
Mito-TIPTP Increases Mitochondrial Function by Repressing the Rubicon-p22phox Interaction in Colitis-Induced Mice

 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice and Cell Culture
2.2. Reagents and Antibodies
2.3. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Glutathione S-Transferase (GST) Pulldown, Immunoblot, and Immunoprecipitation Analysis
2.5. Tissue Distribution of Rubicon
2.6. Subcellular Fractionation
2.7. Fractionation of Mitochondria
2.8. Flow Cytometric Measurement of ROS Production
2.9. Metabolic Assays
2.10. Confocal Fluorescence Microscopy
2.11. Flow Cytometry
2.12. Study Population of Human Normal and UC Patients
2.13. Adenovirus Construction
2.14. Injection of Recombinant Adenoviruses for Depletion of Rubicon in Mice
2.15. Mouse Model of Colitis
2.16. Clinical Score and Histology
2.17. Protein Purification and Mass Spectrometry
2.18. Myeloperoxidase Activity Assay
2.19. Synthesis of Mito-TIPTP
2.20. Statistical Analysis
3. Result
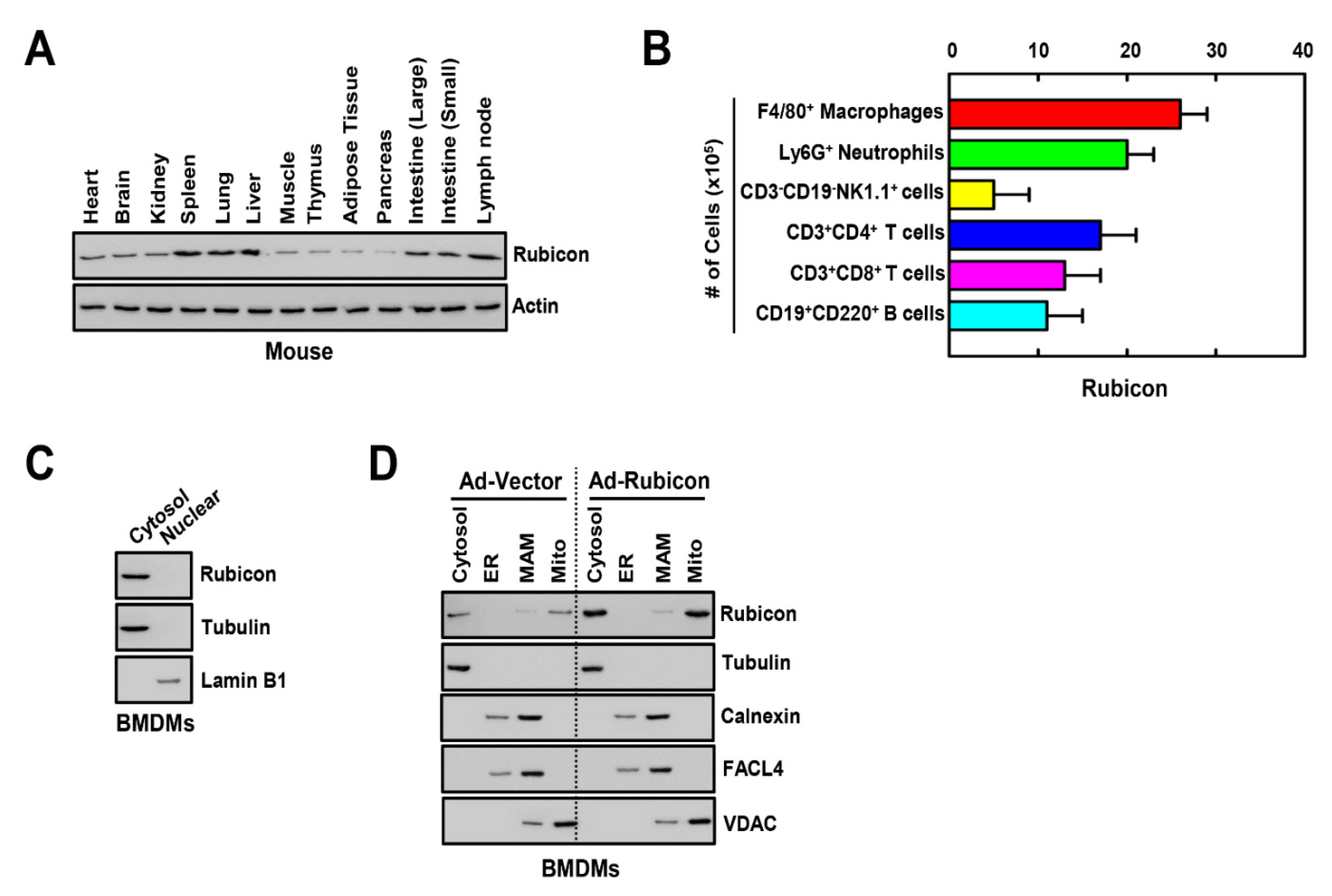
3.1. Rubicon Localizes to the Cytosol and Mitochondria
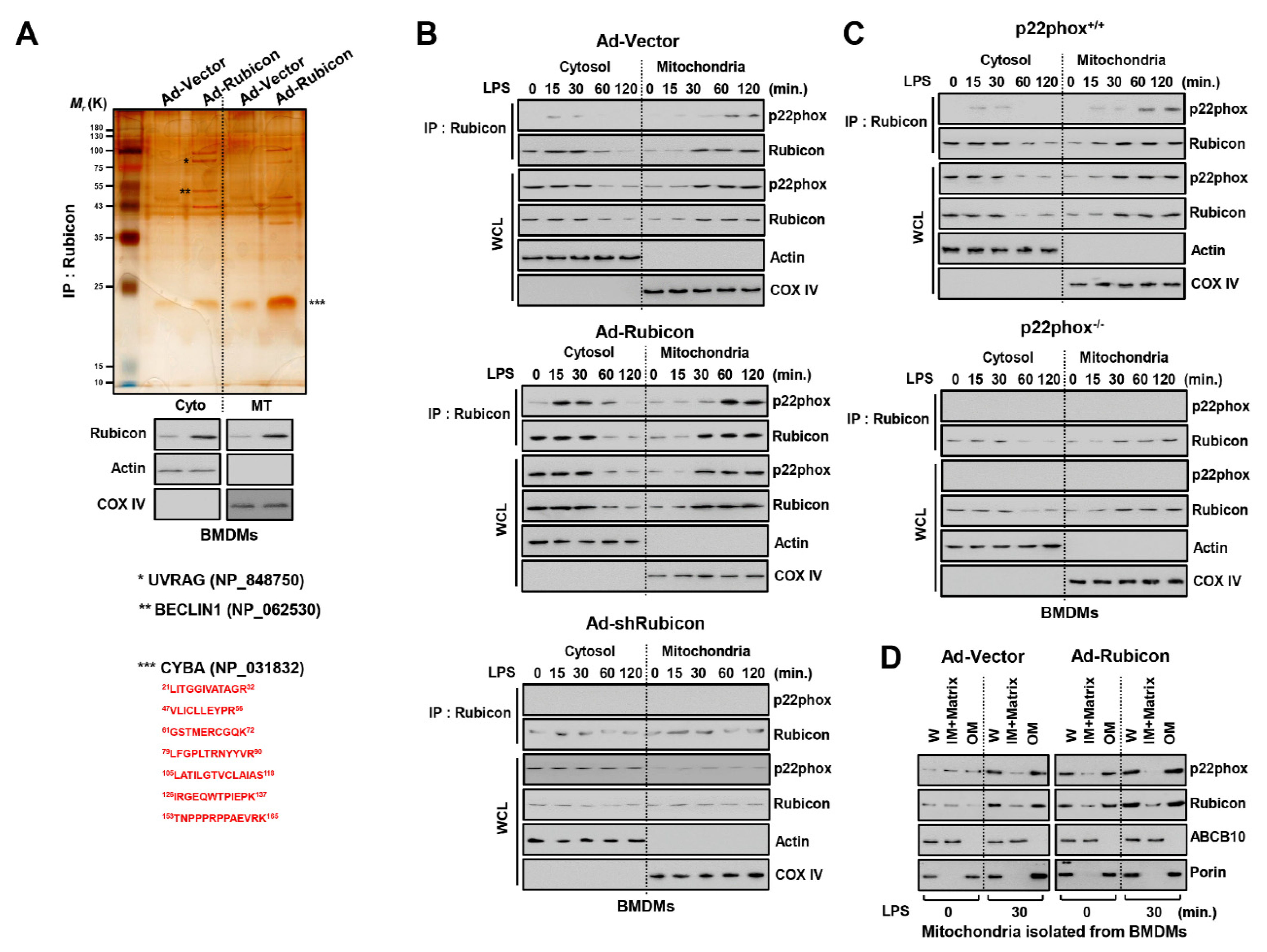
3.2. Rubicon Interacts with P22phox in the Mitochondrial Outer Membrane
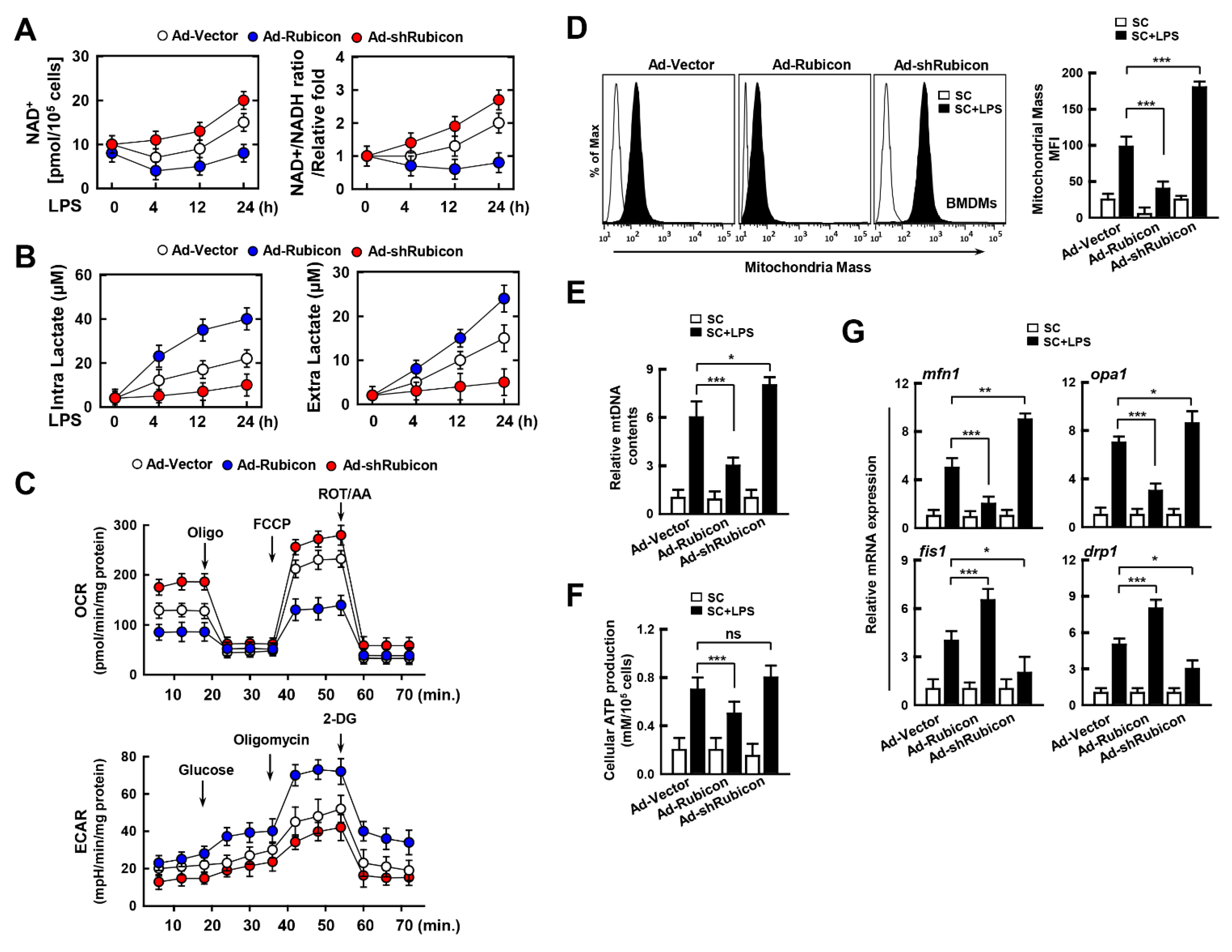
3.3. Interaction of Rubicon-p22phox Enhances Both Cytosolic ROS and mtROS and Reduces Mitochondrial Activity and Biogenesis
3.4. Rubicon Inhibits Mitochondrial Metabolism by Upregulating Glycolysis
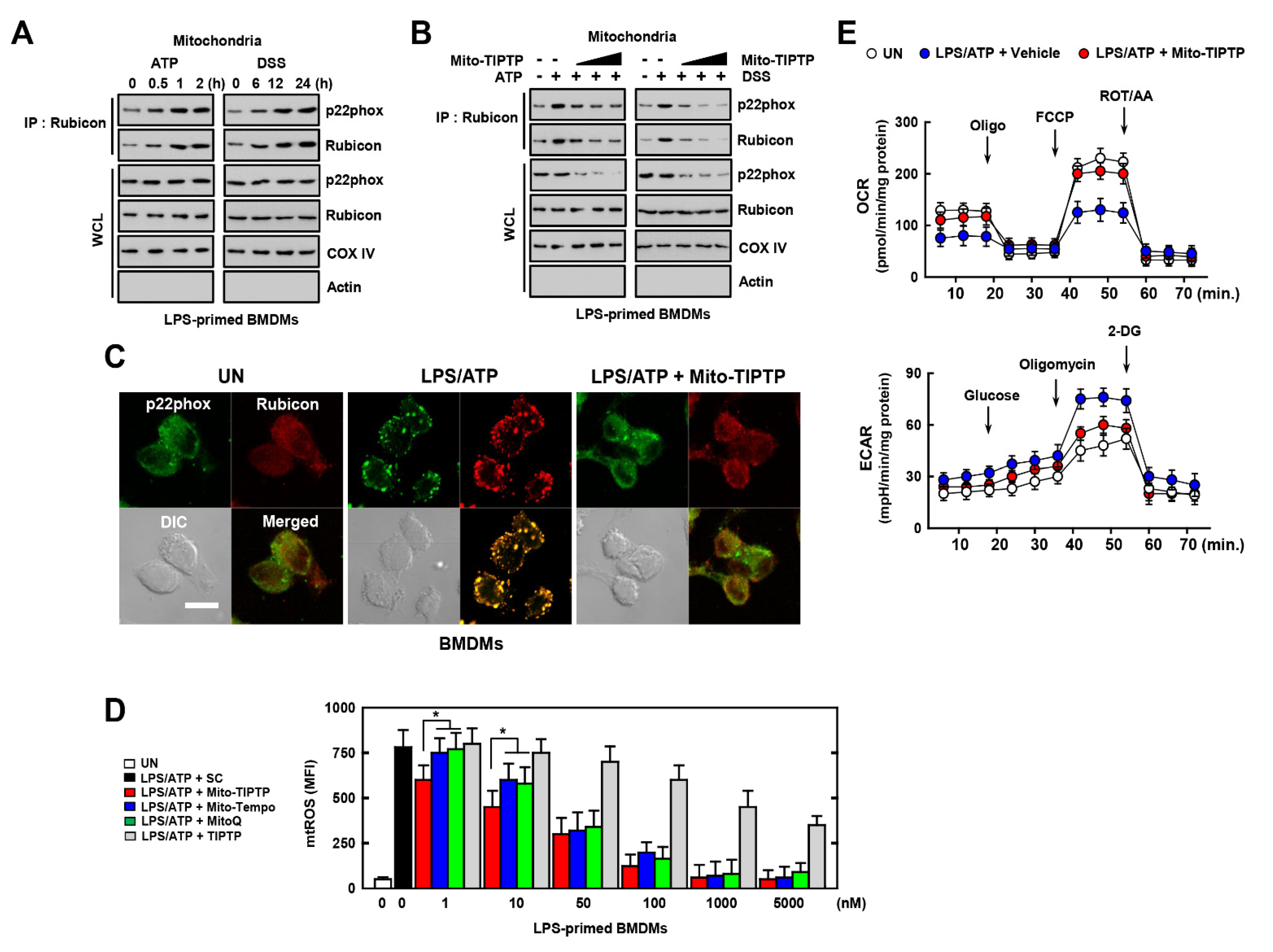
3.5. Mito-TIPTP Enhances Mitochondrial Function through Inhibition of the Rubicon-p22phox Interaction
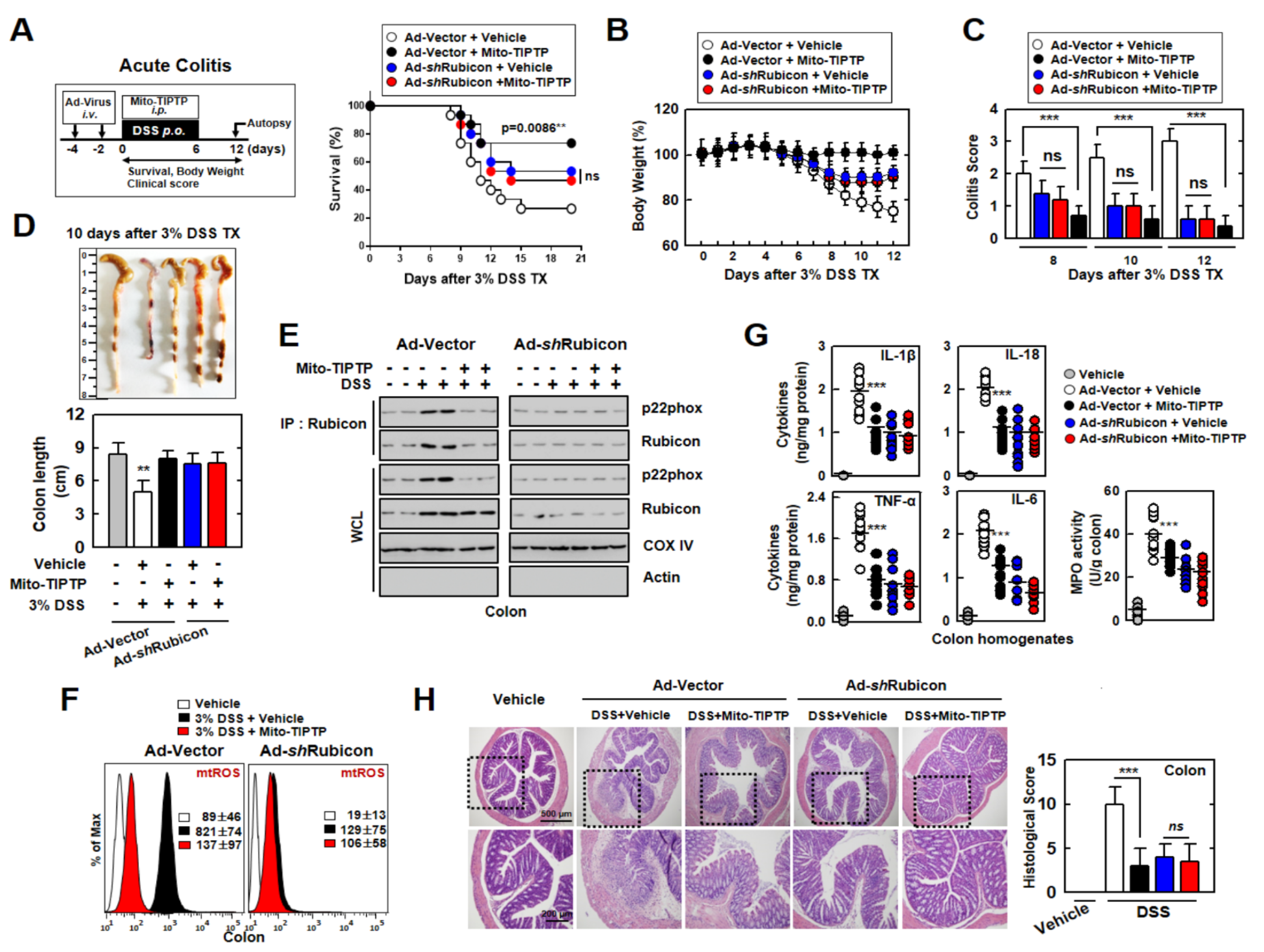
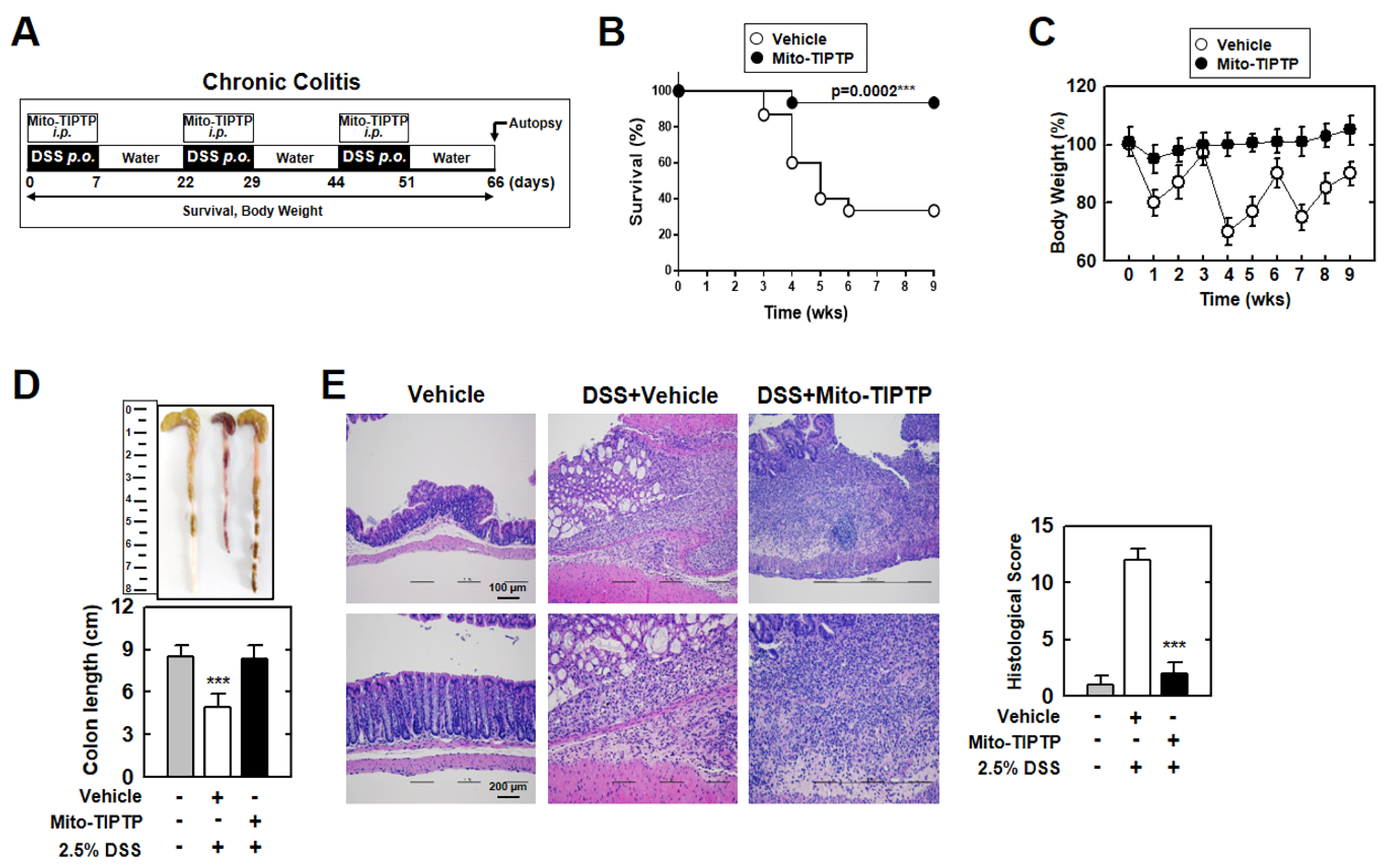
3.6. Mito-TIPTP Alleviates Acute and Chronic DSS-Induced Colitis in Mice
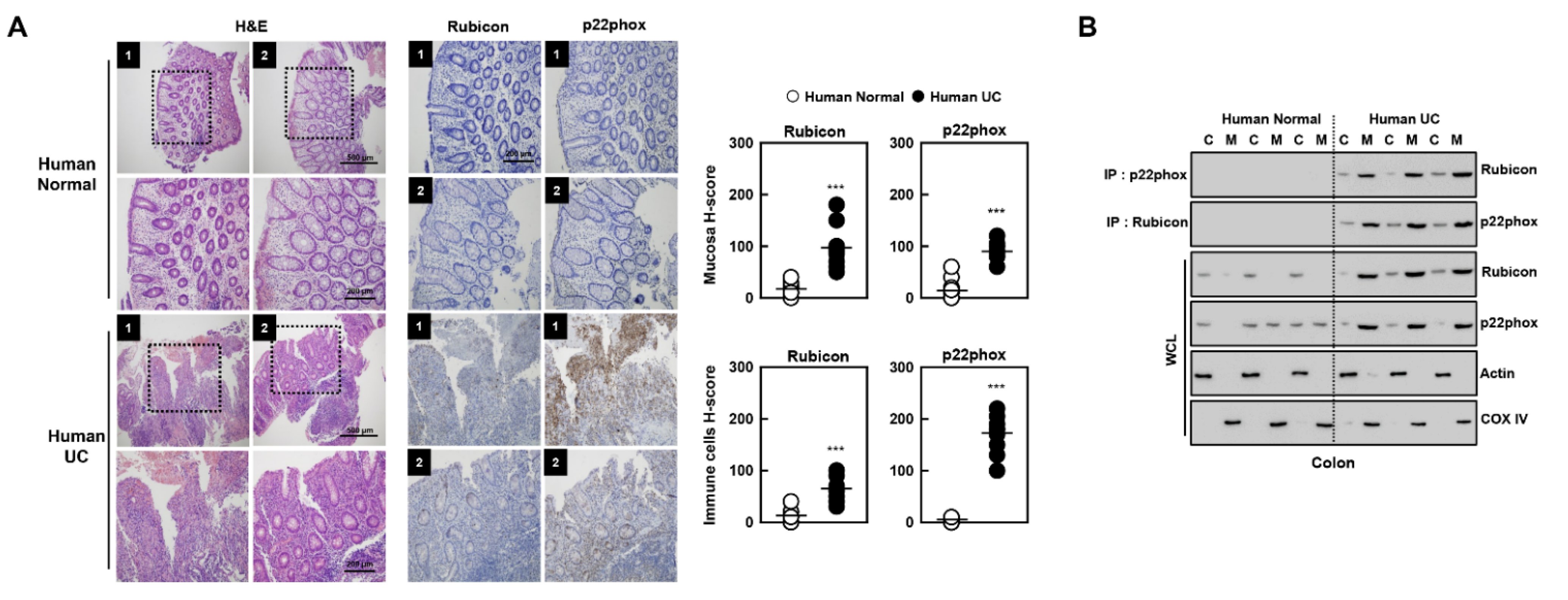
3.7. Novel Biomarkers of Patients with Ulcerative Colitis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Rubicon | Run/cysteine-rich-domain-containing Beclin1-interacting autophagy protein |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| ROS | Reactive oxygen species |
| mtROS | Mitochondrial ROS |
| LPS | Lipopolysaccharide |
| BMDMs | Bone marrow-derived macrophages |
| ATP | Adenosine triphosphate |
| DSS | Dextran sulfate sodium |
| IBDs | Inflammatory bowel diseases |
| TIPTP | 2-(tetrahydroindazolyl)phenoxy-N-(thiadiazolyl) propenamide 2 |
| CLP | Cecal ligation procedure |
| TPP | Triphenylphosphonium |
| LAP | LC3-associated phagocytosis |
| OXPHOS | Oxidative phosphorylation |
| UQCRC2 | Cytochrome bc1 complex subunit 2 |
| UQCRQ | Cytochrome bc1 complex subunit 8 |
| PGC | Peroxisome proliferator-activated receptor gamma coactivator |
| NRF | Nuclear respiratory factor |
| Tfam | Mitochondrial transcription factor A |
| OCR | Oxygen consumption rate |
| ECAR | Extracellular acidification rate |
| Oligo | Oligomycin |
| FCCP | Carbonyl cyanide-p-trifluoromethoxy-phenylhydrazone |
| ROT/AA | Rotenone/antimycin A |
| 2-DG | 2-deoxy-d-glucose |
| IL-1β | Interleukin-1β |
| TLRs | Toll-like receptors |
| FcRs | Fc receptors |
| Mo-DC | Monocyte-derived dendritic cells |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| IB | Immunoblotting |
| IP | Immunoprecipitation |
| ELISA | Enzyme-linked immunosorbent assay |
| H&E staining | hematoxylin and eosin staining |
References
- Balmus, I.M.; Ciobica, A.; Trifan, A.; Stanciu, C. The implications of oxidative stress and antioxidant therapies in Inflammatory Bowel Disease: Clinical aspects and animal models. Saudi. J. Gastroenterol. 2016, 22, 3–17. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Schneider, A.M.; Özsoy, M.; Zimmermann, F.A.; Feichtinger, R.G.; Mayr, J.A.; Kofler, B.; Sperl, W.; Weghuber, D.; Mörwald, K. Age-Related Deterioration of Mitochondrial Function in the Intestine. Oxid. Med. Cell Longev. 2020, 2020, 4898217. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—Gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.P.; Hadley, G.; Fratter, C.; Cullen, S.N.; Bax, B.E.; Bain, M.D.; Sapsford, R.A.; Poulton, J.; Travis, S.P. Unexplained gastrointestinal symptoms: Think mitochondrial disease. Dig. Liver. Dis. 2014, 46, 1–8. [Google Scholar] [CrossRef]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Keita, A.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Geuking, M.B.; McCoy, K.D. Innate and adaptive immunity in host-microbiota mutualism. Front. Biosci. 2012, 4, 685–698. [Google Scholar] [CrossRef]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [PubMed]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic. Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef]
- Yang, C.S.; Lee, J.S.; Rodgers, M.; Min, C.K.; Lee, J.Y.; Kim, H.J.; Lee, K.H.; Kim, C.J.; Oh, B.; Zandi, E.; et al. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host. Microbe. 2012, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Koh, H.J.; Kim, J.S.; Yun, J.S.; Jang, K.; Lee, J.Y.; Jung, J.U.; Yang, C.S. Peptide inhibition of p22phox and Rubicon interaction as a therapeutic strategy for septic shock. Biomaterials 2016, 101, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Kim, J.S.; Gu, S.J.; Jo, S.; Kim, S.; Young Kim, S.; Lee, D.; Jang, K.; Choo, H.; Kim, T.H.; et al. Identification of highly potent and selective inhibitor, TIPTP, of the p22phox-Rubicon axis as a therapeutic agent for rheumatoid arthritis. Sci. Rep. 2020, 10, 4570. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, D.; Kim, S.; Lee, D.; Mun, S.J.; Cho, E.; Son, W.; Jang, K.; Yang, C.S. Mycobacterium tuberculosis Rv2626c-derived peptide as a therapeutic agent for sepsis. EMBO Mol. Med. 2020, 12, e12497. [Google Scholar] [CrossRef]
- Yang, C.S.; Kim, J.J.; Kim, T.S.; Lee, P.Y.; Kim, S.Y.; Lee, H.M.; Shin, D.M.; Nguyen, L.T.; Lee, M.S.; Jin, H.S.; et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6115. [Google Scholar] [CrossRef]
- Williamson, C.D.; Wong, D.S.; Bozidis, P.; Zhang, A.; Colberg-Poley, A.M. Isolation of Endoplasmic Reticulum, Mitochondria, and Mitochondria-Associated Membrane and Detergent Resistant Membrane Fractions from Transfected Cells and from Human Cytomegalovirus-Infected Primary Fibroblasts. Curr. Protoc. Cell Biol. 2015, 68, 3.27.1–3.27.33. [Google Scholar] [CrossRef]
- Walker, L.R.; Hussein, H.A.; Akula, S.M. Subcellular fractionation method to study endosomal trafficking of Kaposi’s sarcoma-associated herpesvirus. Cell Biosci. 2016, 6, 1. [Google Scholar] [CrossRef]
- Annunziata, I.; Weesner, J.A.; d’Azzo, A. Isolation of Mitochondria-Associated ER Membranes (MAMs), Synaptic MAMs, and Glycosphingolipid Enriched Microdomains (GEMs) from Brain Tissues and Neuronal Cells. Methods Mol. Biol. 2021, 2277, 357–370. [Google Scholar] [PubMed]
- Nishimura, N.; Gotoh, T.; Oike, Y.; Yano, M. TMEM65 is a mitochondrial inner-membrane protein. PeerJ 2014, 2, e349. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Hwang, J.; Koh, H.J.; Jang, K.; Lee, J.D.; Choi, J.; Yang, C.S. The targeted delivery of the c-Src peptide complexed with schizophyllan to macrophages inhibits polymicrobial sepsis and ulcerative colitis in mice. Biomaterials 2016, 89, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, Y.; Horinouchi, H.; Asao, T.; Kitahara, S.; Goto, Y.; Kanda, S.; Fujiwara, Y.; Nokihara, H.; Yamamoto, N.; Watanabe, S.; et al. Expression of programmed death 1 (PD-1) and its ligand (PD-L1) in thymic epithelial tumors: Impact on treatment efficacy and alteration in expression after chemotherapy. Lung Cancer 2016, 99, 4–10. [Google Scholar] [CrossRef]
- Wong, S.W.; Sil, P.; Martinez, J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J. 2018, 285, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 Inflammasome in Inflammatory Bowel Disease: Putting out the Fire of Inflammation. Inflammation 2019, 42, 1147–1159. [Google Scholar] [CrossRef]
- Chen, Q.L.; Yin, H.R.; He, Q.Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef]
- Wang, J.Y.; Li, J.Q.; Xiao, Y.M.; Fu, B.; Qin, Z.H. Triphenylphosphonium (TPP)-Based Antioxidants: A New Perspective on Antioxidant Design. ChemMedChem 2020, 15, 404–410. [Google Scholar] [CrossRef]
- Martinez, J. LAP it up, fuzz ball: A short history of LC3-associated phagocytosis. Curr. Opin. Immunol. 2018, 55, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Saito, T. CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 2009, 30, 234–242. [Google Scholar] [CrossRef]
- Yang, C.S.; Rodgers, M.; Min, C.K.; Lee, J.S.; Kingeter, L.; Lee, J.Y.; Jong, A.; Kramnik, I.; Lin, X.; Jung, J.U. The autophagy regulator Rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host. Microbe. 2012, 11, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Tlili, A.; Marie, J.C.; Monteiro, R.C.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. NOX5 and p22phox are 2 novel regulators of human monocytic differentiation into dendritic cells. Blood 2017, 130, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox. Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Mungai, P.T.; Waypa, G.B.; Jairaman, A.; Prakriya, M.; Dokic, D.; Ball, M.K.; Schumacker, P.T. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell Biol. 2011, 31, 3531–3545. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- García-Nogales, P.; Almeida, A.; Bolaños, J.P. Peroxynitrite protects neurons against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. J. Biol. Chem. 2003, 278, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Sorrell, M.F.; Batra, S.K.; Dhawan, P.; Singh, A.B. Gut permeability and mucosal inflammation: Bad, good or context dependent. Mucosal. Immunol. 2017, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Stange, E.F.; Schroeder, B.O. Microbiota and mucosal defense in IBD: An update. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 963–976. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Experim. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef]
- Friedrich, M.; Diegelmann, J.; Schauber, J.; Auernhammer, C.J.; Brand, S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal. Immunol. 2015, 8, 943–958. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Bond, S.T.; Kim, J.; Calkin, A.C.; Drew, B.G. The Antioxidant Moiety of MitoQ Imparts Minimal Metabolic Effects in Adipose Tissue of High Fat Fed Mice. Front. Physiol. 2019, 10, 543. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry 2007, 72, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver. Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-S.; Kim, Y.-R.; Jang, S.; Wang, S.G.; Cho, E.; Mun, S.-J.; Jeon, H.-I.; Kim, H.-K.; Min, S.-J.; Yang, C.-S. Mito-TIPTP Increases Mitochondrial Function by Repressing the Rubicon-p22phox Interaction in Colitis-Induced Mice. Antioxidants 2021, 10, 1954. https://doi.org/10.3390/antiox10121954
Kim J-S, Kim Y-R, Jang S, Wang SG, Cho E, Mun S-J, Jeon H-I, Kim H-K, Min S-J, Yang C-S. Mito-TIPTP Increases Mitochondrial Function by Repressing the Rubicon-p22phox Interaction in Colitis-Induced Mice. Antioxidants. 2021; 10(12):1954. https://doi.org/10.3390/antiox10121954
Chicago/Turabian StyleKim, Jae-Sung, Ye-Ram Kim, Sein Jang, Sang Geon Wang, Euni Cho, Seok-Jun Mun, Hye-In Jeon, Hyo-Keun Kim, Sun-Joon Min, and Chul-Su Yang. 2021. "Mito-TIPTP Increases Mitochondrial Function by Repressing the Rubicon-p22phox Interaction in Colitis-Induced Mice" Antioxidants 10, no. 12: 1954. https://doi.org/10.3390/antiox10121954
APA StyleKim, J.-S., Kim, Y.-R., Jang, S., Wang, S. G., Cho, E., Mun, S.-J., Jeon, H.-I., Kim, H.-K., Min, S.-J., & Yang, C.-S. (2021). Mito-TIPTP Increases Mitochondrial Function by Repressing the Rubicon-p22phox Interaction in Colitis-Induced Mice. Antioxidants, 10(12), 1954. https://doi.org/10.3390/antiox10121954