
Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts

 , ,
, ,
Abstract
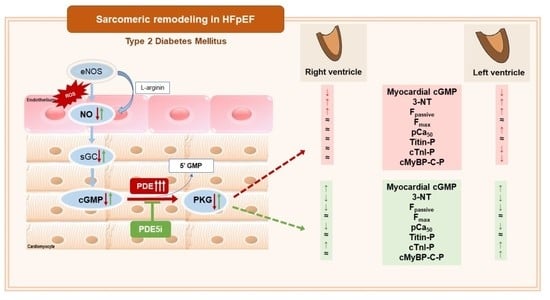
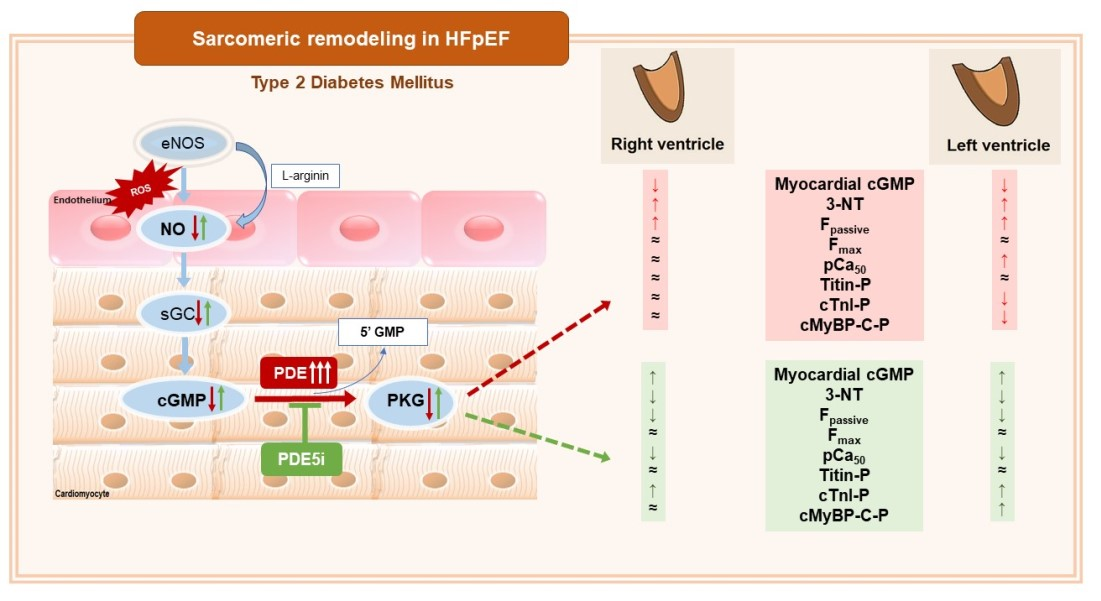
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Mechanical Measurements in Permeabilized Myocyte-Sized Preparations
2.3. SDS-PAGE and Western Immunoblot Analysis
2.4. Histology and Immunohistochemistry
2.5. Data Analysis and Statistics
3. Results
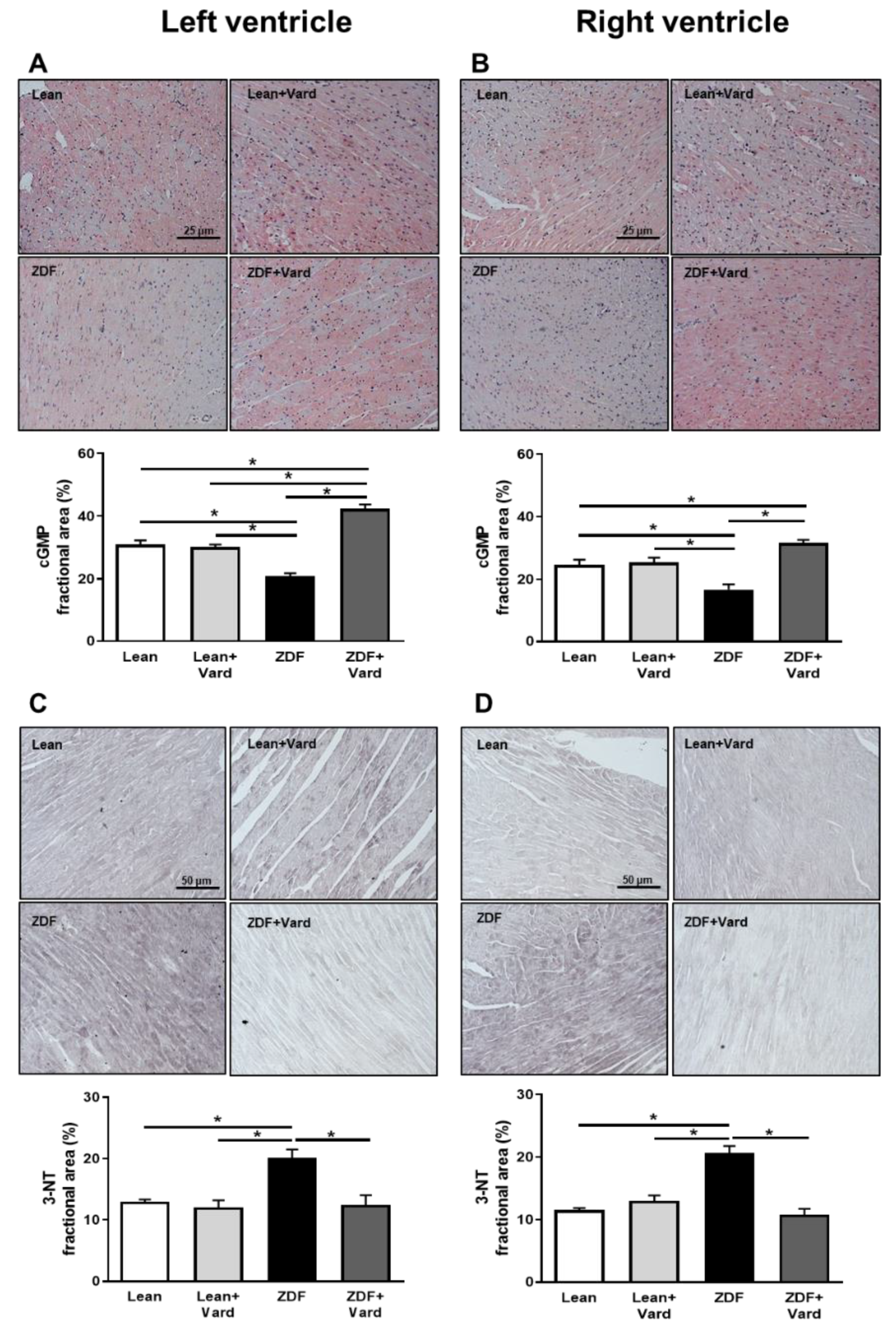
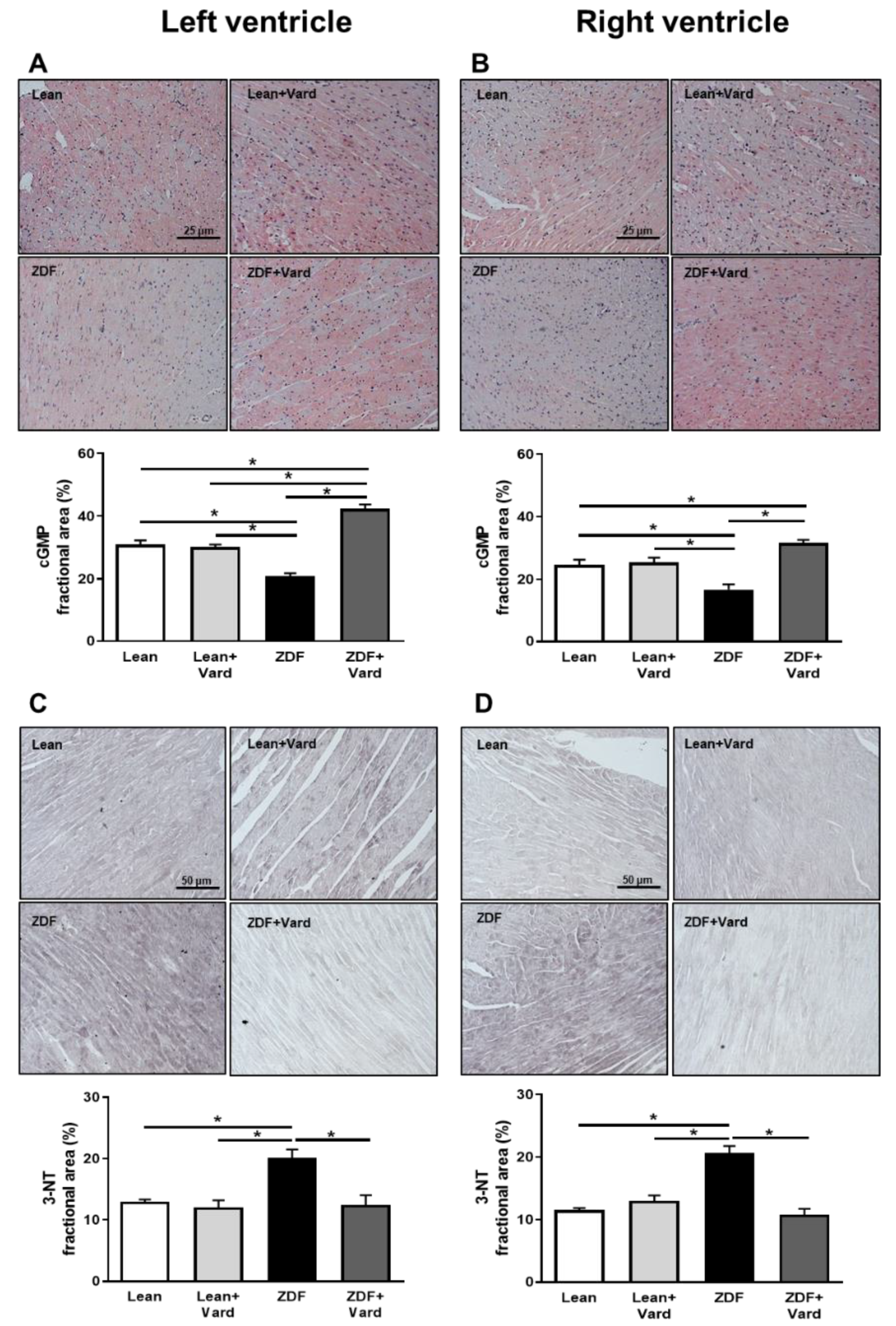
3.1. Vardenafil Prevented the Reduction of Tissue cGMP Levels and the Increase in 3-NT Generation in ZDF Hearts
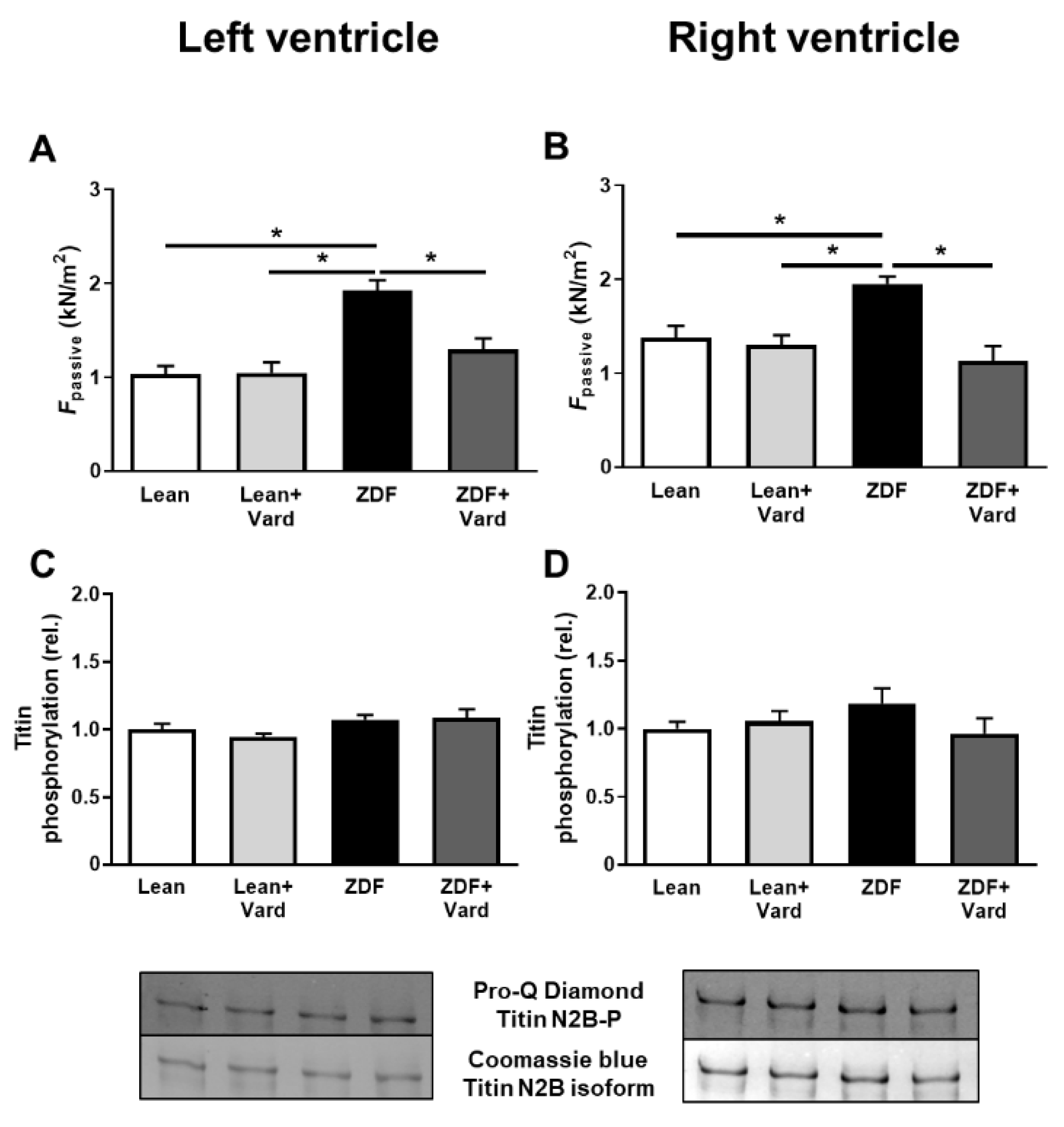
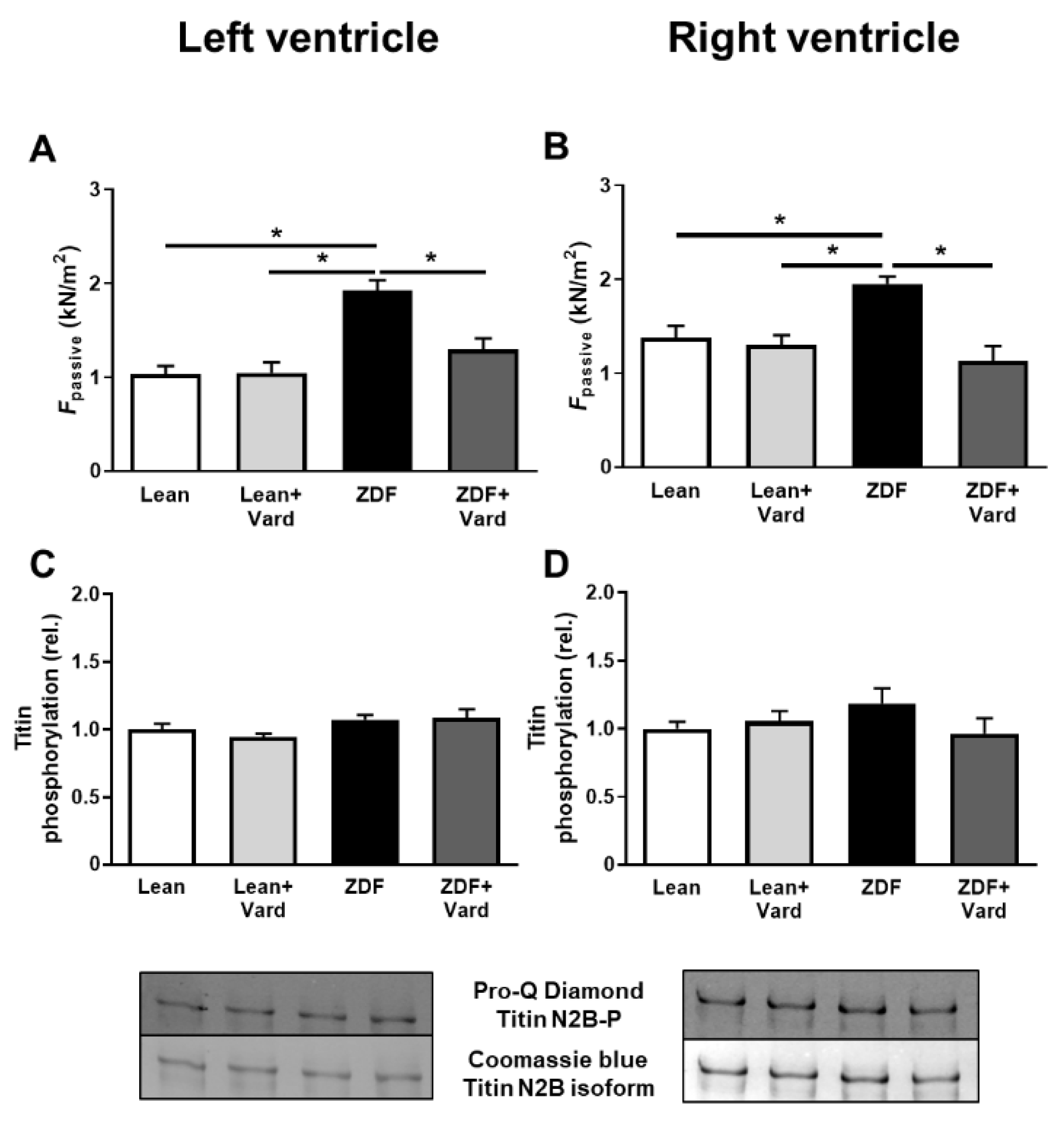
3.2. PDE-5A Inhibition Prevented the Increase in Passive Tension (Fpassive) in LV and RV Cardiomyocytes in ZDF Animals
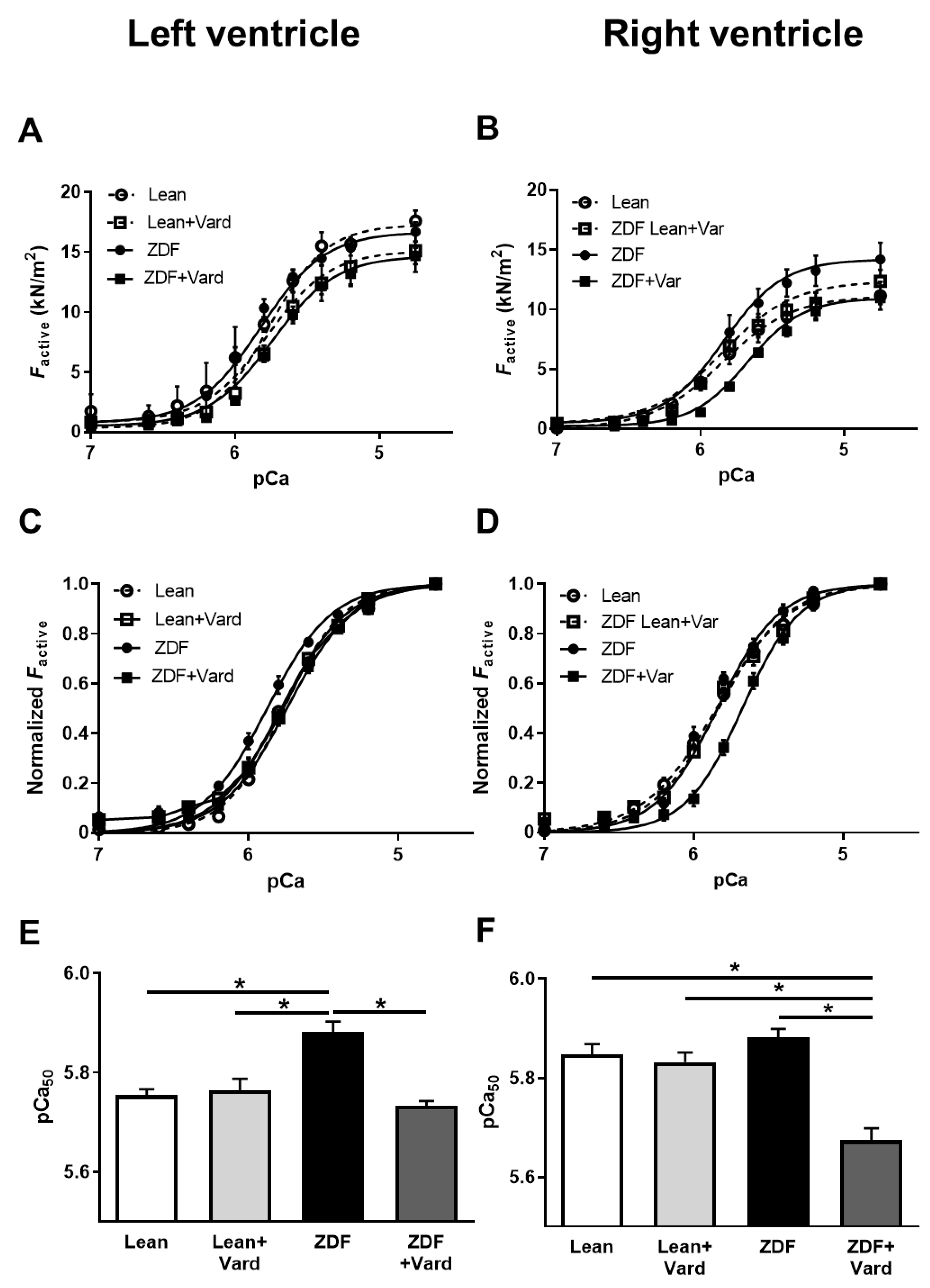
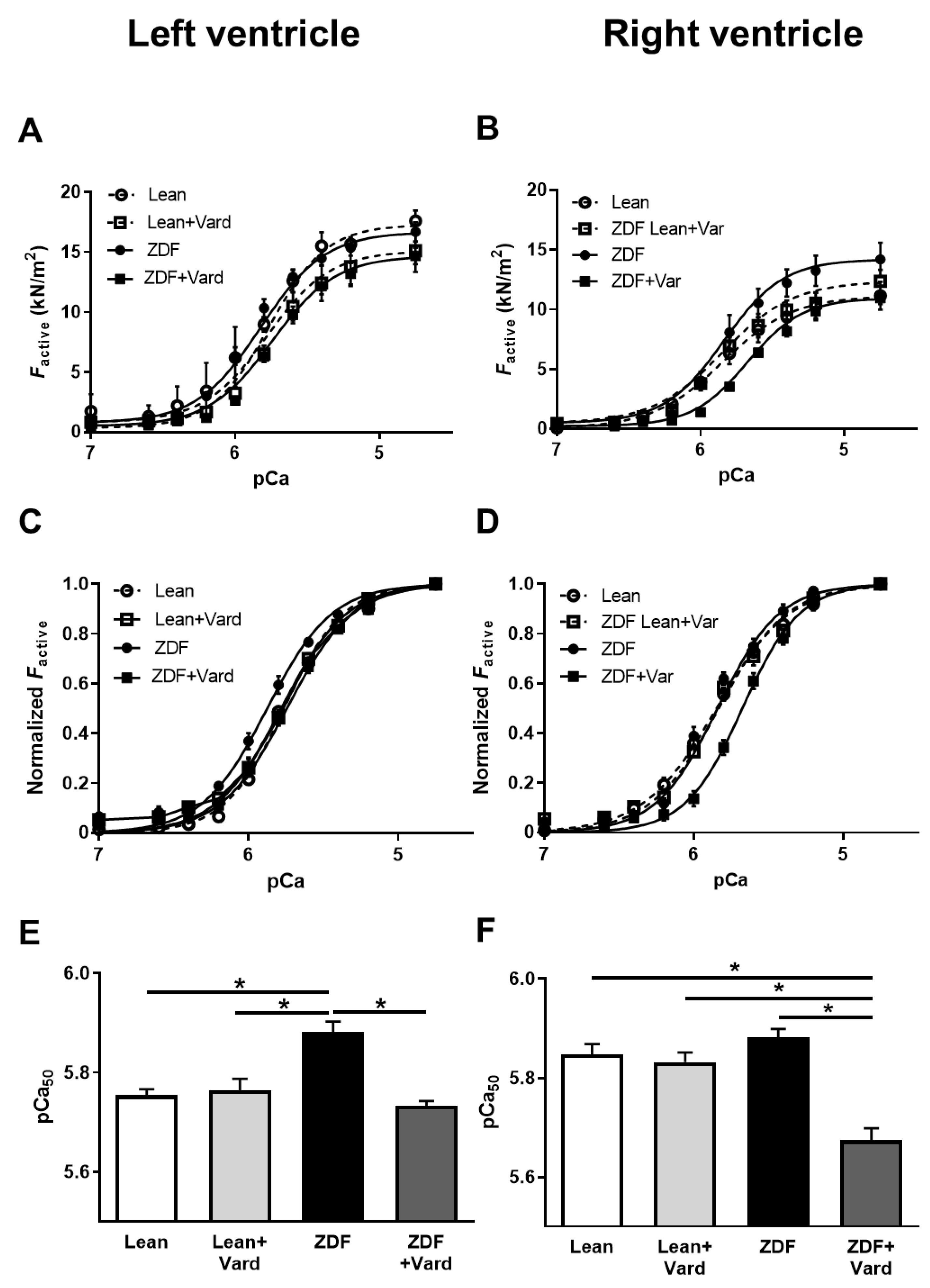
3.3. Ca2+-Sensitivity of Force Production Showed Interventricular Differences in ZDF Rats
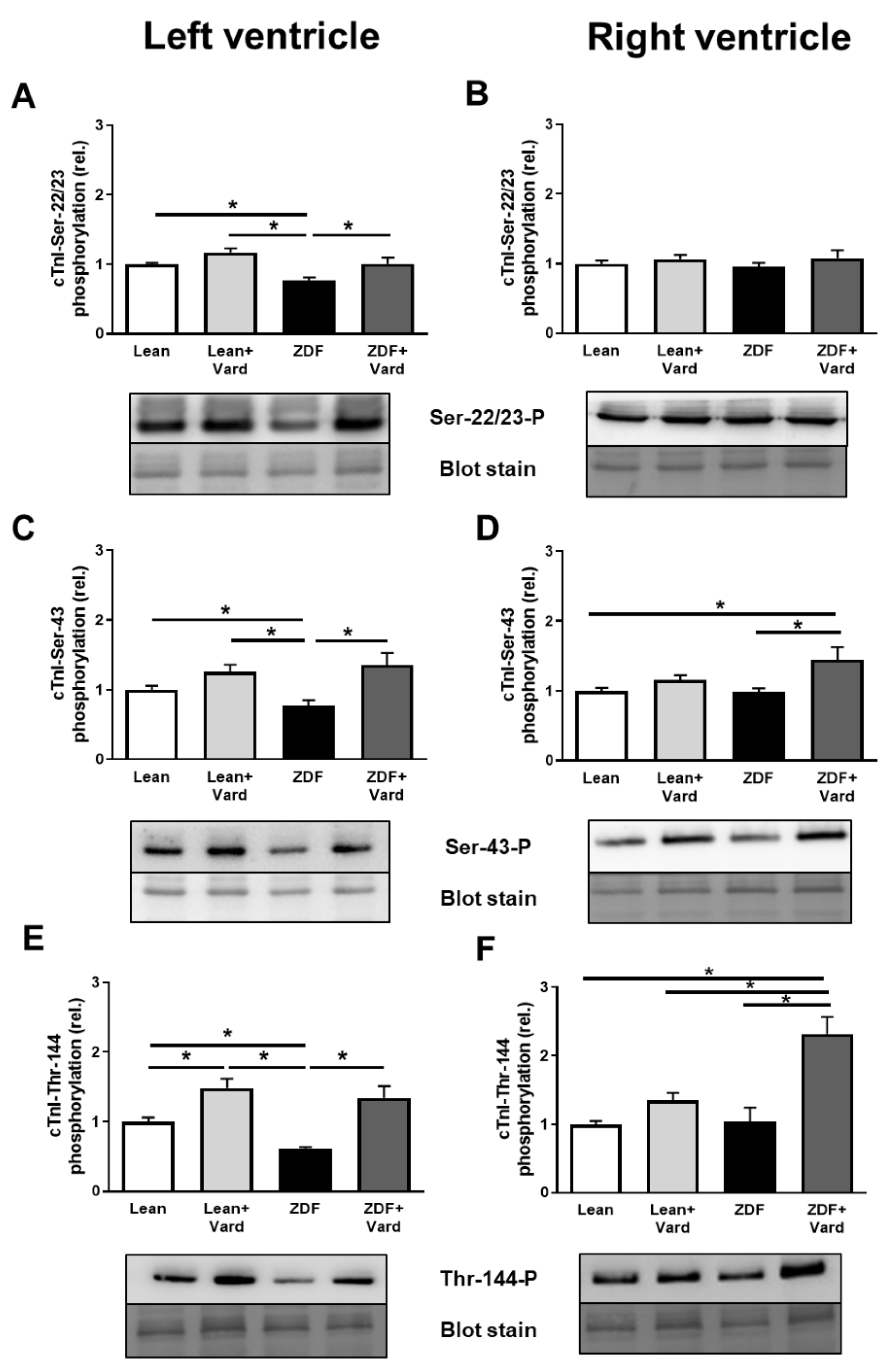
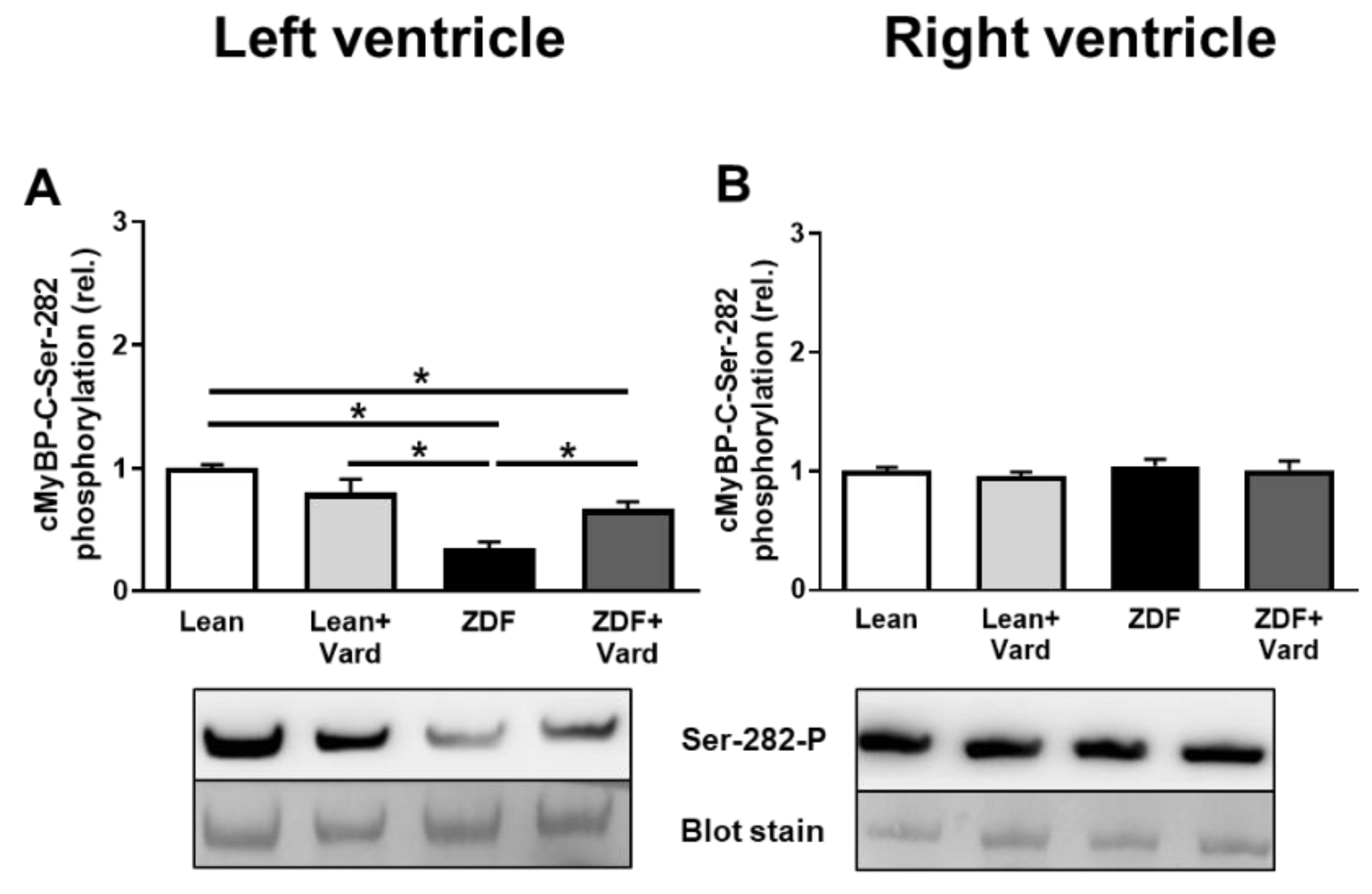
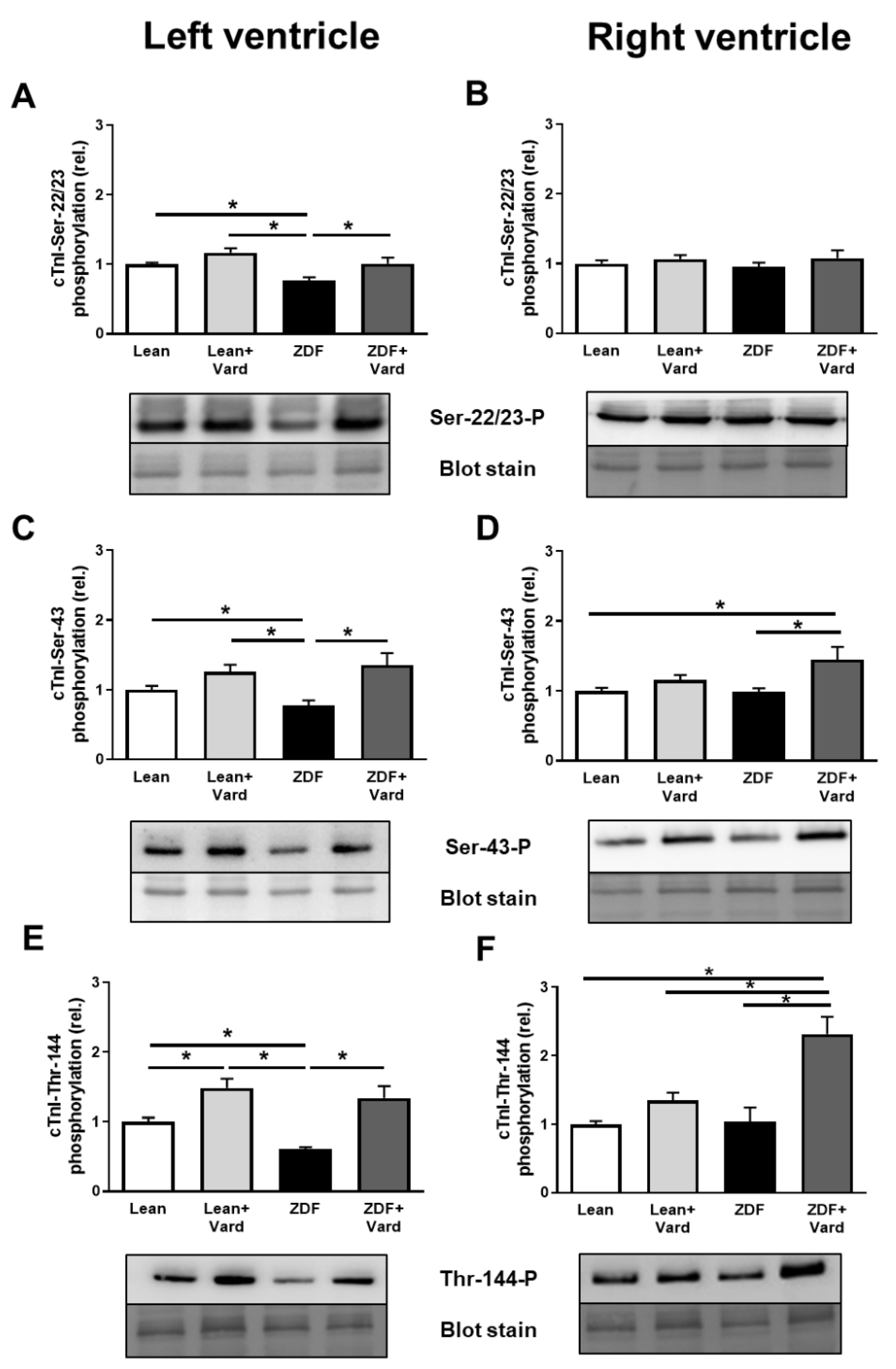
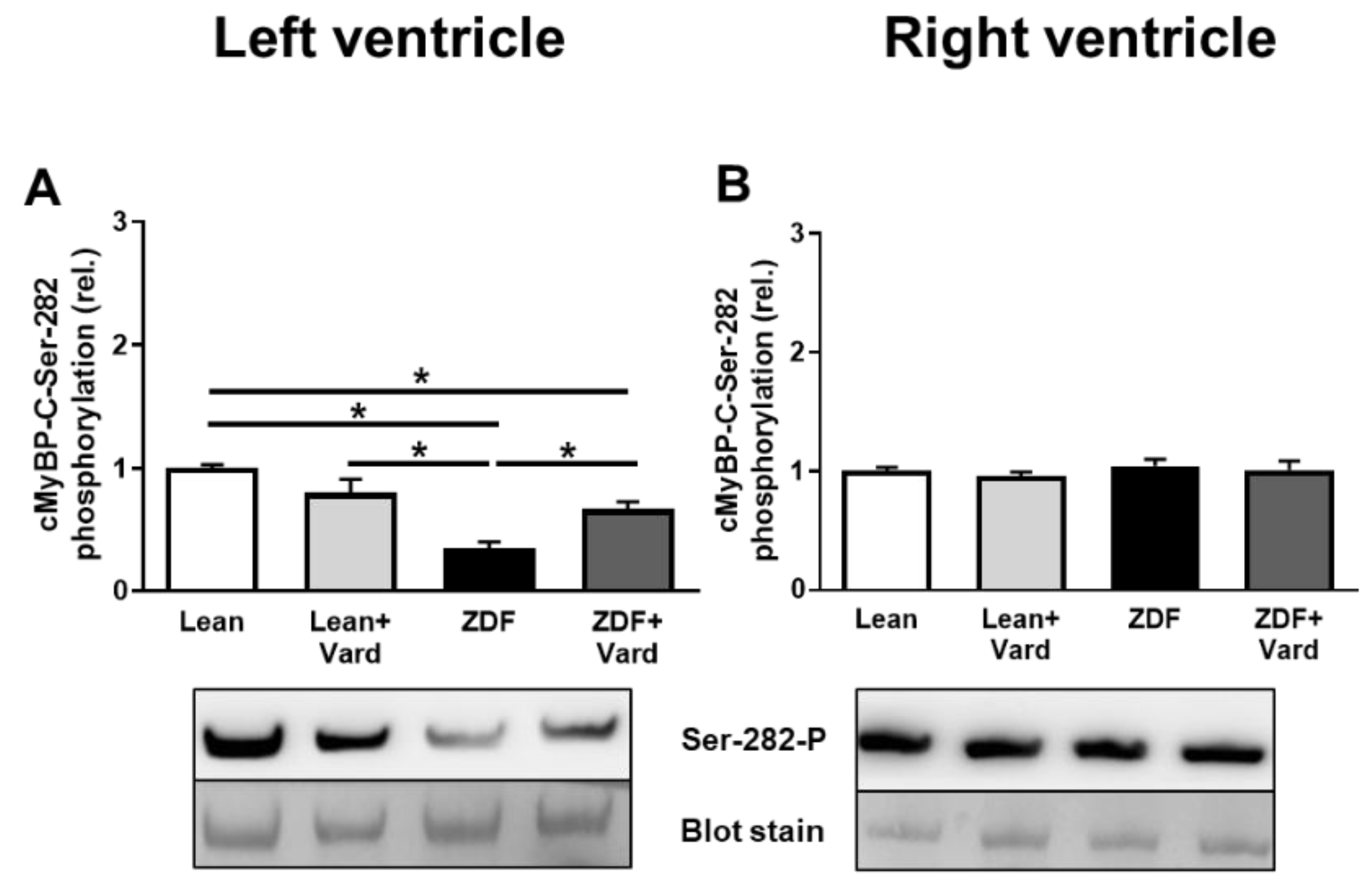
3.4. Interventricular Differences of Myofilament Protein Phosphorylations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Papp, Z.; Radovits, T.; Paulus, W.J.; Hamdani, N.; Seferović, P.M. Molecular and pathophysiological links between heart failure with preserved ejection fraction and type 2 diabetes mellitus. Eur. J. Heart Fail. 2018, 20, 1649–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, W.J. Unfolding Discoveries in Heart Failure. N. Engl. J. Med. 2020, 382, 679–682. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, I.; AbouEzzeddine, O.F.; Takahama, H.; Kwon, S.H.; Forfia, P.; Roger, V.L.; Redfield, M.M. Right ventricular function in heart failure with preserved ejection fraction: A community-based study. Circulation 2014, 130, 2310–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melenovsky, V.; Hwang, S.J.; Lin, G.; Redfield, M.M.; Borlaug, B.A. Right heart dysfunction in heart failure with preserved ejection fraction. Eur. Heart J. 2014, 35, 3452–3462. [Google Scholar] [CrossRef] [Green Version]
- Borlaug, B.A.; Kane, G.C.; Melenovsky, V.; Olson, T.P. Abnormal right ventricular-pulmonary artery coupling with exercise in heart failure with preserved ejection fraction. Eur. Heart J. 2016, 37, 3293–3302. [Google Scholar] [CrossRef] [Green Version]
- Reddy, Y.N.V.; Obokata, M.; Wiley, B.; Koepp, K.E.; Jorgenson, C.C.; Egbe, A.; Melenovsky, V.; Carter, R.E.; Borlaug, B.A. The haemodynamic basis of lung congestion during exercise in heart failure with preserved ejection fraction. Eur. Heart J. 2019, 40, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Berglund, F.; Piña, P.; Herrera, C.J. Right ventricle in heart failure with preserved ejection fraction. Heart 2020, 106, 1798–1804. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Melenovsky, V.; Pislaru, S.; Borlaug, B.A. Deterioration in right ventricular structure and function over time in patients with heart failure and preserved ejection fraction. Eur. Heart J. 2019, 40, 689–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Xu, X.; Hu, X.; Lee, S.; Traverse, J.H.; Zhu, G.; Fassett, J.; Tao, Y.; Zhang, P.; dos Remedios, C.; et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 2010, 121, 1474–1483. [Google Scholar] [CrossRef] [Green Version]
- Kovács, Á.; Alogna, A.; Post, H.; Hamdani, N. Is enhancing cGMP-PKG signalling a promising therapeutic target for heart failure with preserved ejection fraction? Neth. Heart J. 2016, 24, 268–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef]
- Hussain, I.; Mohammed, S.F.; Forfia, P.R.; Lewis, G.D.; Borlaug, B.A.; Gallup, D.S.; Redfield, M.M. Impaired Right Ventricular-Pulmonary Arterial Coupling and Effect of Sildenafil in Heart Failure With Preserved Ejection Fraction: An Ancillary Analysis From the Phosphodiesterase-5 Inhibition to Improve Clinical Status And Exercise Capacity in Diastolic Heart Failure (RELAX) Trial. Circ. Heart Fail. 2016, 9, e002729. [Google Scholar]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef]
- Mátyás, C.; Németh, B.T.; Oláh, A.; Török, M.; Ruppert, M.; Kellermayer, D.; Barta, B.A.; Szabó, G.; Kökény, G.; Horváth, E.M.; et al. Prevention of the development of heart failure with preserved ejection fraction by the phosphodiesterase-5A inhibitor vardenafil in rats with type 2 diabetes. Eur. J. Heart Fail. 2017, 19, 326–336. [Google Scholar] [CrossRef]
- Papp, Z.; Szabó, A.; Barends, J.P.; Stienen, G.J. The mechanism of the force enhancement by MgADP under simulated ischaemic conditions in rat cardiac myocytes. J. Physiol. 2002, 543, 177–189. [Google Scholar] [CrossRef]
- Oláh, A.; Németh, B.T.; Mátyás, C.; Horváth, E.M.; Hidi, L.; Birtalan, E.; Kellermayer, D.; Ruppert, M.; Merkely, G.; Szabó, G.; et al. Cardiac effects of acute exhaustive exercise in a rat model. Int. J. Cardiol. 2015, 182, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Poels, E.M.; da Costa Martins, P.A.; van Empel, V.P. Adaptive capacity of the right ventricle: Why does it fail? Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H803–H813. [Google Scholar] [CrossRef]
- Borbély, A.; van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Linke, W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006, 27, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Kötter, S.; Gout, L.; Von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Krüger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Gorter, T.M.; Streng, K.W.; van Melle, J.P.; Rienstra, M.; Dickinson, M.G.; Lam, C.S.P.; Hummel, Y.M.; Voors, A.A.; Hoendermis, E.S.; van Veldhuisen, D.J. Diabetes Mellitus and Right Ventricular Dysfunction in Heart Failure With Preserved Ejection Fraction. Am. J. Cardiol. 2018, 121, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.I.; Hahn, V.S.; Jani, V.; Hsu, S.; Sharma, K.; Kass, D.A. Reduced Right Ventricular Sarcomere Contractility in Heart Failure With Preserved Ejection Fraction and Severe Obesity. Circulation 2021, 143, 965–967. [Google Scholar] [CrossRef]
- Solaro, R.J.; Henze, M.; Kobayashi, T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 2013, 112, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Solaro, R.J. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J. Biol. Chem. 2008, 283, 26829–26833. [Google Scholar] [CrossRef] [Green Version]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Noland, T.A., Jr.; Guo, X.; Raynor, R.L.; Jideama, N.M.; Averyhart-Fullard, V.; Solaro, R.J.; Kuo, J.F. Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J. Biol. Chem. 1995, 270, 25445–25454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; de Tombe, P.P. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ. Res. 2007, 101, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Sumandea, C.A.; Chen, Y.C.; Garcia-Cazarin, M.L.; Zhang, J.; Balke, C.W.; Sumandea, M.P.; Ge, Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J. Biol. Chem. 2012, 287, 848–857. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Kirk, J.A.; Ji, W.; dos Remedios, C.G.; Kass, D.A.; Van Eyk, J.E.; Murphy, A.M. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 2012, 126, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Burkart, E.M.; Sumandea, M.P.; Kobayashi, T.; Nili, M.; Martin, A.F.; Homsher, E.; Solaro, R.J. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 2003, 278, 11265–11272. [Google Scholar] [CrossRef] [Green Version]
- Avner, B.S.; Hinken, A.C.; Yuan, C.; Solaro, R.J. H2O2 alters rat cardiac sarcomere function and protein phosphorylation through redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H723–H730. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Barefield, D.; Cuello, F.; Avkiran, M.; Lasko, V.M.; Lorenz, J.N.; Maillet, M.; Martin, J.L.; et al. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 2011, 109, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Kooij, V.; Holewinski, R.J.; Murphy, A.M.; Van Eyk, J.E. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. J. Mol. Cell Cardiol. 2013, 60, 116–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czuriga, D.; Tóth, A.; Pásztor, E.T.; Balogh, A.; Bodnár, A.; Nizsalóczki, E.; Lionetti, V.; Recchia, F.A.; Czuriga, I.; Edes, I.; et al. Cell-to-cell variability in troponin I phosphorylation in a porcine model of pacing-induced heart failure. Basic Res. Cardiol. 2012, 107, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, A.; Santer, D.; Pásztor, E.T.; Tóth, A.; Czuriga, D.; Podesser, B.K.; Trescher, K.; Jaquet, K.; Erdodi, F.; Edes, I.; et al. Myofilament protein carbonylation contributes to the contractile dysfunction in the infarcted LV region of mouse hearts. Cardiovasc. Res. 2014, 101, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovács, Á.; Herwig, M.; Budde, H.; Delalat, S.; Kolijn, D.; Bódi, B.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Balogh, Á.; et al. Interventricular Differences of Signaling Pathways-Mediated Regulation of Cardiomyocyte Function in Response to High Oxidative Stress in the Post-Ischemic Failing Rat Heart. Antioxidants 2021, 10, 964. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bódi, B.; Kovács, Á.; Gulyás, H.; Mártha, L.; Tóth, A.; Mátyás, C.; Barta, B.A.; Oláh, A.; Merkely, B.; Radovits, T.; et al. Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts. Antioxidants 2021, 10, 1776. https://doi.org/10.3390/antiox10111776
Bódi B, Kovács Á, Gulyás H, Mártha L, Tóth A, Mátyás C, Barta BA, Oláh A, Merkely B, Radovits T, et al. Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts. Antioxidants. 2021; 10(11):1776. https://doi.org/10.3390/antiox10111776
Chicago/Turabian StyleBódi, Beáta, Árpád Kovács, Hajnalka Gulyás, Lilla Mártha, Attila Tóth, Csaba Mátyás, Bálint András Barta, Attila Oláh, Béla Merkely, Tamás Radovits, and et al. 2021. "Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts" Antioxidants 10, no. 11: 1776. https://doi.org/10.3390/antiox10111776
APA StyleBódi, B., Kovács, Á., Gulyás, H., Mártha, L., Tóth, A., Mátyás, C., Barta, B. A., Oláh, A., Merkely, B., Radovits, T., & Papp, Z. (2021). Long-Term PDE-5A Inhibition Improves Myofilament Function in Left and Right Ventricular Cardiomyocytes through Partially Different Mechanisms in Diabetic Rat Hearts. Antioxidants, 10(11), 1776. https://doi.org/10.3390/antiox10111776