; Chollet, C.; Therville, N.; Fougeray, T.; Tatin, F.; Franchet, C.; Gomez-Brouchet, A.; Vaysse, C.; Martinez, L.O.; et al. Paclitaxel induces lymphatic endothelial cells autophagy to promote metastasis. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Meijles, D.N.; Sahoo, S.; Al Ghouleh, I.; Amaral, J.H.; Bienes-Martinez, R.; Knupp, H.E.; Attaran, S.; Sembrat, J.C.; Nouraie, S.M.; Rojas, M.M.; et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci. Signal. 2017, 10, eaaj1784. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, E.; Coma, S.; Luks, V.L.; Greene, A.K.; Klagsbrun, M.; Warman, M.L.; Bischoff, J. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis 2015, 18, 151–162. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.; Ahn, J.H.; Hong, Y.-K.; Mäkinen, T.; Lim, D.-S.; Koh, G.Y. YAP and TAZ Negatively Regulate Prox1 During Developmental and Pathologic Lymphangiogenesis. Circ. Res. 2019, 124, 225–242. [Google Scholar] [CrossRef]
- Chong, C.R.; Xu, J.; Lu, J.; Bhat, S.; Sullivan, J.D.J.; Liu, J.O. Inhibition of Angiogenesis by the Antifungal Drug Itraconazole. ACS Chem. Biol. 2007, 2, 263–270. [Google Scholar] [CrossRef]
- Kalén, M.; Wallgard, E.; Asker, N.; Nasevicius, A.; Athley, E.; Billgren, E.; Larson, J.D.; Wadman, S.A.; Norseng, E.; Clark, K.J.; et al. Combination of Reverse and Chemical Genetic Screens Reveals Angiogenesis Inhibitors and Targets. Chem. Biol. 2009, 16, 432–441. [Google Scholar] [CrossRef]
- Schulz, M.M.P.; Reisen, F.; Zgraggen, S.; Fischer, S.; Yuen, D.; Kang, G.J.; Chen, L.; Schneider, G.; Detmar, M. Phenotype-based high-content chemical library screening identifies statins as inhibitors of in vivo lymphangiogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2665–E2674. [Google Scholar] [CrossRef] [PubMed]
- Ogata, F.; Fujiu, K.; Matsumoto, S.; Nakayama, Y.; Shibata, M.; Oike, Y.; Koshima, I.; Watabe, T.; Nagai, R.; Manabe, I.; et al. Excess Lymphangiogenesis Cooperatively Induced by Macrophages and CD4+ T Cells Drives the Pathogenesis of Lymphedema. J. Investig. Dermatol. 2016, 136, 706–714. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singla, B.; Lin, H.-P.; Ahn, W.; White, J.; Csányi, G. Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling. Antioxidants 2021, 10, 331. https://doi.org/10.3390/antiox10020331
Singla B, Lin H-P, Ahn W, White J, Csányi G. Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling. Antioxidants. 2021; 10(2):331. https://doi.org/10.3390/antiox10020331
Chicago/Turabian StyleSingla, Bhupesh, Hui-Ping Lin, WonMo Ahn, Joseph White, and Gábor Csányi. 2021. "Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling" Antioxidants 10, no. 2: 331. https://doi.org/10.3390/antiox10020331
APA StyleSingla, B., Lin, H.-P., Ahn, W., White, J., & Csányi, G. (2021). Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling. Antioxidants, 10(2), 331. https://doi.org/10.3390/antiox10020331