
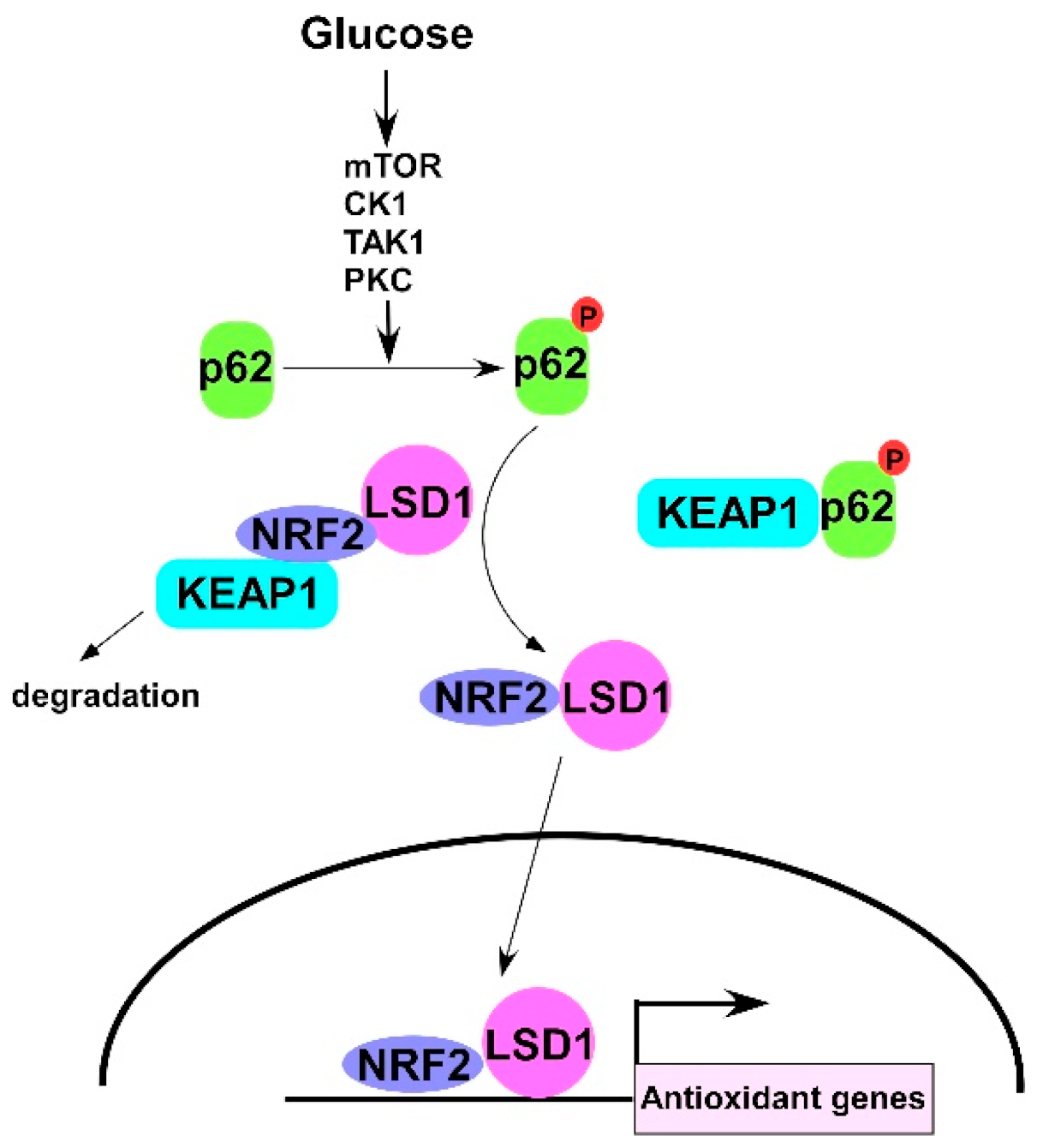
Glucose Activates Lysine-Specific Demethylase 1 through the KEAP1/p62 Pathway

 , , ,
, , ,
Abstract
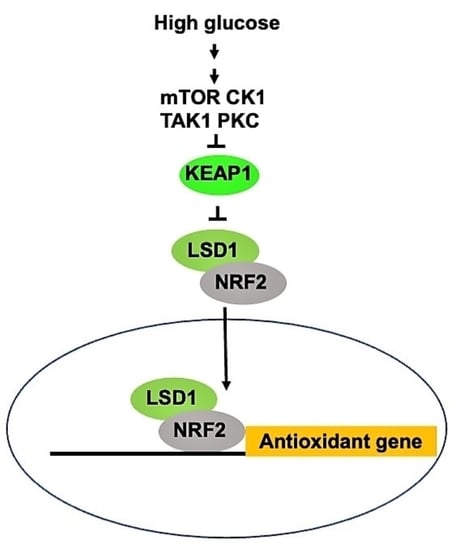
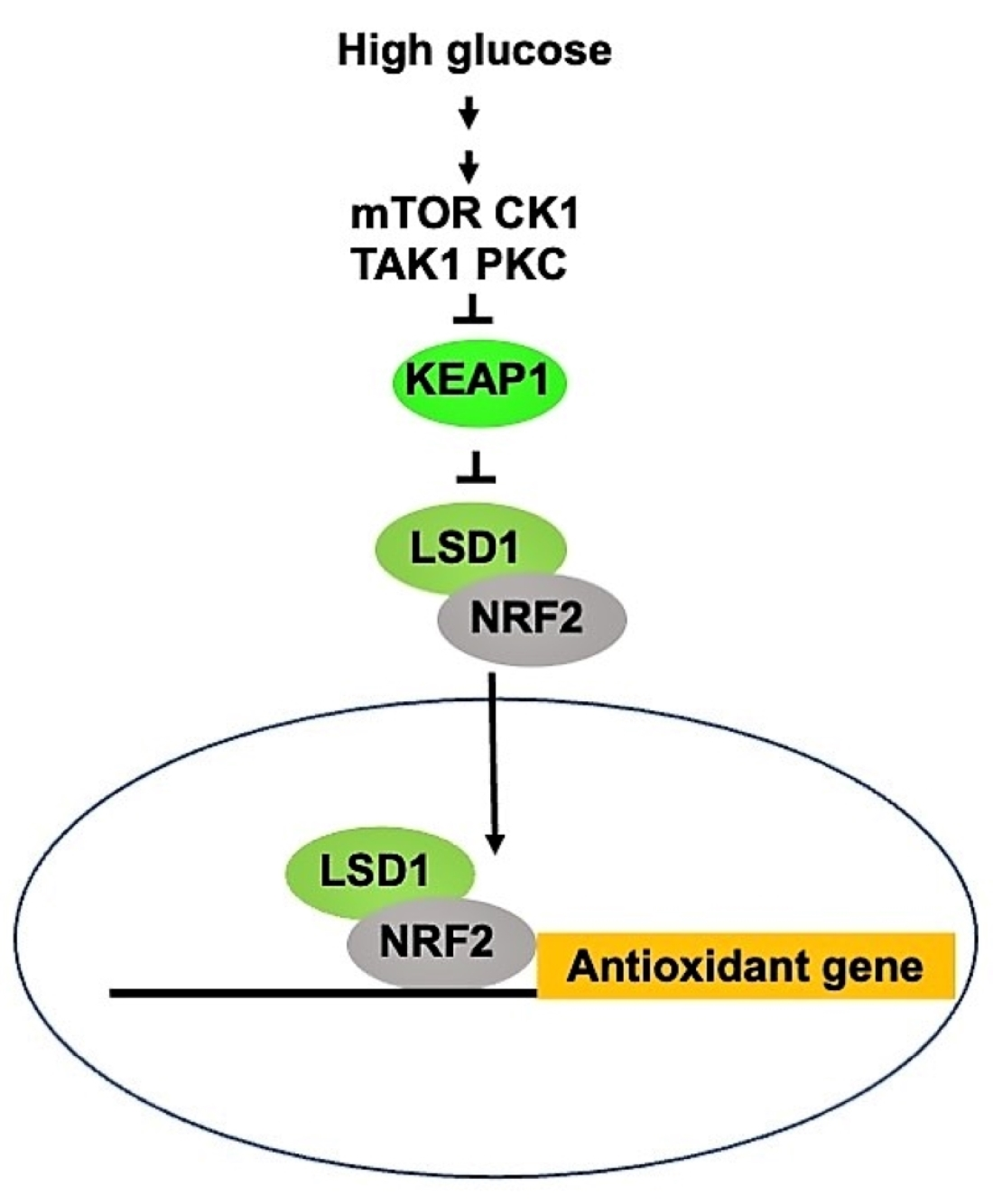{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment of Cell Lines
2.2. Tissue Specimens
2.3. Isolation and Maintenance of Primary Endometrial Stroma Cells
2.4. DNA Construction
2.5. DNA and siRNA Transfection
2.6. RNA Extraction and Real-Time Q-PCR
2.7. Cell Proliferation Assay
2.8. Western Blot
2.9. Immunoprecipitation
2.10. Confocal Microscopy
2.11. Proximity Ligation Assay
2.12. Animal Experiments and Immunohistochemistry
3. Results
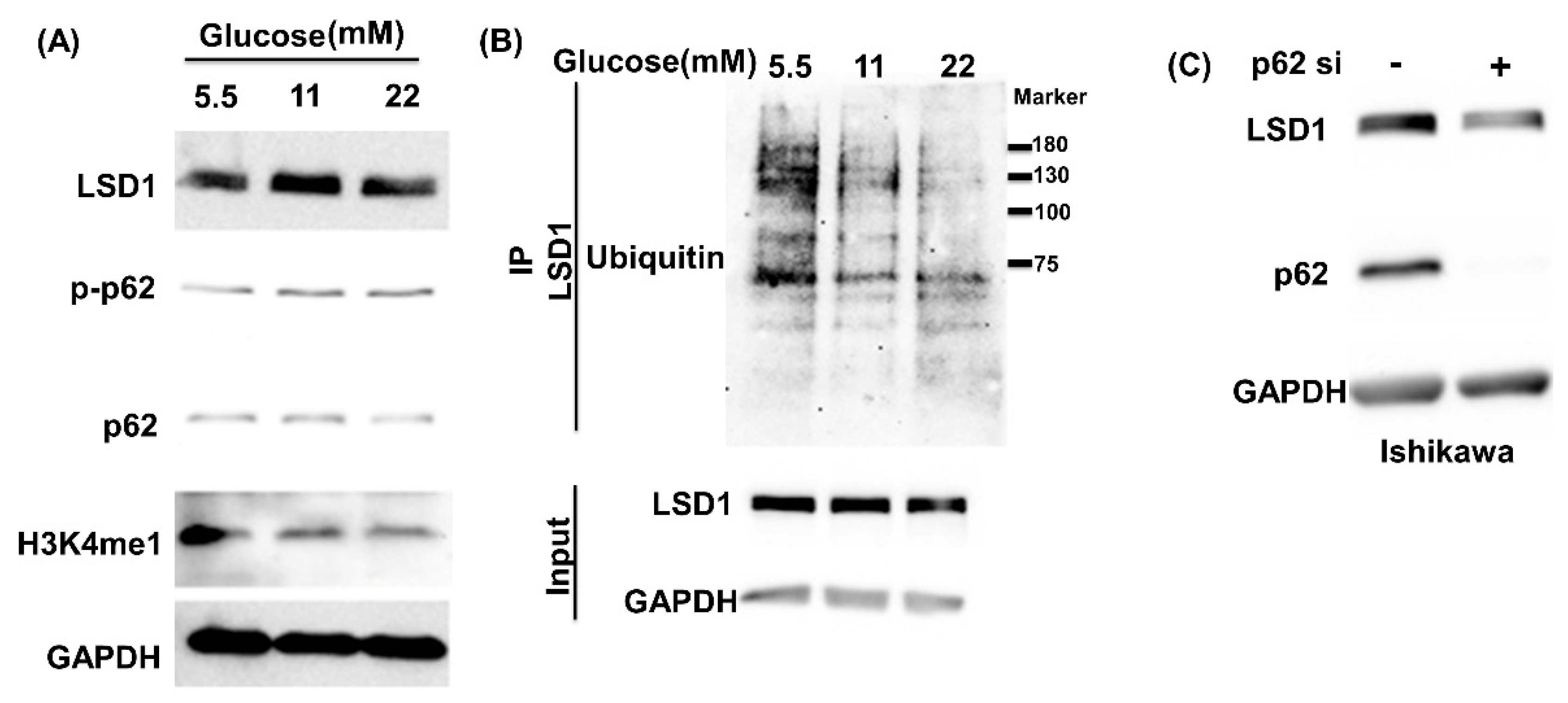
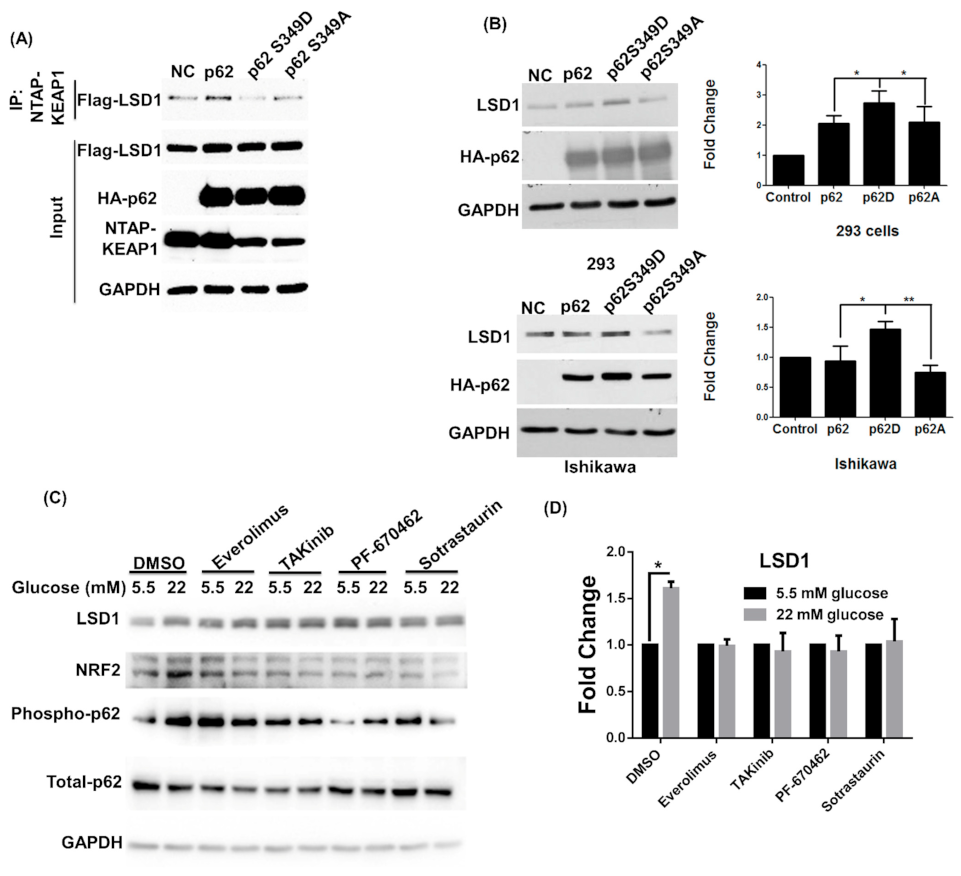
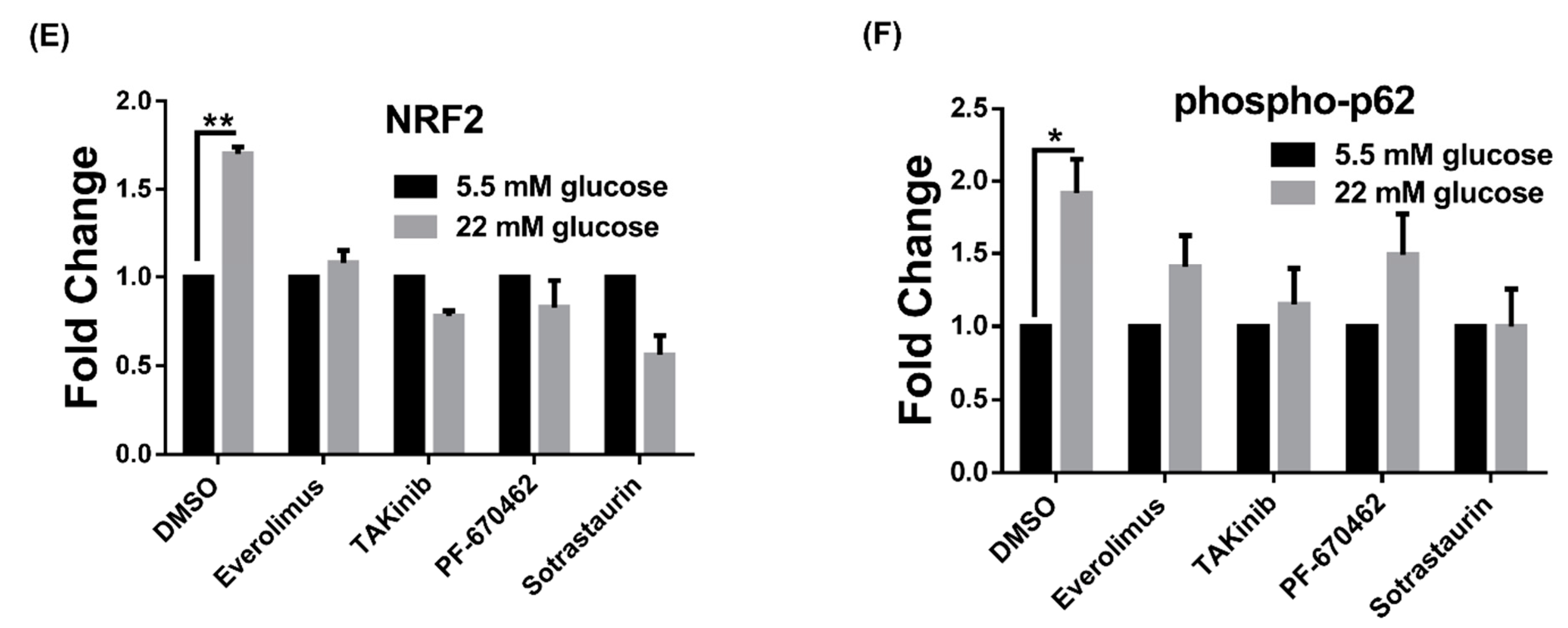
3.1. High Glucose Induces LSD1 Expression and p62 Phosphorylation
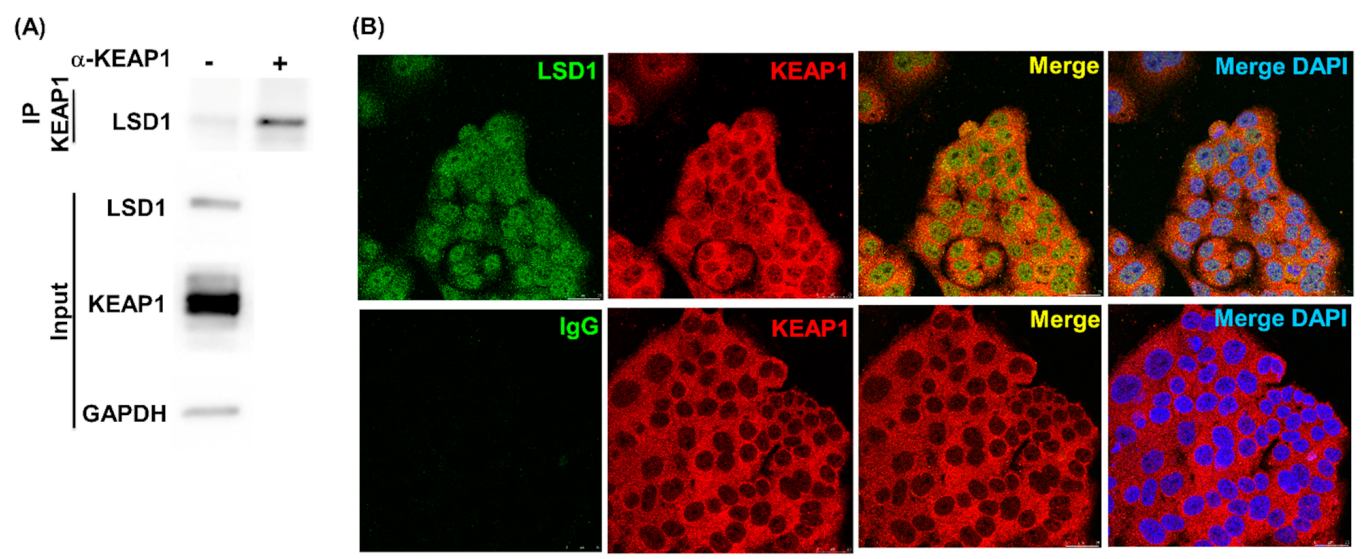
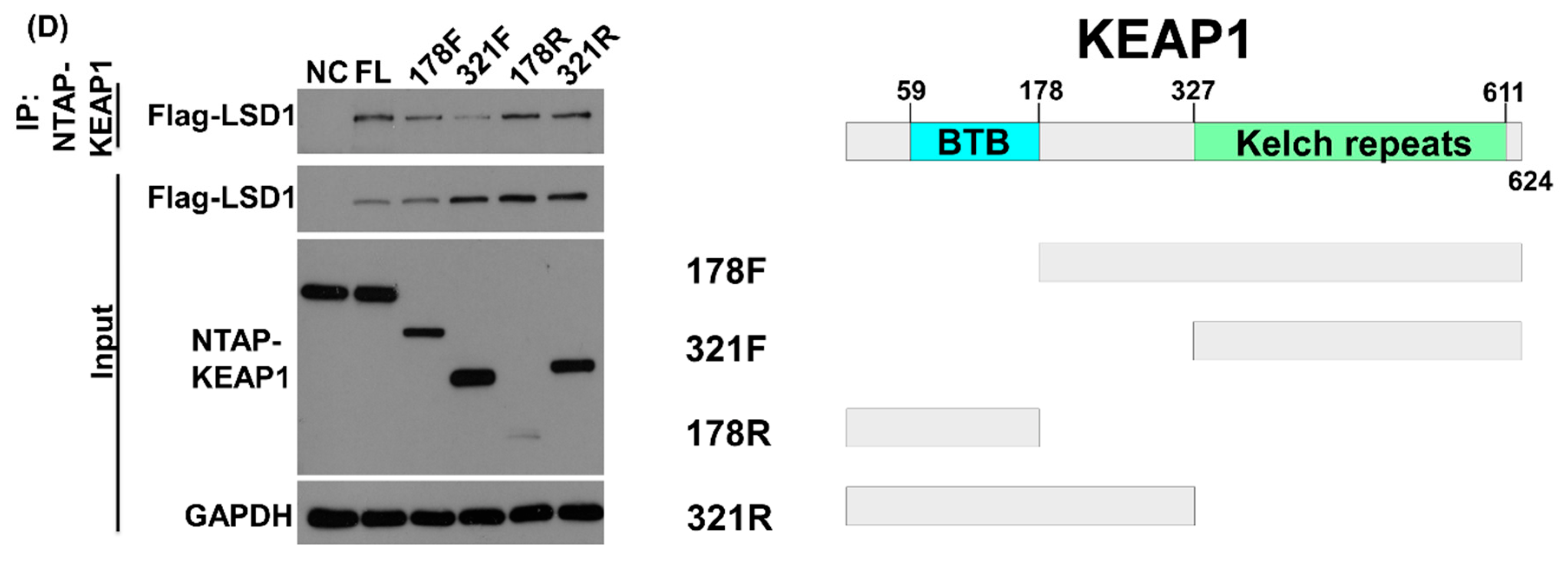
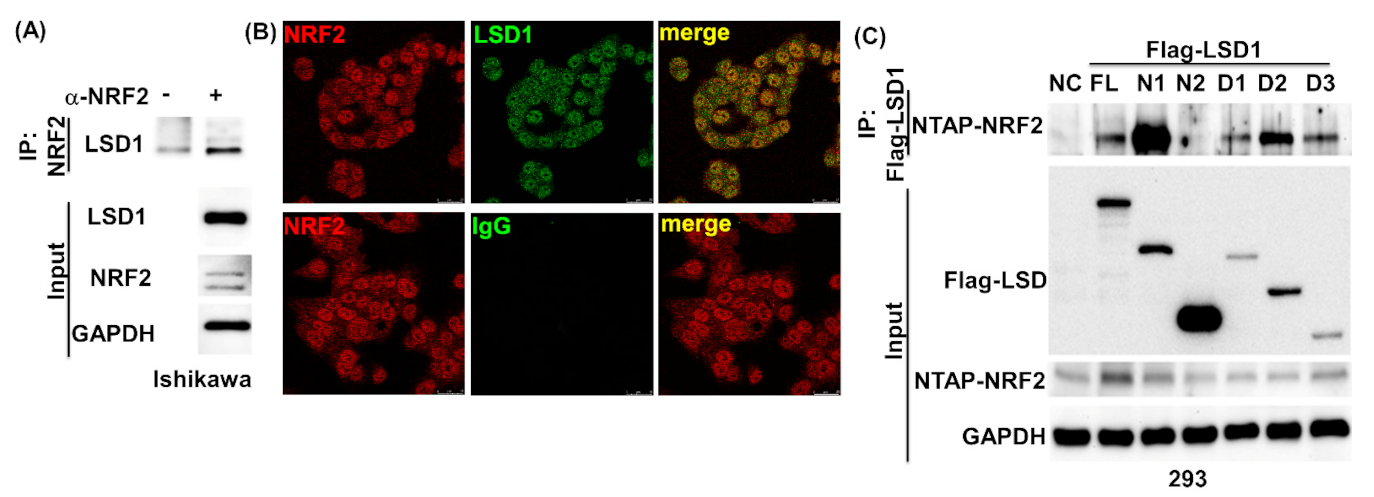
3.2. LSD1 Interacts with KEAP1
3.3. LSD1 Is Upregulated in Phospho-p62 Mimic–Expressed Cells
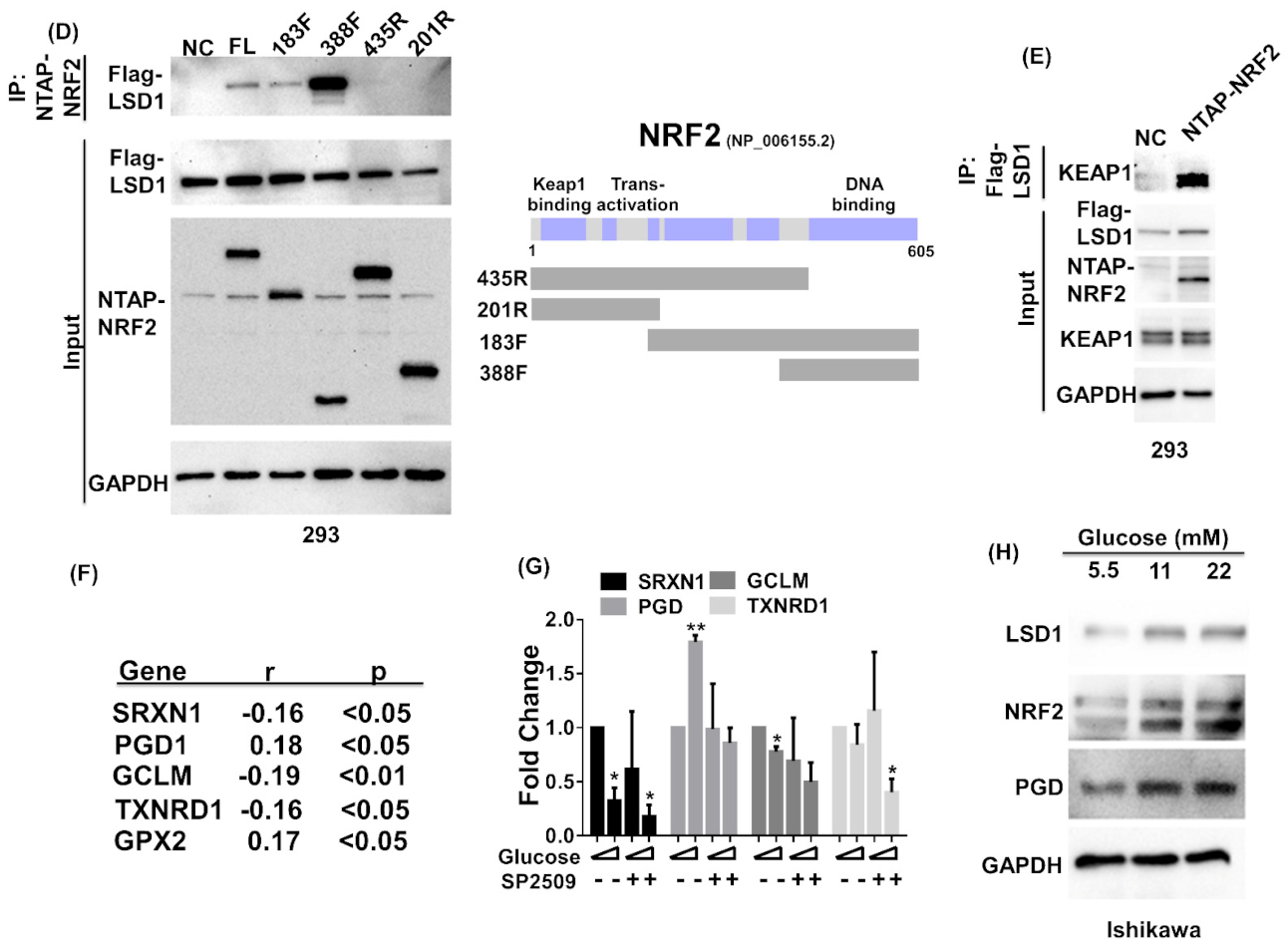
3.4. NRF2 Connects KEPA1 to LSD1
3.5. LSD1 Participates in NRF2-Regulated Gene Expression
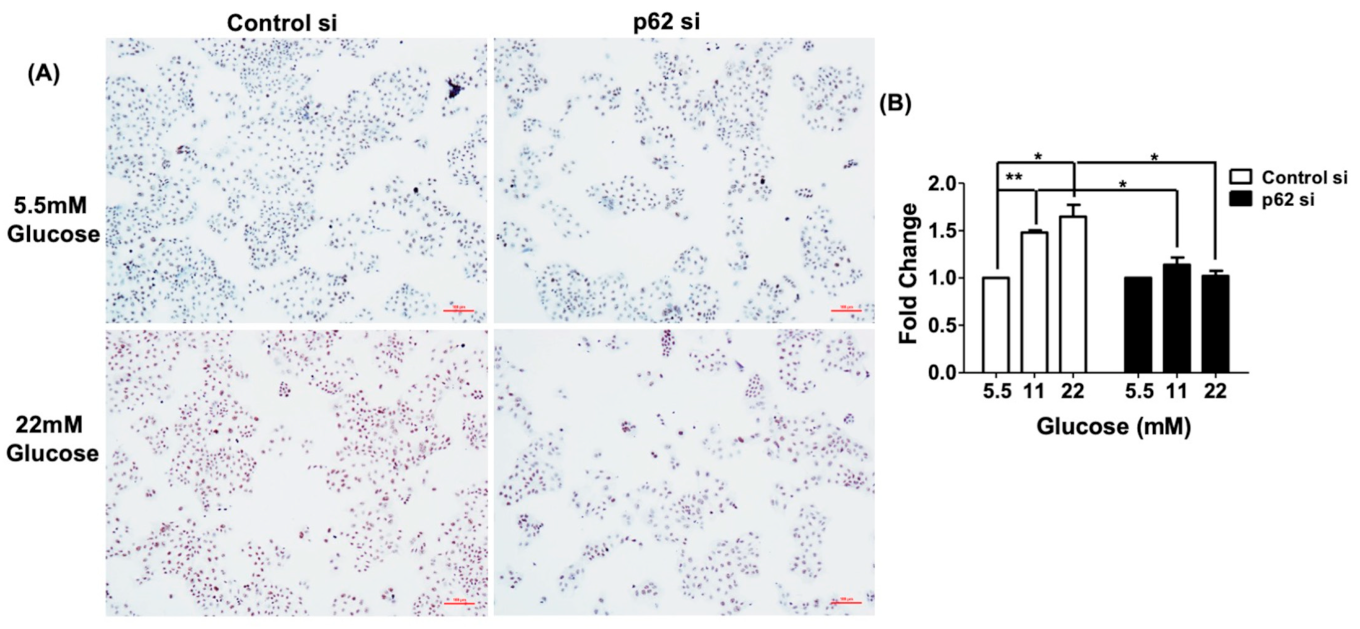
3.6. Cell Proliferation Induced by Glucose Is Blocked by p62 siRNA
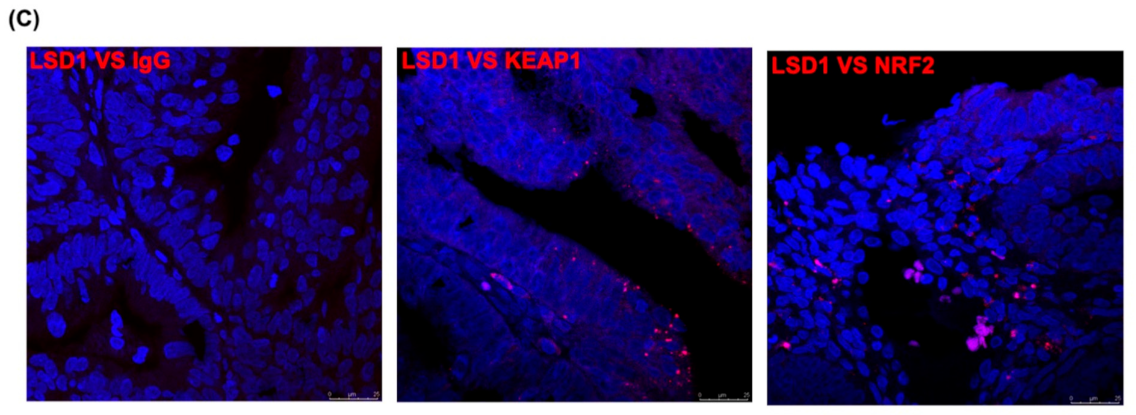
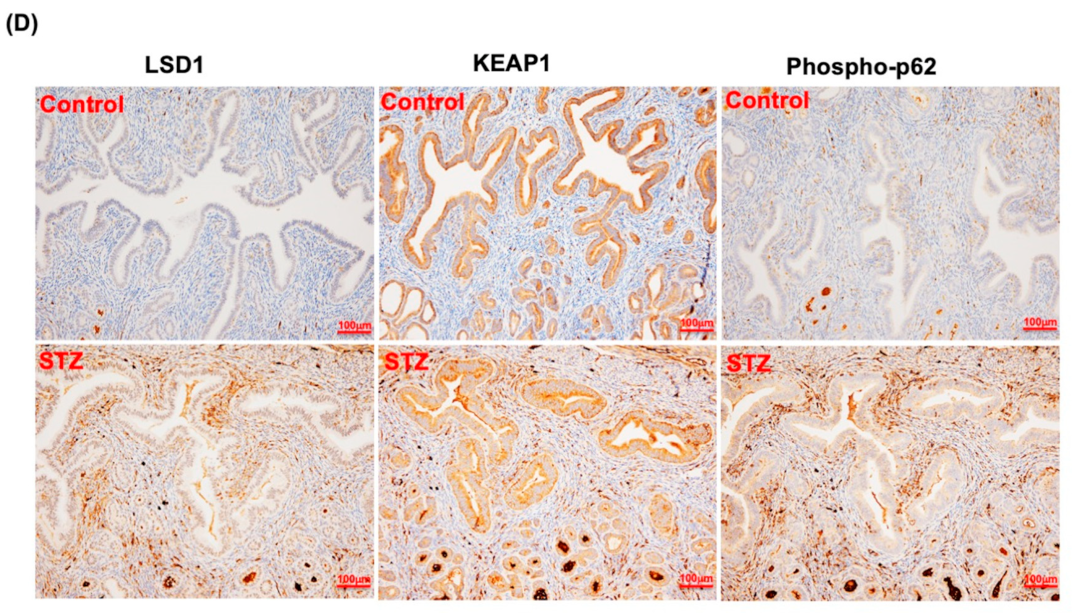
3.7. LSD1 Interactions with KEAP1, p62, and NRF2 In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, K.H.; Broaddus, R.R. Endometrial Cancer. N. Engl. J. Med. 2020, 383, 2053–2064. [Google Scholar] [CrossRef]
- Garg, K.; Soslow, R.A. Endometrial carcinoma in women aged 40 years and younger. Arch. Pathol. Lab. Med. 2014, 138, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I. Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol. Oncol. 2004, 94, 256–266. [Google Scholar] [CrossRef]
- Byrne, F.L.; Martin, A.R.; Kosasih, M.; Caruana, B.T.; Farrell, R. The Role of Hyperglycemia in Endometrial Cancer Pathogenesis. Cancers 2020, 12, 1191. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Sen, A.; Vatten, L.J. Hypertension and the risk of endometrial cancer: A systematic review and meta-analysis of case-control and cohort studies. Sci. Rep. 2017, 7, 44808. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Zucchetto, A.; Bosetti, C.; Dal Maso, L.; Montella, M.; Pelucchi, C.; Negri, E.; Franceschi, S.; La Vecchia, C. Metabolic syndrome and endometrial cancer risk. Ann. Oncol. 2011, 22, 884–889. [Google Scholar] [CrossRef]
- Garg, S.K.; Maurer, H.; Reed, K.; Selagamsetty, R. Diabetes and cancer: Two diseases with obesity as a common risk factor. Diabetes Obes. Metab. 2014, 16, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Masur, K.; Vetter, C.; Hinz, A.; Tomas, N.; Henrich, H.; Niggemann, B.; Zanker, K.S. Diabetogenic glucose and insulin concentrations modulate transcriptome and protein levels involved in tumour cell migration, adhesion and proliferation. Br. J. Cancer 2011, 104, 345–352. [Google Scholar] [CrossRef]
- Hou, Y.; Zhou, M.; Xie, J.; Chao, P.; Feng, Q.; Wu, J. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer Targets Ther. 2017, 9, 429–436. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Gu, C.J.; Xie, F.; Zhang, B.; Yang, H.L.; Cheng, J.; He, Y.Y.; Zhu, X.Y.; Li, D.J.; Li, M.Q. High Glucose Promotes Epithelial-Mesenchymal Transition of Uterus Endometrial Cancer Cells by Increasing ER/GLUT4-Mediated VEGF Secretion. Cell. Physiol. Biochem. 2018, 50, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Phoomak, C.; Vaeteewoottacharn, K.; Silsirivanit, A.; Saengboonmee, C.; Seubwai, W.; Sawanyawisuth, K.; Wongkham, C.; Wongkham, S. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci. Rep. 2017, 7, 43842. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.C.; Yang, W.V. Hyperglycemia, tumorigenesis, and chronic inflammation. Crit. Rev. Oncol. Hematol. 2016, 108, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Tien, S.C.; Hsieh, P.K.; Jeng, Y.M.; Chang, M.C.; Chang, Y.T.; Chen, Y.J.; Chen, Y.J.; Lee, E.Y.P.; Lee, W.H. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metab. 2019, 29, 1334–1349.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Sang, H.; Zhou, Y.; Shang, C.; Wang, Y.; Zhu, H. Effects of hyperglycemia on the progression of tumor diseases. J. Exp. Clin. Cancer Res. 2019, 38, 327. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Blenis, J.; Gomes, A.P. Beyond the Warburg Effect: How Do Cancer Cells Regulate One-Carbon Metabolism? Front. Cell Dev. Biol. 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Mossenta, M.; Busato, D.; Dal Bo, M.; Toffoli, G. Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies. Cancers 2020, 12, 1668. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef]
- Tanji, K.; Miki, Y.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Y.; Wang, S.; Liu, Y.; Zhang, J.; Xu, Y.; Zhang, Z.; Bao, W.; Wu, S. LSD1 sustains estrogen-driven endometrial carcinoma cell proliferation through the PI3K/AKT pathway via di-demethylating H3K9 of cyclin D1. Int. J. Oncol. 2017, 50, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Majello, B.; Gorini, F.; Sacca, C.D.; Amente, S. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers 2019, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Chao, A.S.; Jung, S.M.; Lin, C.Y.; Chao, A.; Wang, T.H. Stress-induced phosphoprotein 1 acts as a scaffold protein for glycogen synthase kinase-3 beta-mediated phosphorylation of lysine-specific demethylase 1. Oncogenesis 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Suzuki, T.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget 2015, 6, 16939–16950. [Google Scholar] [CrossRef]
- Liu, J.; Feng, J.; Li, L.; Lin, L.; Ji, J.; Lin, C.; Liu, L.; Zhang, N.; Duan, D.; Li, Z.; et al. Arginine methylation-dependent LSD1 stability promotes invasion and metastasis of breast cancer. EMBO Rep. 2020, 21, e48597. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: Role of histone methylation. Investig. Opthalmol. Vis. Sci. 2013, 54, 244–250. [Google Scholar] [CrossRef]
- Nishida, M. The Ishikawa cells from birth to the present. Hum. Cell 2002, 15, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.F.; Kim, S.H.; Liao, S.K.; Atabekoglu, C.; Uckac, S.; Arici, A.; Arlier, S.; Lee, C.L.; Wang, H.S.; Kayisli, U.A. Increased expression of integrin-linked kinase during decidualization regulates the morphological transformation of endometrial stromal cells. Fertil. Steril. 2017, 107, 803–812. [Google Scholar] [CrossRef]
- Chao, A.; Lin, C.Y.; Chao, A.N.; Tsai, C.L.; Chen, M.Y.; Lee, L.Y.; Chang, T.C.; Wang, T.H.; Lai, C.H.; Wang, H.S. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 2017, 8, 74434–74450. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Chao, A.; Tsai, C.L.; Chang, C.L.; Chen, S.H.; Lee, Y.S.; Chen, J.K.; Lin, Y.J.; Chang, P.Y.; Wang, C.J.; et al. Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell. Proteom. 2010, 9, 1873–1884. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chao, A.; Jung, S.M.; Tsai, C.N.; Lin, C.Y.; Chen, S.H.; Sue, S.C.; Wang, T.H.; Wang, H.S.; Lai, C.H. Stress-induced phosphoprotein-1 maintains the stability of JAK2 in cancer cells. Oncotarget 2016, 7, 50548–50563. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef]
- Watanabe, Y.; Tsujimura, A.; Taguchi, K.; Tanaka, M. HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis. Autophagy 2017, 13, 133–148. [Google Scholar] [CrossRef]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid. Redox Signal. 2016, 25, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Tramontano, A.; Pezone, A.; Migliaccio, A. LSD1: More than demethylation of histone lysine residues. Exp. Mol. Med. 2020, 52, 1936–1947. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, L.; Guo, H.; Wysham, W.Z.; Roque, D.R.; Willson, A.K.; Sheng, X.; Zhou, C.; Bae-Jump, V.L. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol. Oncol. 2015, 138, 668–675. [Google Scholar] [CrossRef]
- Wallbillich, J.J.; Josyula, S.; Saini, U.; Zingarelli, R.A.; Dorayappan, K.D.; Riley, M.K.; Wanner, R.A.; Cohn, D.E.; Selvendiran, K. High Glucose-Mediated STAT3 Activation in Endometrial Cancer Is Inhibited by Metformin: Therapeutic Implications for Endometrial Cancer. PLoS ONE 2017, 12, e0170318. [Google Scholar] [CrossRef]
- Wu, K.K.; Huan, Y. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 2008, 40, 5.47.1–5.47.14. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef]
- Tseng, C.H. Metformin and endometrial cancer risk in Chinese women with type 2 diabetes mellitus in Taiwan. Gynecol. Oncol. 2015, 138, 147–153. [Google Scholar] [CrossRef] [PubMed]
- King, M.G.; Chandran, U.; Olson, S.H.; Demissie, K.; Lu, S.E.; Parekh, N.; Bandera, E.V. Consumption of sugary foods and drinks and risk of endometrial cancer. Cancer Causes Control 2013, 24, 1427–1436. [Google Scholar] [CrossRef]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015, 5, 8926. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Relic, B.; Charlier, E.; Deroyer, C.; Malaise, O.; Crine, Y.; Neuville, S.; Gillet, P.; de Seny, D.; Malaise, M.G. Serum starvation raises turnover of phosphorylated p62/SQSTM1 (Serine 349), reveals expression of proteasome and N-glycanase1 interactive protein RAD23B and sensitizes human synovial fibroblasts to BAY 11-7085-induced cell death. Oncotarget 2018, 9, 35830–35843. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsai, C.L.; Lin, C.Y.; Chao, A.; Lee, Y.S.; Wu, R.C.; Tsai, C.N.; Yen, C.F.; Chao, A.S. GPR30 Activation by 17beta-Estradiol Promotes p62 Phosphorylation and Increases Estrogen Receptor alpha Protein Expression by Inducing Its Release from a Complex Formed with KEAP1. J. Pers. Med. 2021, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Tu, Y.K.; Wu, M.S.; Lin, J.T.; Wang, H.P.; Chien, K.L. Blood glucose concentration and risk of pancreatic cancer: Systematic review and dose-response meta-analysis. BMJ 2015, 350, g7371. [Google Scholar] [CrossRef]
- Stattin, P.; Bjor, O.; Ferrari, P.; Lukanova, A.; Lenner, P.; Lindahl, B.; Hallmans, G.; Kaaks, R. Prospective study of hyperglycemia and cancer risk. Diabetes Care 2007, 30, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhang, T.; Jin, Z.; Guo, H.; Wei, X.; Liu, Y.; Chen, Q.; He, J. Blood glucose concentration and risk of liver cancer: Systematic review and meta-analysis of prospective studies. Oncotarget 2017, 8, 50164–50173. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-Y.; Chang, C.-B.; Wu, R.-C.; Chao, A.; Lee, Y.-S.; Tsai, C.-N.; Chen, C.-H.; Yen, C.-F.; Tsai, C.-L. Glucose Activates Lysine-Specific Demethylase 1 through the KEAP1/p62 Pathway. Antioxidants 2021, 10, 1898. https://doi.org/10.3390/antiox10121898
Lin C-Y, Chang C-B, Wu R-C, Chao A, Lee Y-S, Tsai C-N, Chen C-H, Yen C-F, Tsai C-L. Glucose Activates Lysine-Specific Demethylase 1 through the KEAP1/p62 Pathway. Antioxidants. 2021; 10(12):1898. https://doi.org/10.3390/antiox10121898
Chicago/Turabian StyleLin, Chiao-Yun, Chen-Bin Chang, Ren-Chin Wu, Angel Chao, Yun-Shien Lee, Chi-Neu Tsai, Chih-Hao Chen, Chih-Feng Yen, and Chia-Lung Tsai. 2021. "Glucose Activates Lysine-Specific Demethylase 1 through the KEAP1/p62 Pathway" Antioxidants 10, no. 12: 1898. https://doi.org/10.3390/antiox10121898
APA StyleLin, C.-Y., Chang, C.-B., Wu, R.-C., Chao, A., Lee, Y.-S., Tsai, C.-N., Chen, C.-H., Yen, C.-F., & Tsai, C.-L. (2021). Glucose Activates Lysine-Specific Demethylase 1 through the KEAP1/p62 Pathway. Antioxidants, 10(12), 1898. https://doi.org/10.3390/antiox10121898