
Lack of Endothelial α1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling

 , , ,
, , ,  , , ,
, , ,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Materials
2.3. Determination of Vascular Reactivity and NO Levels
2.4. Vascular Reactive Oxygen Species (ROS) Production
2.5. Isolation of Mouse Lung Endothelial Cells
2.6. Reverse Transcription Real-Time PCR (qRT-PCR)
2.7. Immunoblotting
2.8. Serum Antioxidant Capacity
2.9. Statistical Analysis
3. Results
3.1. Mouse Model and Phenotype
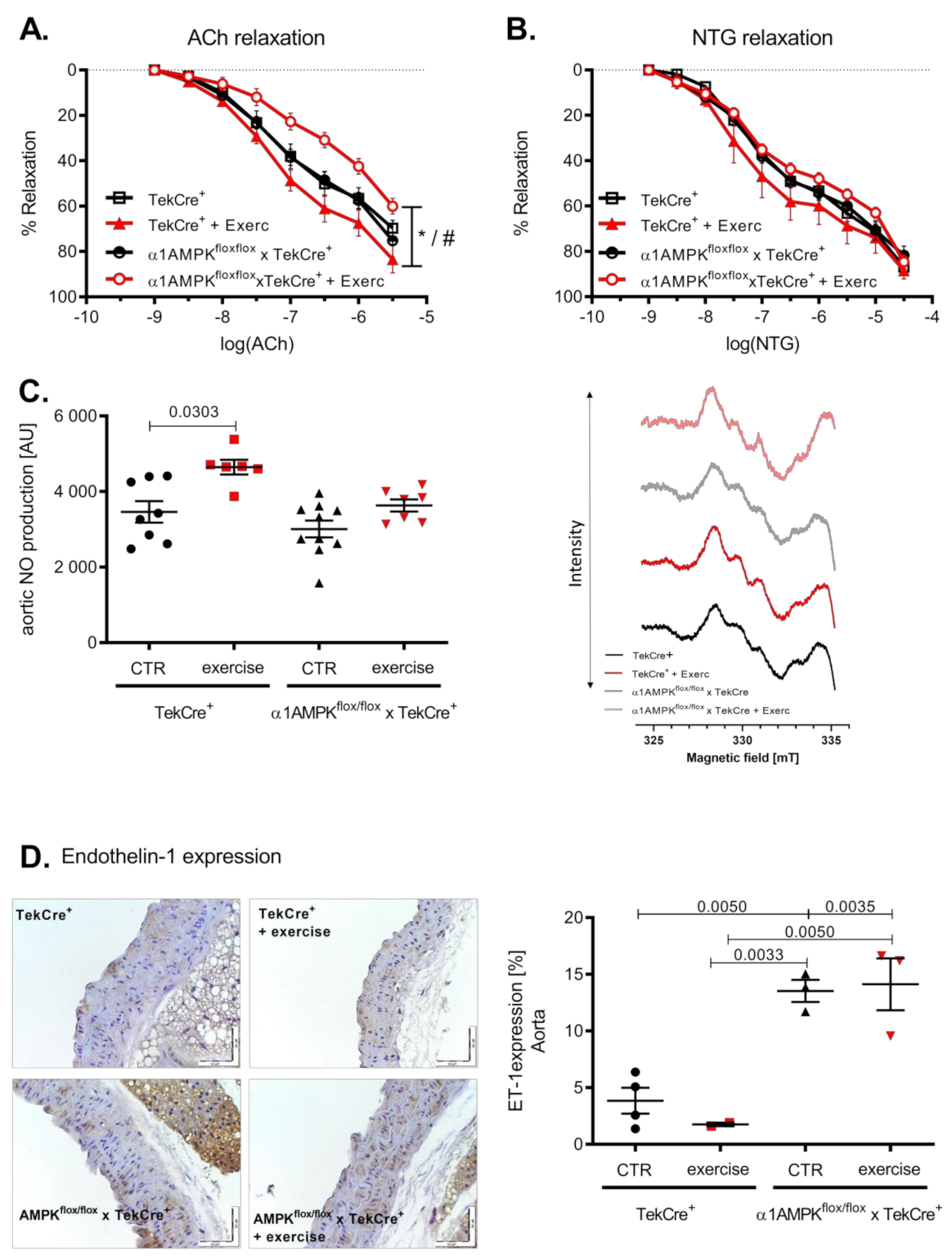
3.2. Exercise Training Induces Endothelial Dysfunction in Endothelial α1AMPK-Knockout Mice
3.3. Endothelial α1AMPK Deletion Results in Impaired eNOS Expression
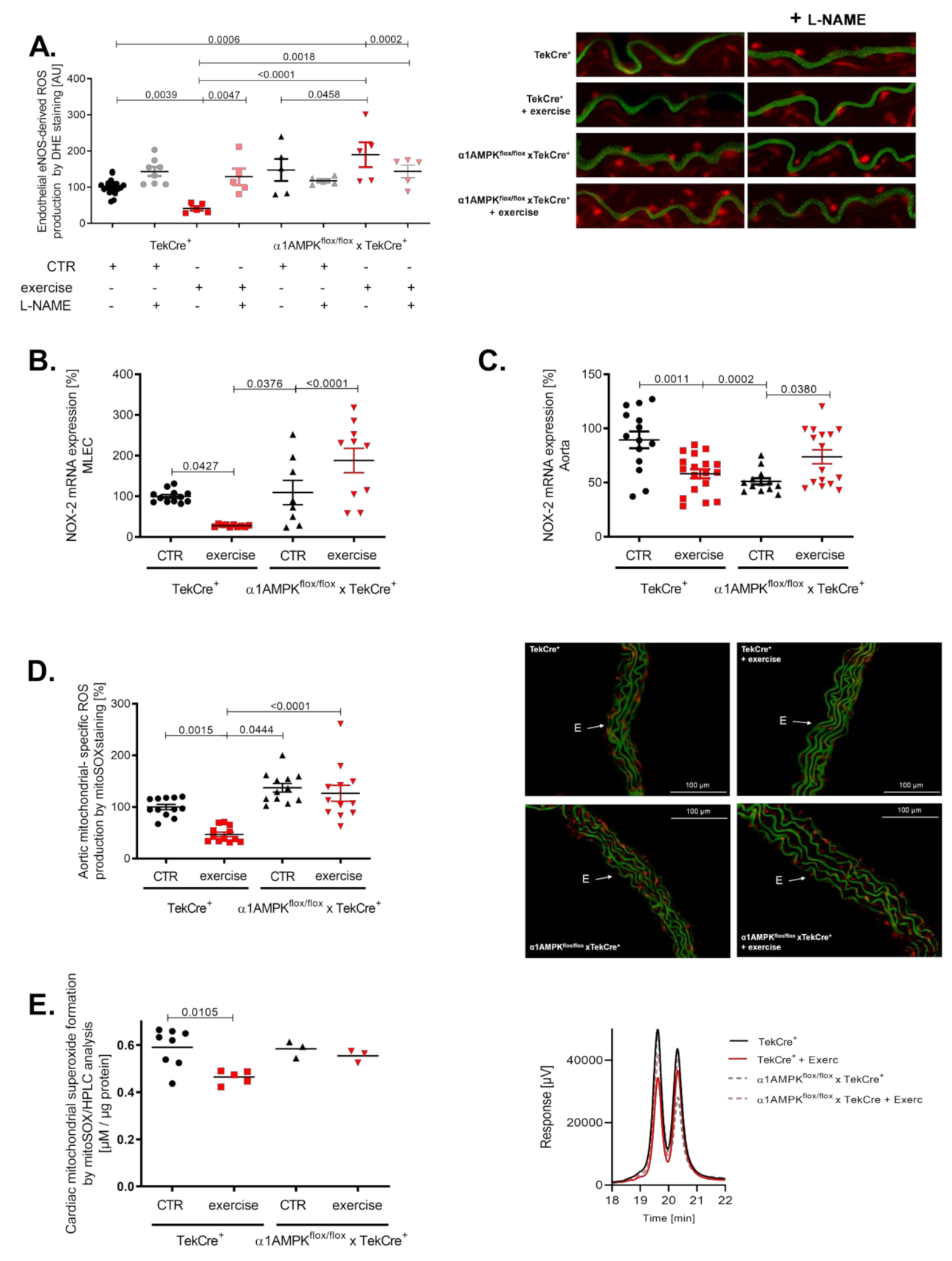
3.4. Endothelial α1AMPK Disruption Aggravates Vascular ROS Production during Exercise
3.5. Voluntary Exercise Training Induces eNOS Uncoupling in α1AMPKflox/flox x TekCre+ Mice
3.6. Endothelial α1AMPK Deletion Results in Diminished Anti-Oxidant Capacities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Okunrintemi, V.; Benson, E.A.; Tibuakuu, M.; Zhao, D.; Ogunmoroti, O.; Valero-Elizondo, J.; Gulati, M.; Nasir, K.; Michos, E.D. Trends and Costs Associated With Suboptimal Physical Activity Among US Women With Cardiovascular Disease. JAMA Netw. Open 2019, 2, e191977. [Google Scholar] [CrossRef]
- Bachmann, J.M.; DeFina, L.F.; Franzini, L.; Gao, A.; Leonard, D.S.; Cooper, K.H.; Berry, J.D.; Willis, B.L. Cardiorespiratory Fitness in Middle Age and Health Care Costs in Later Life. J. Am. Coll. Cardiol. 2015, 66, 1876–1885. [Google Scholar] [CrossRef] [Green Version]
- Garrett, N.A.; Brasure, M.; Schmitz, K.H.; Schultz, M.M.; Huber, M.R. Physical inactivity: Direct cost to a health plan. Am. J. Prev. Med. 2004, 27, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelton, S.P.; Chin, A.; Xin, X.; He, J. Effect of aerobic exercise on blood pressure: A meta-analysis of randomized, controlled trials. Ann. Intern. Med. 2002, 136, 493–503. [Google Scholar] [CrossRef]
- Tanasescu, M.; Leitzmann, M.F.; Rimm, E.B.; Willett, W.C.; Stampfer, M.J.; Hu, F.B. Exercise type and intensity in relation to coronary heart disease in men. JAMA 2002, 288, 1994–2000. [Google Scholar] [CrossRef]
- Green, D.J.; Maiorana, A.; O’Driscoll, G.; Taylor, R. Effect of exercise training on endothelium-derived nitric oxide function in humans. J. Physiol. 2004, 561, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and its alterations in cardiovascular diseases: Life style intervention. Biomed. Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef] [Green Version]
- Di Francescomarino, S.; Sciartilli, A.; Di Valerio, V.; Di Baldassarre, A.; Gallina, S. The effect of physical exercise on endothelial function. Sports Med. 2009, 39, 797–812. [Google Scholar] [CrossRef]
- Lavie, C.J.; Arena, R.; Swift, D.L.; Johannsen, N.M.; Sui, X.; Lee, D.C.; Earnest, C.P.; Church, T.S.; O’Keefe, J.H.; Milani, R.V.; et al. Exercise and the cardiovascular system: Clinical science and cardiovascular outcomes. Circ. Res. 2015, 117, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.; Linke, A. Impact of exercise training on cardiovascular disease and risk. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Jansen, T.; Hauptmann, F.; Schuler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; Keaney, J.F., Jr.; Wenzel, P.; et al. alpha1AMP-activated protein kinase mediates vascular protective effects of exercise. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmacher, S.; Foretz, M.; Knorr, M.; Jansen, T.; Hortmann, M.; Wenzel, P.; Oelze, M.; Kleschyov, A.L.; Daiber, A.; Keaney, J.F., Jr.; et al. alpha1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation. Arterioscler Thromb. Vasc. Biol. 2011, 31, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; An, X.; Chen, D.; Ye, M.; Shen, W.; Han, W.; Zhang, Y.; Gao, P. Chronic Exercise Training Improved Aortic Endothelial and Mitochondrial Function via an AMPKalpha2-Dependent Manner. Front. Physiol. 2016, 7, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, T.; Kroller-Schon, S.; Schonfelder, T.; Foretz, M.; Viollet, B.; Daiber, A.; Oelze, M.; Brandt, M.; Steven, S.; Kvandova, M.; et al. alpha1AMPK deletion in myelomonocytic cells induces a pro-inflammatory phenotype and enhances angiotensin II-induced vascular dysfunction. Cardiovasc. Res. 2018, 114, 1883–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Giaid, A.; Kurz, S.; Stewart, D.J.; Harrison, D.G. Evidence for a Role of Endothelin-1 and Protein-Kinase-C in Nitroglycerin Tolerance. Proc. Natl. Acad. Sci. USA 1995, 92, 5244–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummala, P.E.; Chen, X.L.; Sundell, C.L.; Laursen, J.B.; Hammes, C.P.; Alexander, R.W.; Harrison, D.G.; Medford, R.M. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation 1999, 100, 1223–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [Green Version]
- Kroller-Schon, S.; Daiber, A.; Steven, S.; Oelze, M.; Frenis, K.; Kalinovic, S.; Heimann, A.; Schmidt, F.P.; Pinto, A.; Kvandova, M.; et al. Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 2018, 39, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Steven, S.; Tran, L.P.; Ullmann, E.; Kossmann, S.; Schmidt, F.P.; Oelze, M.; Xia, N.; Li, H.; et al. Effects of noise on vascular function, oxidative stress, and inflammation: Mechanistic insight from studies in mice. Eur. Heart J. 2017, 38, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; August, M.; Baldus, S.; Wendt, M.; Oelze, M.; Sydow, K.; Kleschyov, A.L.; Munzel, T. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic. Biol. Med. 2004, 36, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; August, M.; Wendt, M.; Sydow, K.; Wieboldt, H.; Kleschyov, A.L.; Munzel, T. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radic. Res. 2004, 38, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; McEachern, G.E.; Myint, A.T.; Robinson, B.H. Superoxides from mitochondrial complex III: The role of manganese superoxide dismutase. Free Radic. Biol. Med. 2000, 29, 170–180. [Google Scholar] [CrossRef]
- Kalinovic, S.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Vujacic-Mirski, K.; Kvandova, M.; Schmal, I.; Al Zuabi, A.; Munzel, T.; Daiber, A. Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress. Antioxidants 2019, 8, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimasaki, Y.; Pan, N.; Messina, L.M.; Li, C.; Chen, K.; Liu, L.; Cooper, M.P.; Vita, J.A.; Keaney, J.F., Jr. Uncoupling protein 2 impacts endothelial phenotype via p53-mediated control of mitochondrial dynamics. Circ. Res. 2013, 113, 891–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlencordt, P.J.; Rosel, E.; Gerszten, R.E.; Morales-Ruiz, M.; Dombkowski, D.; Atkinson, W.J.; Han, F.; Preffer, F.; Rosenzweig, A.; Sessa, W.C.; et al. Role of endothelial nitric oxide synthase in endothelial activation: Insights from eNOS knockout endothelial cells. Am. J. Physiol. Cell Physiol. 2004, 286, C1195–C1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, B.D.; Mock, J.R.; D’Alessio, F.R.; Aggarwal, N.R.; Mandke, P.; Johnston, L.; Damarla, M. Flow-cytometric method for simultaneous analysis of mouse lung epithelial, endothelial, and hematopoietic lineage cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L796–L801. [Google Scholar] [CrossRef]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by NADPH oxidase and endothelial dysfunction in angiotensin II-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Ben-Ari, J.; Wolach, O.; Gavrieli, R.; Wolach, B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti. Infect. Ther. 2012, 10, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Chrzczanowicz, J.; Gawron, A.; Zwolinska, A.; de Graft-Johnson, J.; Krajewski, W.; Krol, M.; Markowski, J.; Kostka, T.; Nowak, D. Simple method for determining human serum 2,2-diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity - possible application in clinical studies on dietary antioxidants. Clin. Chem. Lab. Med. 2008, 46, 342–349. [Google Scholar] [CrossRef]
- Bourque, S.L.; Davidge, S.T.; Adams, M.A. The interaction between endothelin-1 and nitric oxide in the vasculature: New perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1288–R1295. [Google Scholar] [CrossRef] [Green Version]
- Perez-Torres, I.; Manzano-Pech, L.; Rubio-Ruiz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Calay, D.; Mason, J.C. The multifunctional role and therapeutic potential of HO-1 in the vascular endothelium. Antioxid. Redox Signal. 2014, 20, 1789–1809. [Google Scholar] [CrossRef] [PubMed]
- Ghadieh, A.S.; Saab, B. Evidence for exercise training in the management of hypertension in adults. Can. Fam. Physician 2015, 61, 233–239. [Google Scholar]
- Ivy, J.L. Role of exercise training in the prevention and treatment of insulin resistance and non-insulin-dependent diabetes mellitus. Sports Med. 1997, 24, 321–336. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical Activity/Exercise and Diabetes: A Position Statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrieri, D.; Moon, H.Y.; van Praag, H. Exercise in a Pill: The Latest on Exercise-Mimetics. Brain Plast. 2017, 2, 153–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, C.; Contreras, C.; Saenz-Medina, J.; Munoz, M.; Corbacho, C.; Carballido, J.; Garcia-Sacristan, A.; Hernandez, M.; Lopez, M.; Rivera, L.; et al. Activation of the AMP-related kinase (AMPK) induces renal vasodilatation and downregulates Nox-derived reactive oxygen species (ROS) generation. Redox Biol. 2020, 34, 101575. [Google Scholar] [CrossRef] [PubMed]
- Shafique, E.; Torina, A.; Reichert, K.; Colantuono, B.; Nur, N.; Zeeshan, K.; Ravichandran, V.; Liu, Y.; Feng, J.; Zeeshan, K.; et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc. Res. 2017, 113, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafique, E.; Choy, W.C.; Liu, Y.; Feng, J.; Cordeiro, B.; Lyra, A.; Arafah, M.; Yassin-Kassab, A.; Zanetti, A.V.; Clements, R.T.; et al. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging 2013, 5, 515–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, C.; Dong, B.; Xing, F.; Huang, H.; Yao, F.; Ma, Y.; He, J.; Dong, Y. AMPK attenuates ventricular remodeling and dysfunction following aortic banding in mice via the Sirt3/Oxidative stress pathway. Eur. J. Pharmacol. 2017, 814, 335–342. [Google Scholar] [CrossRef]
- Song, P.; Zou, M.H. Regulation of NAD(P)H oxidases by AMPK in cardiovascular systems. Free Radic. Biol. Med. 2012, 52, 1607–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroller-Schon, S.; Jansen, T.; Tran, T.L.P.; Kvandova, M.; Kalinovic, S.; Oelze, M.; Keaney, J.F., Jr.; Foretz, M.; Viollet, B.; Daiber, A.; et al. Endothelial alpha1AMPK modulates angiotensin II-mediated vascular inflammation and dysfunction. Basic Res. Cardiol. 2019, 114, 8. [Google Scholar] [CrossRef]
- Zucker, I.H.; Musch, T.I. Benefits of exercise training on cardiovascular dysfunction: Molecular and integrative. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1027–H1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinckard, K.; Baskin, K.K.; Stanford, K.I. Effects of Exercise to Improve Cardiovascular Health. Front. Cardiovasc. Med. 2019, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [Green Version]
- Katusic, Z.S. Vascular endothelial dysfunction: Does tetrahydrobiopterin play a role? Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H981–H986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevers, L.M.; Braam, B.; Post, J.A.; van Zonneveld, A.J.; Rabelink, T.J.; Koomans, H.A.; Verhaar, M.C.; Joles, J.A. Tetrahydrobiopterin, but not L-arginine, decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension 2006, 47, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Williams, S.M.; Brown, R.C.; Meredith-Jones, K.A.; Osborne, H.; Jospe, M.; Taylor, R.W. High-Intensity Interval Training in the Real World: Outcomes from a 12-Month Intervention in Overweight Adults. Med. Sci. Sports Exerc. 2018, 50, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Olguin, C.; Renani, L.B.; Arab-Ceschia, L.; Raun, S.H.; Bhatia, A.; Li, Z.; Knudsen, J.R.; Holmdahl, R.; Jensen, T.E. Adaptations to high-intensity interval training in skeletal muscle require NADPH oxidase 2. Redox. Biol. 2019, 24, 101188. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, A.; Khaw, K.T.; Luben, R.; Wareham, N.; Brage, S. Physical activity trajectories and mortality: Population based cohort study. BMJ 2019, 365, l2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen, T.; Kvandová, M.; Schmal, I.; Kalinovic, S.; Stamm, P.; Kuntic, M.; Foretz, M.; Viollet, B.; Daiber, A.; Oelze, M.; et al. Lack of Endothelial α1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling. Antioxidants 2021, 10, 1974. https://doi.org/10.3390/antiox10121974
Jansen T, Kvandová M, Schmal I, Kalinovic S, Stamm P, Kuntic M, Foretz M, Viollet B, Daiber A, Oelze M, et al. Lack of Endothelial α1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling. Antioxidants. 2021; 10(12):1974. https://doi.org/10.3390/antiox10121974
Chicago/Turabian StyleJansen, Thomas, Miroslava Kvandová, Isabella Schmal, Sanela Kalinovic, Paul Stamm, Marin Kuntic, Marc Foretz, Benoit Viollet, Andreas Daiber, Matthias Oelze, and et al. 2021. "Lack of Endothelial α1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling" Antioxidants 10, no. 12: 1974. https://doi.org/10.3390/antiox10121974
APA StyleJansen, T., Kvandová, M., Schmal, I., Kalinovic, S., Stamm, P., Kuntic, M., Foretz, M., Viollet, B., Daiber, A., Oelze, M., Keaney, J. F., Jr., Münzel, T., Schulz, E., & Kröller-Schön, S. (2021). Lack of Endothelial α1AMPK Reverses the Vascular Protective Effects of Exercise by Causing eNOS Uncoupling. Antioxidants, 10(12), 1974. https://doi.org/10.3390/antiox10121974