Abstract
Coenzyme Q (CoQ) is a vital lipophilic molecule that is endogenously synthesized in the mitochondria of each cell. The CoQ biosynthetic pathway is complex and not completely characterized, and it involves at least thirteen catalytic and regulatory proteins. Once it is synthesized, CoQ exerts a wide variety of mitochondrial and extramitochondrial functions thank to its redox capacity and its lipophilicity. Thus, low levels of CoQ cause diseases with heterogeneous clinical symptoms, which are not always understood. The decreased levels of CoQ may be primary caused by defects in the CoQ biosynthetic pathway or secondarily associated with other diseases. In both cases, the pathomechanisms are related to the CoQ functions, although further experimental evidence is required to establish this association. The conventional treatment for CoQ deficiencies is the high doses of oral CoQ10 supplementation, but this therapy is not effective for some specific clinical presentations, especially in those involving the nervous system. To better understand the CoQ biosynthetic pathway, the biological functions linked to CoQ and the pathomechanisms of CoQ deficiencies, and to improve the therapeutic outcomes of this syndrome, a variety of animal models have been generated and characterized in the last decade. In this review, we show all the animal models available, remarking on the most important outcomes that each model has provided. Finally, we also comment some gaps and future research directions related to CoQ metabolism and how the current and novel animal models may help in the development of future research studies.
1. Introduction
Coenzyme Q (CoQ) or ubiquinone is an essential lipid present in almost all living organisms. CoQ is composed of a benzoquinone ring and a polyisoprene chain of variable length. Each specie has a major CoQ form attending to the length of the polyisoprene chain, i.e., 10 isoprene units (CoQ10) in humans, zebrafish (Danio rerio), and Schizosaccharomyces pombe, 9 units (CoQ9) in mice, Caenorhabditis elegans and plants, 8 units (CoQ8) in Escherichia coli, and 6 units (CoQ6) in Saccharomyces cerevisiae [1,2]. Although one form of CoQ is dominant in each organism, a minor form of CoQ can be also detected in some species, e.g., CoQ9 in humans or CoQ10 in mice [3].
Thanks to its redox chemistry, CoQ participates in the mitochondrial respiration, accepting electrons from complex I or complex II and transferring them to complex III at the same time that a proton gradient is produced in the intermembrane space [4,5]. This proton motive force is used by the ATP synthase to produce the final ATP molecule. Moreover, CoQ is a cofactor for different mitochondrial dehydrogenases involved in different metabolic pathways [1,4,6], including the following: the dihydroorotate dehydrogenase (DHODH), involved in the pyrimidine biosynthesis [7,8]; the mitochondrial glycerol-3-phosphate dehydrogenase (G3PDH), linking glycolysis, oxidative phosphorylation, and fatty acid metabolism [9,10]; the electron transport flavoprotein dehydrogenase (ETFDH), involved in fatty acid β-oxidation, the catabolism of several amino acids, and sarcosine metabolism [11,12]; the proline dehydrogenase (PRODH) and proline dehydrogenase 2 (PRODH2) required for proline, glyoxylate, and arginine metabolism [6,13]; the choline dehydrogenase (CHDH) related to glycine metabolism [14]; and the sulfide-quinone oxidoreductase (SQOR) that catalyzes the first reaction required for the detoxification of hydrogen sulfide (H2S) [15] (Figure 1a).
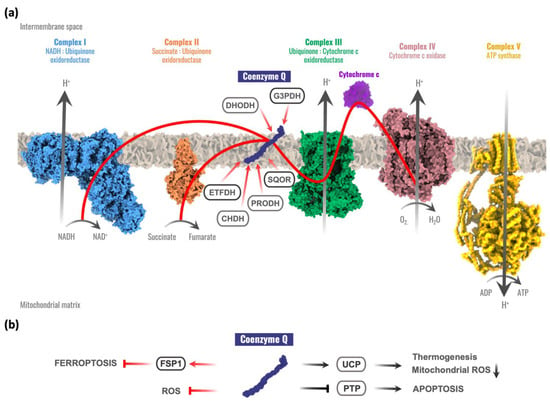
Figure 1.
Coenzyme Q functions in the cell. (a) The role of coenzyme Q (CoQ) in the mitochondrial respiratory chain and its relationships with other mitochondrial enzymes. Red arrows represent electron flow. CoQ accepts electrons from complex I and complex II, sulfide:quinone oxidoreductase (SQOR), proline dehydrogenase and proline dehydrogenase 2 (PRODH), choline dehydrogenase (CHDH), mitochondrial glycerol-3-phosphate dehydrogenase (G3PDH), dihydroorotate dehydrogenase (DHOH), and electron transport flavoprotein dehydrogenase (ETFDH). (b) CoQ extramitochondrial functions. Red arrows represent electron flow. FSP1 = Ferroptosis Suppressor Protein 1; UCP = Uncoupling Protein; PTP = Permeability Transition Pore; ROS = Reactive Oxygen Species.
Uncoupling proteins (UCPs) are other mitochondrial components susceptible to be regulated by redox reactions. Some experimental evidence has suggested that CoQ is a cofactor for UCPs, connecting CoQ with the control of thermogenesis [16,17] and with an alternative mechanism for decreasing reactive oxygen species (ROS) inside mitochondria via UCPs–superoxide interaction [18]. However, controversial results have been reported on the involvement of CoQ in the regulation of the UCPs [17,19]. Other studies investigated the regulation of the mitochondrial permeability transition pore (PTP) by ubiquinone analogues [20,21]. Experimental evidence suggests that quinones modulate the PTP through a common binding site rather than through redox reactions. CoQ seems also to prevent PTP opening [22], but further studies are required to understand whether CoQ has a primary function in the PTP. Recently, two studies reported that CoQ could also act as a cofactor for the ferroptosis suppressor protein 1 (FSP1) (previously known as apoptosis-inducing factor mitochondrial 2, AIFM2), thus contributing to the suppression of ferroptosis, an iron-dependent form of cell death with increased oxidative damage [23,24] (Figure 1b).
In addition to these metabolic roles related to mitochondria, CoQ is a crucial antioxidant that specially acts in the lipid membranes. The antioxidant capacity of CoQ is due to its capability to directly reduce reactive oxygen species (ROS) but also to regenerate other antioxidants, e.g., tocopherol and ascorbate [2,6,25,26]. Moreover, different non-mitochondrial functions of CoQ have been identified, and extensive reviews of these actions have been published elsewhere [1,27,28].
The synthesis of CoQ mainly occurs in the mitochondrial inner membrane by a set of nuclear-encoded COQ proteins through a biochemical pathway that is not completely understood [2,4,10,25,29] (Figure 2a). However, some authors have also located the synthesis of CoQ outside the mitochondria, specifically in the endoplasmic reticulum (ER)–Golgi system [30,31]. Moreover, a study in yeast demonstrated the role of the endoplasmic reticulum–mitochondria encounter structure (ERMES) in the coordination of CoQ biosynthesis in mitochondria, highlighting the importance of the communication between these two organelles [32]. The CoQ biosynthetic process in the mitochondria is similar in prokaryotes and eukaryotes: a long polyisoprenoid lipid tail is coupled to a benzenoid precursor, and the benzenoid ring is further modified through successive steps to yield the final product [2,33,34]. 4-Hydroxybenzoic acid (4-HB) is the precursor of the benzoquinone ring of CoQ [35], although some other alternative ring precursors are being postulated [2]. For the biosynthesis of 4-HB, human cells utilize phenylalanine or tyrosine as ring precursors [29]. The isoprene carbon units for making the CoQ side-chain are derived from the mevalonate pathway in eukaryotes and some prokaryotes [36]. The lipophilic polyprenyl tail is synthesized by the addition of dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP) by PDSS1/PDSS2 in humans and mice. Consequently, the heterotetramer PDSS1/PDSS2 is responsible for the length of the polyprenyl tail [29,36,37]. Then, COQ2 in mammals catalyze the attachment of the polyisoprenoid tail to the ring precursor. From this step, a total of seven reactions (one decarboxylation, three hydroxylation, and three methylation) produce the fully substituted benzoquinone ring of CoQ [35]: C5-hydroxylation (human COQ6), O-methylations (human COQ3), C1-hydroxilation and C1-decarboxylation (unidentified), C2-methylation (human COQ5), and C6-hydroxilation (human COQ7) [4,38,39,40,41]. In addition to the enzymes with catalytic activity in the CoQ biosynthetic pathway, there are additional proteins necessary for CoQ synthesis, although their exact functions are not well established. However, the disruption in any of them produce a reduction in the CoQ levels. In humans, these proteins are COQ4, COQ8A (also known as ADCK3), COQ8B (also known as ADCK4), COQ9, COQ10A, and COQ10B [42,43,44,45,46,47]. Some of the biosynthetic proteins are assembled into a high molecular mass complex, named complex Q, with the objective of improving the catalytic efficiency of the CoQ biosynthetic pathway and minimizing the escape of intermediates [4,29,37] (Figure 2b). The benzoquinone ring of CoQ can be found in its completely oxidized form (ubiquinone, CoQ), in the semireduced form (ubisemiquinone, CoQH●), and in the completely reduced form (ubiquinol, CoQH2) after receiving two electrons [1,4] (Figure 2c).
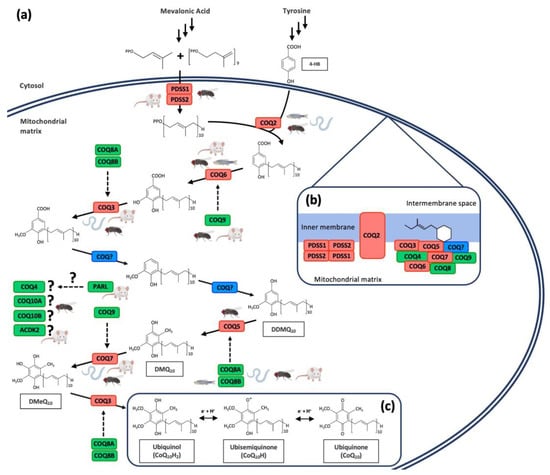
Figure 2.
Coenzyme Q10 biosynthetic pathway and CoQ10 redox cycle. (a) Schematic model of human CoQ10 biosynthetic pathway. The red color indicates proteins with enzymatic activity. The green color indicates proteins with regulatory function. The blue color shows currently unidentified enzymes. Figures of mouse, C. elegans, D. melanogaster, and zebrafish illustrate animal models with mutation in each specific protein. (b) Model of human CoQ10 biosynthetic complex, containing at least COQ3–COQ9 and lipids, such as CoQ itself. (c) Chemical structure of coenzyme Q10 and its redox cycle. 4-HB = 4-hydroxybenzoic Acid; DDMQ = demethoxy-demethyl-ubiquinone; DMQ = demethoxyubiquinone; DMeQ = demethylubiquinone.
CoQ levels can be severely reduced in a group of mitochondrial diseases named CoQ deficiencies, which are clinically and genetically heterogeneous disorders. Five major phenotypes have been described: (1) encephalomyopathy (recurrent myoglobinuria, encephalopathy, and mitochondrial myopathy); (2) cerebellar ataxia (cerebellar atrophy associated with other neurologic manifestations and, occasionally, endocrine dysfunctions); (3) infantile multisystemic form; (4) isolated myopathy, characterized by muscle weakness, myoglobinuria, exercise intolerance, and elevated creatine kinase (CK); and (5) nephropathy. Growth retardation, deafness, hearing loss, and cardiomyopathy have also been described in CoQ10-deficient patients [48]. This heterogeneity in the clinical presentations suggests the existence of multiple pathomechanisms, which could be theoretically due to the multiple functional roles of CoQ and the complexity of the CoQ biosynthetic pathway, although further experimental evidences are required to understand the diseases’ conditions and tissue-specific pathomechanisms. Primary CoQ deficiency is caused by autosomal recessive mutations in CoQ genes [48], while secondary CoQ deficiency is produced by mutations in genes unrelated to CoQ biosynthesis or is derived from other physiological processes or pharmacological treatments [49,50]. Mutations or deletions in 10 genes involved in the CoQ biosynthetic pathway have been reported in humans: PSSS1 [51], PDSS2 [52], COQ2 [51,53,54], COQ4 [55], COQ5 [56], COQ6 [57], COQ7 [58,59], COQ8A (or ADCK3) [60,61], COQ8B (or ADCK4) [62,63], and COQ9 [64,65]. The secondary forms are probably more frequent, and they include patients with ataxia and oculomotor apraxia due to mutations in the aprataxin (APTX) gene [66,67,68]; with isolated myopathy due to mutations in the electron-transferring-flavoprotein dehydrogenase gene (ETFDH) [69]; and with cardiofaciocutaneous (CFC) syndrome due to mutations in BRAF [70]. Moreover, CoQ10 deficiency has been reported in association with pathogenic mitochondrial DNA (mtDNA) depletion, deletions, or point mutations [71,72,73]. In addition, secondary CoQ deficiency has been linked to a decrease in the levels of proteins of the complex Q in various mouse models of mitochondrial diseases [74], as well as in the muscle and adipose tissue of patients and a mouse model with insulin resistance [75]. Furthermore, the levels of CoQ are one of the mitochondrial factors involved in aging, and a specific review about this topic was recently published [29].
Supplementation with exogenous CoQ10 is the main therapeutic strategy for these pathologies. However, the therapeutic efficacy of CoQ10 supplementation is quite variable [48,50,76,77], highlighting the need to look for new options. Thus, this review is focused on describing the available animal models for CoQ deficiency that have been used to date in the study of CoQ deficiencies (Table 1), how they have contributed to expand our knowledge about CoQ metabolism and the pathomechanisms of CoQ deficiency, and how they may provide optimal therapeutic alternatives for patients suffering from CoQ deficiencies.

Table 1.
Overview of the animal models of CoQ deficiency mentioned, remarking their contribution to expand the knowledge about the CoQ biosynthetic pathway and functions, and the pathomechanisms and treatments related to CoQ deficiency.
2. Invertebrate Models of CoQ Deficiency
Invertebrate models have the great advantage of being easily generated and characterized compared to vertebrate models. Thus, worms and flies lacking CoQ have been produced, and they highlighted important functions of CoQ. However, invertebrate systems are not sufficient to reproduce the complexity of mammalian systems [125,126].
2.1. Fruit Fly Models: Drosophila Melanogaster
Drosophila melanogaster is commonly known as the fruit fly or vinegar fly [126]. It has been widely used because it offers several advantages for the investigation of molecular and cellular mechanisms due to its short lifespan, large number of offspring, and having a genome that is easy to modulate [127]. Drosophila contains three CoQ forms with the following approximated proportions: 5% CoQ8, 82% CoQ9, and 13% CoQ10. Those proportions change depending on the age of the fly and the developmental stage [83].
Mutations in the Drosophila qless (cg31005) gene, an orthologue of the human PDSS1, induce an upregulation of markers of mitochondrial stress and caspase-dependent apoptosis in neurons. Full rescue of the qless neural phenotype was achieved by dietary supplementation with CoQ4, CoQ9, or CoQ10 [78]. Additionally, mutations in the Drosophila sbo (cg9613 or coq2) gene, homolog of the human COQ2, leads to a small larvae phenotype. The sbo null mutants are developmentally arrested at the first instar larval stage. Flies that are heterozygous for sbo show reduced CoQ9 and CoQ10 production and a controversial extended lifespan [79]. Coq2 mutant flies are more susceptible to bacterial and fungal infections, while they are more resistant to viruses. The supplementation with CoQ10 partially rescues the impaired immune functions of coq2 mutants because it restores the gene expression of anti-microbial genes but increases the susceptibility to viral infection [80]. Moreover, another Drosophila model with mutation in the coq2 gene was generated employing garland cell nephrocytes (GCN) to model human COQ2 nephropathy. This study found that coq2 is required for slit diaphragm morphology and function, since coq2 silencing causes large areas without slit diaphragms, while slit diaphragms densely populate the surface of control GCN. Following, it was proved that the pathogenesis of coq2 is linked to mitochondrial reactive oxygen species (ROS) formation because of the increased ROS formation in coq2 loss-of-function. According to that, the phenotype was rescued after the treatment of coq2 flies with the ROS scavenger glutathione for 5 days, remarking the importance of oxidative stress in the renal pathology associated to CoQ deficiency. Interestingly, this coq2 phenotype was also partially rescued by the supplementation with vanillic acid (VA) [81]. VA is an analog of the CoQ natural precursor, the 4-hydroxybenzoic acid (4-HB), which has previously demonstrated its therapeutic activity in other models of CoQ deficiency [35,128], although the therapeutic mechanism for defects in coq2 have not been clearly identified.
The UAS-GAL4 system for gene silencing with RNAi has been also used in Drosophila to interfere with the expression of each of the CoQ biosynthetic genes [83]. The results demonstrate that flies with RNAi against CoQ genes show a decrease in CoQ levels. The percentage of decrease depends on the affected gene and the intensity of the gene silencing. RNAi against the gene cg10585 (human PDSS2) showed the hardest phenotype with flies arresting their development cycle after egg hatching. Gene interference of qless (cg31005), coq2, coq3, coq5, coq7, coq8, coq9, or coq10 produced lethality at larvae or pupae development stages. When the silencing gene was coq6, flies managed to achieve the adult fly stage but suffered from severe CoQ deficiency. Demethoxyubiquinone (DMQ) was detected when coq3, coq6, coq7, and coq9 were silenced, supporting the idea of a multi-enzymatic complex for CoQ biosynthesis [82,83].
2.2. Worm Models: Caenorhabditis Elegans
Caenorhabditis elegans (C. elegans) has been extensively used as a model organism in different fields of research [29,126]. The ability to inactivate a target gene transiently by RNAi has greatly accelerated the analysis of loss-of-function phenotypes in C. elegans [129].
A model of secondary CoQ deficiency in C. elegans is the mev-1 mutant strain, which has a defect in complex II. This model shows a decrease in the CoQ9 levels and altered rates of the reduced and oxidized forms of CoQ [84,126]. Mev-1 mutant also manifests a disruption of the superoxide dismutase activity, leading to an accumulation of reactive oxygen species (ROS) and a shorter lifespan compared to the wild type [85]. The supplementation with CoQ10 rescues the phenotype by the reduction of superoxide anion level, suggesting the link between lifespan and oxidative stress in mitochondria [86]. Models of primary CoQ deficiency in C. elegans will be described below.
C. elegans clk-1 mutants are worms with primary CoQ deficiency. The clk-1 gene is the homologous of the COQ7 gene in humans. As a result of this mutation, the clk-1 mutant shows a decrease in the CoQ9 levels and an accumulation of the intermediate demethoxyubiquinone (DMQ9), the substrate of COQ7 [87], thus confirming the catalytic reaction of this enzyme. Intriguingly, the phenotype is characterized by an increase in the lifespan together with a slowed pharyngeal pumping and abnormalities in defecation, movement, embryogenesis, and larvae development [89,130,131]. The reason for the life extension seems to be a lower production of reactive oxygen species (ROS) due to a reduction in the electron flow of the respiratory chain because of the inhibition of complex I [126]. This hypothesis is based on (1) Clk-1 mutants showing a profound defect in complex I+III activity, which contributes to ROS production and its link to lifespan [88]; and (2) the accumulated DMQ9 in clk-1 mutant could inhibit complex I+III activity [90]. Nevertheless, it is also important to take into account that either the small amount of CoQ9 synthesized by clk-1 mutant and/or the CoQ8 coming from the diet would stabilize complex III and determine the longevity of this model [87,126,132]. The supplementation with CoQ10 to clk-1 mutant rescued the phenotype and returned the lifespan to wild-type levels [91,126]. Other studies in the clk-1 mutant indicated that CoQ10 is the CoQ isoform with higher antioxidant properties because the clk-1 mutant feed with genetically engineered bacteria that produce CoQ10 rescued the phenotype by lowering oxidative stress, while those fed with bacteria that produce CoQ6, CoQ7, CoQ8, or CoQ9 had no effect [86,89,92,126].
C. elegans mutant strains VC479, VC752, and VC614 are available heterozygous mutant strains with deletions in the genes coq-1 (allele ok749), coq-2 (allele ok1066), and coq-8 (allele ok840), respectively. Coq-1, coq-2, and coq-8 knockouts arrested development during larval stages L1–L2 (coq-1 mutants) or L4-adult (coq-2 and coq-8 mutants) [93]. Larval development arrest was also reported for C. elegans coq-3 knockouts, pointing out the importance of CoQ in the early stages of development [95]. The first generation of coq-1 and coq-2 knockouts had muscular atrophy due to cell death, apoptosis corpses, and eventual tissue disorganization with paralysis of the posterior half of the larval body, which was probably mimicking the encephalomyopathic form of CoQ deficiency [93]. Other coq-1, coq-2, and coq-3 knockdown models mimic the cerebellar involvement of CoQ deficiency with an age-dependent loss of motor coordination correlated with the progressive degeneration of GABA neurons. However, other types of neurons in motor and sensory circuits that use other neurotransmitters (dopamine, acetylcholine, glutamate, serotonin) and body muscle cells were less sensitive to CoQ depletion [94]. The supplementation with exogenous CoQ10 or feeding worms with bacteria containing its own CoQ8 did not have a great impact on phenotype improvement of coq-1, coq-2, or coq-3 mutants, suggesting that exogenous CoQ has therapeutic limitations due to its limited bioavailability [92]. A full rescue of the coq-3 mutant phenotype was achieved by an extra-chromosomal array containing the own C. elegans coq-3 gene, illustrating the crucial role that the endogenous synthesized CoQ9 isoform plays in fertility and development [92,126]. Alternatively, coq-8 mutants increased their lifespan if they were fed with a CoQ-rich diet with wild-type E. coli, but no improvement was observed in fertility [96].
3. Vertebrate Models of CoQ Deficiency
Vertebrate models of CoQ deficiency, especially the mammalian ones, are helpful to study the physiopathology of CoQ deficiencies and to understand the heterogeneity of these syndromes because they mimic the cellular functions of CoQ, the biosynthesis, its regulation, and the tissue specificity that may exist in humans. That is the reason why these models are also useful in preclinical studies to test new therapies [125].
3.1. Zebrafish Models of CoQ Deficiency
Zebrafish biology allows ready access to all developmental stages, and the optical clarity of embryos and larvae allow real-time imaging of developing pathologies. Sophisticated mutagenesis and screening strategies on a large scale, and with a cost that is not possible in other vertebrate systems, have allowed the generation of zebrafish models for a wide variety of human diseases [133].
The generation of a zebrafish model with a null allele in ubiad1, called barolo (bar), produced a phenotype with specific cardiovascular failure due to oxidative stress and ROS-mediated cellular damage [97]. Ubiad1 is considered as a nonmitochondrial prenyltransferase responsible for the synthesis of CoQ10 in the Golgi membrane compartment in zebrafish [31,97]. As a result of that, the bar mutants showed a depletion in the CoQ10 levels in the cytosol. The accumulation of ROS reported in the bar mutants increased the lipid peroxidation in vascular cells, leading to the cardiovascular oxidative damage characteristic of this model. Moreover, the inhibition of eNOS rescued the oxidative damage previously described, suggesting a specific role of Ubiad1 on the regulation of NO signaling. Additionally, in the same study, they knocked-down Coq2 expression during zebrafish development. Loss of Coq2, the mitochondrial prenyltransferase involved in CoQ10 biosynthesis, leads to oxidative stress, but it does not significantly affect vascular integrity and survival. Comparing both zebrafish models, the authors found that the Coq2-mediated CoQ10 production is mainly for mitochondrial respiratory chain function and energy production; whereas Ubiad1-mediated CoQ10 production is important for membrane redox signaling and protection from lipid peroxidation [97]. UBIAD1 is also a novel vitamin K2 biosynthetic enzyme screened and identified from the human genome database [134]. Another ubiad1 mutant (reddishs587) was generated to demonstrate the essential role of the vitamin K2 generated by Ubiad1 in the maintenance of the endothelial cell function and overall vascular homeostasis. The treatment with vitamin K2 rescued the vascular phenotype of ubiad1 mutant reddishs587 but not the cardiac phenotype, suggesting that an alternative Ubiad1/vitamin K-independent pathway may participate in the cardiac function [135].
In other series of experiments, morpholino oligonucleotide (MO) knockdown of Coq6 or Coq8b (=Adck4) in zebrafish caused apoptosis in the Coq6 knockdown zebrafish embryos [57] and nephrosis phenotype of periorbital and total body edema in the Coq8b knockdown zebrafish embryos [63], mimicking the nephrotic syndrome phenotype of the patients.
In addition to those already characterized models, the Zebrafish Mutation Project seems to have generated different zebrafish models with mutations in Coq genes (https://zmp.buschlab.org/search/coq; accessed on 26 September 2021), although none of these models have been characterized so far.
3.2. Mouse Models of CoQ Deficiency
3.2.1. Mouse Models with Spontaneous Mutation
The polyisoprenyl diphosphate synthase is the enzyme responsible for the formation of the isoprenyl side chain of CoQ in mice and humans. It is a heterotetramer composed of two protein subunits. The genes that encode these subunits are designated Pdss1 and Pdss2 in mice, and PDSS1 and PDSS2 in humans [105,136]. A mutation in Pdss2 was reported in mice with inherited kidney disease. The mutation arose spontaneously in an inbred strain of mice, which appeared to be due to an autosomal recessive gene, and it was designated ‘kidney disease’ (kd) [125,137]. The first clinical manifestations were developed at 10 weeks of age, and it was characterized by increased proteinuria. This was followed over several weeks by excessive drinking, dilute urine, loss of weight, anemia, and death in 5 to 7 months. Histological examination of kidneys at earlier age reveals kidney damage due to mononuclear cell infiltrate and tubular dilatation with proteinaceous casts in cortical areas, which with time expands throughout the entire kidney and leads to renal failure [137]. Initial studies prior to the identification of the genetic defect suggested an autoimmune mechanism as the pathophysiological mechanism behind this syndrome [138,139]. However, this hypothesis was discarded, as it was proved that the immune response was a secondary consequence of the genetic defect of kd/kd mice [140]. Kd/kd mice developed a typical nephrotic syndrome with the characteristic biochemical perturbations in serum accompanied by albuminuria and visceral epithelial abnormalities, including hyperplasia and podocyte effacement [125,141].
A positional cloning approach demonstrated that the kd allele is a missense mutation in Pdss2 gene (Pdss2kd/kd), showing that the failure in the coenzyme Q biosynthetic pathway is the cause of a lethal kidney disease in mice [142]. The kd/kd mutation (V117M) occurs within the conserved domain I of Pdss2, and the levels of CoQ9 and CoQ10 in kidney homogenates from Pdss2kd/kd were significantly lower than in control mice [105]. The Pdss2 mutant mice manifested widespread CoQ deficiency and abnormalities in the mitochondrial respiratory chain. However, other parameters such as ROS production, oxidative stress, mitochondrial DNA depletion, and citrate synthase activity, an index of mitochondrial mass, appeared only in affected organs. Those data suggested that kidney-specific loss of mitochondria triggered by oxidative stress may be the cause of renal failure in Pdss2kd/kd mice [98]. Additionally, the impairment of the sulfide oxidation pathway induced by decreased levels of CoQ has been proposed as a pathophysiological mechanism under this syndrome. The disruption in the sulfide oxidation pathway caused the following: (1) accumulation of sulfides due to the decreased levels of SQOR and downstream enzymes, (2) low levels of plasma and urine thiosulfate, (3) the inhibition of short-chain acyl-CoA dehydrogenase, and (4) the decreased levels of glutathione, which may partially avoid the scavenge of hydrogen peroxide, thus contributing to oxidative damage and structural and functional alteration of the renal glomerulus [15,99].
Oral supplementation with CoQ10 induces a rescue of proteinuria and interstitial nephritis in the Pdss2kd/kd mutant mice through the normalization of different CoQ functions [100,101]. Additional studies demonstrated that probucol had a more powerful health improvement than high-dose CoQ10 supplementation and was able to restore CoQ9 content in the kidneys of Pdss2kd/kd mice [102]. In addition, rapamycin administration was able to reduce the proteinuria in Pdss2kd/kd mice [103], and the treatment with GDC-0879, a Braf/Mapk-targeting compound, ameliorated renal disease in Pdss2kd/kd mice, which was probably through a mechanism that involves the activity of GPx4 [104].
3.2.2. Conditional Knockout Mouse Models
To further evaluate the nephrotic phenotype associated with CoQ deficiency, tissue-specific conditional Pdss2 knockout (KO) mice were generated [105,125]. The deletion of Pdss2 was targeted to renal glomerular podocytes in Podocin/cre,Pdss2loxP/loxP, renal tubular epithelium and hepatocytes in PEPCK/cre,Pdss2loxP/loxP, monocytes in LysM/cre,Pdss2loxP/loxP, and hepatocytes in Alb/cre,Pdss2loxP/loxP. Interestingly, the kidney disease phenotype appeared only in Podocin/cre,Pdss2loxP/loxP KO mice but not in the other conditional KO, as estimated by albuminuria and morphological evidence of nephritis (dilated tubules and extensive interstitial infiltration). These data suggested that the renal glomerular podocytes are particularly sensitive to the dysfunction of PDSS2. In fact, knocking out the Pdss2 gene in podocytes resulted in a more severe phenotype than that observed in Pdss2kd/kd mice, indicating that the product of the missense allele in Pdss2kd/kd has some residual activity. The liver-conditional Alb/cre,Pdss2loxP/loxP KO mice did not show detectable levels of CoQ9 in the liver, leading to impaired mitochondrial respiration and altered intermediary metabolism as demonstrated by transcriptional profiling and amino acid quantitation [105]. Nevertheless, Alb/cre,Pdss2loxP/loxP KO mice did not develop any symptoms of disease.
In addition to kidney, cerebellum is one of the most often affected organs in CoQ deficiency [143], and cerebellar atrophy has been diagnosed in many infants with this disease. In order to analyze the cerebellum defect in CoQ deficiency, a Pdss2 conditional KO (Pax2/cre,Pdss2f/-) was generated crossing a previously generated Pdss2 floxed mouse (Pdss2f/f) with a Pax2-cre deleter mouse, in which Cre recombinase is expressed in the hindbrain region at E9.5 and influences many cells in the cerebellum at birth [106]. Pax2/cre,Pdss2f/- died within the first 36 h of life and suffered from cerebellar hypoplasia and cellular disorganization in the cerebellum, which mimics the cerebellum atrophy commonly observed in CoQ-deficient infants. This macroscopic observation was accompanied by inhibition of cell migration and cell proliferation as well as increased ectopic apoptosis in the cerebellum of Pdss2 KO embryos [106]. A different specific deleter mouse Pcp2-cre [144] was also crossed with a Pdss2f/f mouse to generate another tissue-specific KO mouse (Pcp2/cre,Pdss2f/−). In this last Pdss2 mutant model, the mutation was developed only in cerebellum Purkinje cells after birth. Pcp2/cre,Pdss2f/− was clinically healthy and with normal behavior until 4.5 months. From this time, an ataxic phenotype at old age was manifested due to the progressive decrease of Purkinje cells and initiation of diffusive neuron death by apoptosis, showing at 9.5-month loss in motor coordination and incapability of maintaining body balance on a rod. Thus, this last conditional KO mouse model may be a better model to study the cerebellar defects linked to CoQ deficiency in adulthood [106].
As steroid-resistant nephrotic syndrome (SRNS) has been linked to mutations in several genes encoding CoQ biosynthetic enzymes [52,54,57,63], a podocyte-specific Coq6 KO mouse model was generated by crossing Podocin-cre mice with Coq6loxP/loxP mice in which two loxP sites surround exon 6 in the Coq6 gene. The COQ6 protein is an hydroxylase that seems to catalyze two uncharacterized hydroxylation steps in CoQ biosynthesis [136]. The Coq6 KO model is referred as Nphs2/cre,Coq6loxP/loxP KO mice, also Coq6podKO [107]. Coq6podKO mice appeared to be normal in development and body condition until the age of 5 months, when they deteriorated, gradually developing progressive glomerular sclerosis and proteinuria. They became moribund at 10 months of age with advanced decline of renal function. Interestingly, the administration of 2,4-dihidroxibenzoate acid (2,4-diHB), a CoQ10 precursor analogue, to 5-month-old Coq6podKO mice significantly protected from disease progression, and the survival was comparable to that of control mice. With the 2,4-diHB treatment, proteinuria and renal histology improved dramatically in treated Coq6podKO mice compared with untreated Coq6podKO mice [107]. However, the therapeutic mechanisms of 2,4-diHB in this model are unclear, and the levels of CoQ after the treatment were not reported.
As mutations in ADCK4 (COQ8B) have been also reported in patients with SRNS, a similar study was carried out to evaluate the role of Adck4 in kidney function. For that purpose, a podocyte-specific Adck4 KO mouse was generated. Known as Adck4tm1d or Nphs2/cre, Adck4flox/flox (also Adck4ΔPodocyte) was generated by crossing the Nphs2-cre mouse with the Adck4flox/flox mouse in which two loxP sites surround exons 5 and 6 in the Adck4 gene. Adck4ΔPodocyte developed an increased in adult morbidity, mortality, and weight loss with progressive albuminuria and renal structural abnormalities (focal segmental glomerulosclerosis with extensive interstitial fibrosis and tubular atrophy) and functional decline. Treatment of 3-month-old Adck4 KO mice with 2,4-diHB prevented the development of renal pathology and reversed mitochondrial dysfunction. Again, the CoQ levels after 2,4-diHB therapy were not reported, so the therapeutic mechanisms were unknown. Nevertheless, these data suggest that ADCK4, an uncharacterized mitochondrial protein with no enzymatic activity directly involved in the CoQ biosynthetic pathway, must be a protein component in CoQ biosynthesis [108]. The rescue in the phenotypes of the Coq6podKO and Adck4ΔPodocyte KO mice after the treatment with 2,4-diHB suggests a potential treatment strategy for nephrotic syndrome resulting from COQ6 and ADCK4 mutations.
Separately, the Coq7(=Mclk1)liver-KO mouse was generated. This model carried a liver-specific KO mutation in the Coq7 gene. The quinone measurements in the liver of Coq7liver-KO showed that CoQ9 levels were decreased by 85% and DMQ9 was substantially accumulated, which is consistent with a dysfunctional COQ7 activity. This decrease in CoQ in hepatocytes caused only a mild reduction of the mitochondrial respiratory chain function and no gross abnormalities, suggesting a nonlinear dependence of mitochondrial respiratory capacity on CoQ content. In addition, these results suggest that very little CoQ is required in this process. Moreover, DMQ seems not to interfere with CoQ-mediated mitochondrial electron transport in the liver. The supplementation with CoQ10 to Coq7liver-KO increased the CoQ10 content in the liver and partially rescued the electron transport deficit in this tissue [109]. Later on, a global conditional KO mouse model for the Coq7 gene was generated by expressing tamoxifen TM-dependent CreERT2 transgene. Following the induction of Coq7 KO by TM at two months of age, adult-onset global Coq7 KO (aogCoq7) animals gradually accumulated DMQ and lost CoQ, leading to mitochondrial dysfunction and shortened lifespan. Interestingly, during the time that Coq7 KO mice started to die, the levels of CoQ were very low in the heart. However, cardiac function was not compromised, suggesting that this tissue has a CoQ reservoir. Dietary CoQ10 was ineffective in rescuing the phenotype of Coq7 KO mouse mice, but the supplementation with 2,4-diHB produced a partial restoration of mitochondrial respiration and led to a marked increase in their survival [110]. In this particular case, the therapeutic mechanism was attributed to the increase in CoQ and decrease in DMQ, as 2,4-diHB is able to bypass defects in COQ7. This result highlighted the important clinical implications of the treatment with 2,4-diHB also in COQ7 mutations. Furthermore, a late-onset treatment with 2,4-diHB 6 months after TM induction, a moment in which the untreated KO mice show apparent phenotypes, was also able to increase the survival the Coq7 KO mice [110].
3.2.3. Constitutive Knockout and Knock-In Mouse Models
In addition to the mouse models with spontaneous mutation and the conditional KO mouse models of CoQ deficiency previously described, some constitutive KO and knock-in (KI) models have been generated to date.
The KO for Pdss2 (B6.Ȥp3/cre,Pdss2loxP/loxP) was embryonically lethal, with no homozygous embryos surviving beyond 10.5 days of gestation [105], which is consistent with the hypothesis that ubiquinone is essential for mouse embryonic development [106,112]. Similar results were obtained with the constitutive KO mouse for Coq3 [111]. COQ3 is an O-methyltransferase responsible for the first and last O-methyltransferase steps in CoQ biosynthesis [145]. Coq3−/− resulted in embryonic lethality, as no Coq3 homozygous mice were obtained from heterozygous crosses [111]. However, Coq3 heterozygous mouse (Coq3+/−) showed normal lifespan and no changes in CoQ levels in pure mitochondria isolated from 3-month-old Coq3+/− livers [111].
Similar to Pdss2 and Coq3 KO, two independently generated KO models of Coq7 showed embryonic lethality [112,113]. In one case, the Coq7−/− embryos failed to survive beyond E10.5, exhibiting small-sized body and delayed neural development. These results suggest that COQ7-deficient embryos are able to develop until E8 with the energy generated from anaerobic glycolysis, but they are unable to survive due to the lack of aerobic glycolysis required for further embryogenesis in mice. Electron microscopic analysis showed a loss of organized neuroepithelial structures in Coq7−/− embryos and enlarged mitochondria with vesicular cristae and enlarged lysosomes filled with disrupted membranes, demonstrating that COQ7 is essential for neurogenesis and for the maintenance of mitochondrial integrity [113]. In the other study, the Coq7−/− embryos showed a developmental delay that was evident by E9.5, and all Coq7−/− embryos detected were completely resorbed by E13.5 [112]. Consistent again with a dysfunctional COQ7 protein, the embryos of both KO Coq7 mice showed reduced CoQ levels and accumulation of DMQ, highlighting the crucial role of COQ7 in the CoQ metabolism [112,113].
On the other hand, heterozygous Coq7+/− mice were viable and fertile with no obvious anatomical or behavioral defects [112]. The amounts of CoQ9 and CoQ10 were similar in wild-type and heterozygous embryos. Young Coq7+/− mutants showed reduced mitochondrial oxygen consumption, reduced electron transport, reduced mitochondrial ATP synthesis, reduced mitochondrial and overall ATP levels, and reduced whole-animal oxygen consumption [146]. Moreover, it was discovered that in the livers of very old Coq7+/− mutants, a phenomenon of loss-of-heterozygosity took place because there were areas of complete loss of expression of Coq7. That was followed by a decrease in the CoQ levels in the livers of very old Coq7+/− mutants [111,146]. Furthermore, it has been reported that Coq7+/− mice present a unique mitochondrial CoQ profile that was characterized by decreased CoQ levels in the inner mitochondrial membrane coupled with higher CoQ levels in the outer mitochondrial membrane. The low levels of CoQ in the inner membrane could increase oxidative stress by partially inhibiting the electron transport chain. At the same time, similar amounts of total CoQ were detected in the mitochondrial, peroxisomal, and plasma membrane fractions from Coq7+/− and control mice livers at three months of age. Dietary supplementation of Coq7+/− mice with CoQ10 normalized the CoQ levels in the inner mitochondrial membrane and in the outer mitochondrial membrane as well as the respiratory chain dysfunction [111]. Interestingly, Coq7+/− mice showed an increase in lifespan up to 31%, as it was reported in the mutational inactivation of its orthologue clk-1 in C. elegans, with significantly lower levels of DNA damage [114]. The increase in the lifespan in Coq7+/− mice has been associated with an early hepatic mitochondrial dysfunction, which induces a protective physiological response called mitohormesis [114]. The mechanisms underlying the increased lifespan in Coq7+/− mice include the following:
(1) Despite the early mitochondrial dysfunction, the function of Coq7+/− mitochondria declines less rapidly with age than that of the wild type, and there is a slower accumulation of global oxidative biomarkers of aging in these mutants [147].
(2) The altered mitochondrial phenotype of Coq7+/− mutants enhanced immune reaction to infection by improving basal and stimulated expression of HIF-1α in liver and macrophages, in association with elevated expression of inflammatory cytokines [148].
(3) Coq7+/− mutants have enhanced resistance to neurological damage after ischemia and reperfusion [149].
Finally, a recent study in Coq7+/− mutants suggested that Coq7 regulates microglial metabolic reprogramming participating in neuroinflammation and dopaminergic cell death [150].
In contrast with the Coq gene KOs previously mentioned, the lack of COQ8A in mice (Coq8a−/−) resulted in a mild phenotype with progressive cerebellar ataxia, mild exercise intolerance, and moderate CoQ deficiency, recapitulating the more frequent features of autosomal-recessive cerebellar ataxia type 2 (ARCA2, the most frequent form of hereditary CoQ deficiency in humans) [115]. The pathophysiology of the disease was linked to dysfunctional cerebellar Purkinje cells, defective skeletal muscle, and disruption of complex Q. Coq8a−/− mice develop mild, tissue-specific CoQ deficiency. The levels of CoQ were low in skeletal muscle of Coq8a−/− mice, while normal CoQ levels were observed in whole Coq8a−/− cerebellum. These data suggest that CoQ deficiency may specifically affect the Purkinje cells of the cerebellum, although the specific measurement of CoQ in those cells was not performed. Additionally, complex Q proteins were deficient across multiple Coq8a−/− tissues, showing that COQ8A participates in the stability of complex Q in mammals. Moreover, Coq8a−/− Purkinje cells displayed Golgi morphology defects, but normal mitochondria, providing a model system to study CoQ production across different organelles, cells, and tissues [115].
Another mouse model with intriguing results is the KO model for Coq9 (Coq9Q95X), which had normal development, in contrast with the embryonic lethality described in Pdss2, Coq3, and Coq7 KO mice. The Coq9Q95X mouse model was generated by the Wellcome Trust Sanger Institute from ES cell clone EPD0112_2_A09 obtained from the supported KOMP Repository (www.komp.org; accessed on 26 September 2021). Lack of the COQ9 protein caused moderate CoQ deficiency; i.e., the cerebrum, cerebellum, and heart showed around 50% residual CoQ9 levels, while the kidney and skeletal muscle had 30% of residual CoQ9 levels compared with wild-type mice. This led to a reduction in CI+III activity and mitochondrial respiration in skeletal muscle and late-onset mild mitochondrial myopathy with exercise intolerance in female mice. The COQ9 protein is needed for the stability and activity of COQ7 in the CoQ biosynthetic pathway, so the brain, kidneys, and muscles of Coq9Q95X mice also showed a decrease in the levels of COQ7 [116]. As in other animal models with a decrease in the COQ7 levels [146,151], Coq9Q95X mice showed an increase in lifespan, especially in males because they lived on average 15% longer than their wild-type littermates. The increase in lifespan in Coq9Q95X mice is accompanied by a reduction in the animals’ body weight [117]. However, unlike Coq7+/− mice, the liver of Coq9Q95X mice showed normal levels of CoQ levels; consequently, mitochondrial function was also normal in this tissue. Therefore, unlike Coq7+/− mice, the hepatic mitochondrial dysfunction and increased oxidative stress induced by subphysiological levels of CoQ biosynthetic proteins are not the cause of the increased lifespan in Coq9Q95X mice. These effects could be mediated by mechanisms initiated in the mitochondria from other tissues or by other unknown mechanisms [117].
To better understand the pathomechanisms of primary CoQ deficiency due to a mutation in the Coq9 gene, a knock-in mouse model carrying a homozygous mutation in Coq9 gene was generated (Coq9R239X) [45]. The Coq9R239X mice encoded the mutation CGT > TGA (R239X) within exon 7 in the mouse genome, which is a homologue to the human R244X mutation described in a patient with CoQ10 deficiency [64]. In contrast with the Coq9Q95X mouse, the Coq9R239X model manifested severe CoQ deficiency; i.e., the CoQ9 levels were 15–20% compared to wild-type animals. The deficit in CoQ induced a decrease in mitochondrial respiration, particularly in the brain and kidneys. These effects lead to neuronal death and demyelination with spongiosis and astrogliosis in the brain of Coq9R239X mice, leading to a premature death [45,116]. In tissues from Coq9R239X mice, a truncated version of the COQ9 protein was detected, leading to a reduction in the COQ7 protein and, as a consequence, a widespread CoQ deficiency and accumulation of DMQ [45,116]. The truncated version of COQ9 protein in Coq9R239X mice destabilizes the complex Q and produces a severe phenotype associated with fatal encephalomyopathy. Therefore, the stability of the complex Q clearly influences the CoQ biosynthesis rate and, consequently, the degree of the severity of CoQ deficiency and the development of tissue-specific phenotypes [116]. Moreover, the disruption of mitochondrial sulfide oxidation has been identified as one of the pathomechanisms associated to CoQ deficiency in both Coq9Q95X and Coq9R239X mice [118], similarly to the Pdss2kd/kd mice [15]. Specifically, severe CoQ deficiency caused a remarkable reduction in SQOR levels and activity, and this deficit induced changes in the mitochondrial sulfide metabolism. In the brain of Coq9R239X mice, the low SQOR levels produced an increase in downstream enzymes as well as adjustments in the levels of thiols. As a result, the biosynthetic pathways of glutamate, serotonin, and catecholamines were altered in the cerebrum, and the blood pressure was reduced [118]. Additionally, CoQ deficiency in Coq9R239X mice also affected the sulfide biosynthetic pathway (also known as the transsulfuration pathway), independently of the availability of sulfur amino acids. In the kidneys of Coq9R239X mice, the levels of cystathione-β-synthase, an enzyme of the transsulfuration pathway, marginally increased compared with the control mice, and this variation was maintained under supplementation with N-acetylcysteine or a sulfur amino acids restriction diet, displaying the tight regulation between the biosynthetic and catabolic pathways of sulfide metabolism [119].
Oral supplementation with ubiquinone-10, the oxidized form of CoQ10, had limited efficacy in the treatment of the Coq9R239X mouse model due to its poor absorption and bioavailability. The supplementation with ubiquinol-10, the reduced form of ubiquinone-10, provided better therapeutic results. It increased the levels of CoQ10 in tissue homogenates and cerebral mitochondria, which results in an increase in CoQ-dependent respiratory chain activities, reduction in the spongiosis, astrogliosis, and oxidative stress in different brain areas, and an increase in body weight. These data suggest that water-soluble formulations of ubiquinol-10 are a better option than ubiquinone-10 for the treatment of primary CoQ10 deficiency [120]. Looking for alternative strategies to the classical exogenous CoQ10 supplementation with the aim of increasing the endogenous CoQ biosynthesis, the therapeutic potential of 2,4-diHB (β-resorcylic acid or β-RA in the study) was also tested in Coq9R239X mice [121,152]. Remarkably, the treatment in Coq9R239X mice increased the lifespan to values close to the lifespan in wild-type mice. Moreover, while the maximal survival of Coq9R239X mice treated with ubiquinol-10 was 17 months of age, the lifespan achieved by 2,4-diHB reached a maximum of 25 months of age [121]. 2,4-diHB supplementation rescued the phenotype of Coq9R239X mice, as shown by the reduction in the histopathological signs of the encephalopathy, i.e., the spongiosis and reactive astrogliosis. Those effects were mainly linked to the decrease in the levels DMQ9 and the increase in mitochondrial bioenergetics in peripheral tissues. However, even if the CoQ biosynthesis or the mitochondrial function did not change in the brain after the therapy, the Coq9R239X mice showed an almost absence of cerebral vacuoles and reactive astrocytes after the 2,4-diHB treatment. The authors suggested the hypothesis of a possible tissue–brain cross-talk, but further studies are needed to reveal how the peripheral tissues communicate with the brain in this situation [121,152].
Two other therapeutic strategies have been tested using the Coq9R239X mouse model or cells from this mouse model. Firstly, the use of a lentiviral vector (CCoq9WP) allowed the ectopi over-expression of Coq9 in mouse embryonic fibroblasts (MEFs) and hematopoietic progenitor cells (HPC), leading to the restore of the CoQ biosynthetic pathway and mitochondrial function [122]. Secondly, researchers tested the therapeutic effects of rapamycin administration in Coq9R239X mice [153] based on other studies that had demonstrated the therapeutic benefits of rapamycin therapy in a few mouse models of mitochondrial diseases [154,155]. Neither a low nor a high dose of rapamycin were able to increase the mitochondrial bioenergetics, to reduce the spongiosis and reactive astrogliosis, and to rescue the phenotypic characteristics of Coq9R239X mice, resulting in the lack of efficacy for increasing the survival [153].
Recently, a heterozygous Adck2 KO mouse model (Adck2+/−) has been generated to clarify the role of ADCK2 in CoQ biosynthesis and to explain the pathological mechanisms involved in the disease caused by a mutation in this gene. While Adck2−/− mice are embryonically lethal, Adck2+/− mice partially recapitulated the phenotype of a human patient characterized by an adult-onset myopathy due to CoQ deficiency and an overall defect in mitochondrial lipid metabolism. The quinones determination showed a significant decrease in CoQ9 and CoQ10 levels in the skeletal muscle of Adck2+/−, leading to a decrease in mitochondrial complex I+III and II+III activities and oxygen consumption rate. These results suggest that ADCK2 deficiency impaired the normal mitochondrial respiratory chain function in skeletal muscle, ultimately leading to mitochondrial dysfunction. Further transcriptomics and metabolomics analysis supported the hypothesis that ADCK2 plays an important role in the energy homeostasis in skeletal muscle. Based on these data, it was proposed that ADCK2 is involved in the regulation of mitochondrial fatty acid β-oxidation and CoQ levels in skeletal muscle. The supplementation with CoQ10 partially rescued the phenotype of Adck2+/− mice, indicating the possibility of improving some of the defects stemming from CoQ deficiency due to Adck2 mutation [123].
A mouse model of secondary CoQ deficiency of genetic origin is also available [124]. PARL is a protease located in the inner mitochondrial membrane with relevant but unclear physiological roles. However, it is related to different prevalent human diseases, including cancer and neurodegenerative diseases, highlighting its biological significance in pathological conditions [156]. Parl−/− mice developed a necrotizing encephalomyopathy closely resembling Leigh syndrome. Mitochondria from the brain of Parl−/− mice showed progressive structural alterations and early deficiencies of complex III (CIII) and CoQ, resulting in a disruption of the mitochondrial calcium metabolism. A mitochondrial proteome analysis in the brain of the PARL-deficient mice showed downregulation of the CIII-regulating protein TTC19, several proteins required for CoQ biosynthesis, and SQOR. The disruption in the COQ4 levels was detected in Parl−/− brains already at one week of age. Subsequently, a downregulation of COQ3, COQ5, COQ6, COQ7, COQ9, and SQOR was also observed. As a conclusion, the data indicated that the stabilization of TTC19 and the following regulation of CIII activity is mediated by PARL as well as the maintenance of COQ4 expression in the brain, supporting CoQ biosynthesis in this organ. Nevertheless, further studies are needed to explain the connection between the disruption in PARL and in COQ4 and the resulting CoQ deficiency in the brain [124].
4. Conclusions and Perspectives
Primary CoQ deficiency is caused by mutations in any of the genes involved in the CoQ biosynthetic pathway. Clinically, CoQ deficiency is a heterogeneous disease in humans, which is linked to five major phenotypes and some other clinical symptoms. The clinical heterogeneity and the variety of functions associated with CoQ complicate the description of the pathomechanisms underlying this mitochondrial disease as well as the development of new therapies. For that reason, several animal models with CoQ deficiency have been generated, providing very useful insights, such as: (1) they have confirmed the involvement of the target proteins in CoQ biosynthesis in different species, indicating the evolutionary conservation of the pathway; (2) they have clearly shown the importance of CoQ during development and its role in the mitochondrial respiratory chain, sulfide metabolism, and pyrimidine metabolism; (3) they have revealed that the genotype–phenotype association, the bioenergetics defect, the increased oxidative stress, and the disruption of sulfide and pyrimidine metabolism are key disease pathomechanisms that may explain, at least in part, the tissue specificities and the clinical heterogeneity; (4) they have shown the limitation of the exogenous CoQ10 therapy, especially in some particular phenotypes; and (5) they have opened promising therapeutic strategies based on the supplementation with 4-HB analogs. However, some functional roles of CoQ have not been evaluated yet in the context of CoQ deficiency. In addition, the therapeutic mechanisms of 2,4-diHB in some models seem to be independent of the CoQ biosynthetic pathway, increasing the potential relevance of this compound in the clinical practice. Furthermore, the development of other animal models is required to cover the study of this complex syndrome and the different functions of CoQ, and to elucidate the synthesis of CoQ in mammals. This is especially important, for example, in the case of CoQ deficiency due to mutations in the COQ2 gene. In vitro studies have demonstrated the therapeutic potential of the natural precursor of CoQ, 4-hydroxybenzoic acid (4-HB) [157], but there is no available mouse model with mutation in Coq2 to extend the study, limiting the potential translation of this treatment into the clinic.
Author Contributions
Writing—original draft preparation, P.G.-G. and L.C.L.; writing—review and editing, P.G.-G. and L.C.L.; writing—review, E.B.-C., M.E.D.-C., S.L.-H. and A.H.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the MCIN/AEI/10.13039/501100011033, Spain, and the ERDF (RTI2018-093503-B-100); the Muscular Dystrophy Association (MDA-602322); and from the Junta de Andalucía (grant number P20_00134). P.G.-G. is ‘FPU fellow’ from the Ministerio de Universidades, Spain. A.H.-G. is supported by the “Plan Propio de Investigación” from the University of Granada. S.L.-H. is supported by the “garantía juvenil” program. E.B.-C. is supported by the Consejería de Salud, Junta de Andalucía, Spain.
Conflicts of Interest
The other authors have declared that no conflict of interest exists.
References
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Del-Rio, L.; Clarke, C.F. Coenzyme Q biosynthesis: An update on the origins of the benzenoid ring and discovery of new ring precursors. Metabolites 2021, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Kawamukai, M. Biosynthesis and bioproduction of coenzyme Q10 by yeasts and other organisms. Biotechnol. Appl. Biochem. 2009, 53, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [PubMed]
- Awad, A.M.; Bradley, M.C.; del Rio, L.F.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [PubMed]
- Hidalgo-Gutiérrez, A.; González-García, P.; Díaz-Casado, M.; Barriocanal-Casado, E.; López-Herrador, S.; Quinzii, C.; López, L. Metabolic targets of coenzyme Q10 in mitochondria. Antioxidants 2021, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Luo, F.; Li, X.; Yi, S.; Yang, B.; Jiang, Z. Structural characteristics and catalytic cycle of dihydroorotate dehydrogenase-a review. Sheng Wu Gong Cheng Xue Bao. Chin. J. Biotechnol. 2020, 36, 2732–2740. [Google Scholar]
- Mracek, T.; Drahota, Z.; Houstek, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Navas, P.; Calvo, G.T.B. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta 2010, 1797, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Olsen, R.K.J.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407. [Google Scholar] [CrossRef] [PubMed]
- Blake, R.L.; Hall, J.G.; Russell, E.S. Mitochondrial proline dehydrogenase deficiency in hyperprolinemic PRO/Re mice: Genetic and enzymatic analyses. Biochem. Genet. 1976, 14, 739–757. [Google Scholar] [CrossRef]
- Salvi, F.; Gadda, G. Human choline dehydrogenase: Medical promises and biochemical challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H (+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Jaburek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E.; Ichas, F.; Bernardi, P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 1998, 273, 25734–25740. [Google Scholar] [CrossRef]
- Walter, L.; Miyoshi, H.; Leverve, X.; Bernardi, P.; Fontaine, E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic. Res. 2002, 36, 405–441. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjáková, A.; Low, H.; Sun, I.; Navas, P.; Crane, F. Plasma membrane coenzyme Q: Evidence for a role in autism. Biol. Targets Ther. 2014, 8, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Morre, D.J.; Morre, D.M. Non-mitochondrial coenzyme Q. Biofactors 2011, 37, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Cirilli, I.; Damiani, E.; Dludla, P.V.; Hargreaves, I.; Marcheggiani, F.; Millichap, L.E.; Orlando, P.; Silvestri, S.; Tiano, L. Role of coenzyme Q10 in health and disease: An update on the last 10 years (2010–2020). Antioxidants 2021, 10, 1325. [Google Scholar] [CrossRef]
- Casado, D.; Quiles, J.L.; Casado, B.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The paradox of coenzyme Q10 in aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef]
- Teclebrhan, H.; Jakobsson-Borin, A.; Brunk, U.; Dallner, G. Relationship between the endoplasmic reticulum-Golgi membrane system and ubiquinone biosynthesis. Biochim. Biophys. Acta 1995, 1256, 157–165. [Google Scholar] [CrossRef]
- Mugoni, V.; Medana, C.; Santoro, M.M. 13C-isotope-based protocol for prenyl lipid metabolic analysis in zebrafish embryos. Nat. Protoc. 2013, 8, 2337–2347. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; del Rio, L.F.; Bradley, M.C.; Dunn, C.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The endoplasmic reticulum-mitochondria encounter structure complex coordinates coenzyme Q biosynthesis. Contact 2019, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. The complexity of making ubiquinone. Trends Endocrinol. Metab. 2019, 30, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef]
- Pierrel, F. Impact of chemical analogs of 4-hydroxybenzoic acid on coenzyme Q Biosynthesis: From inhibition to bypass of coenzyme q deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Ozeir, M.; Mühlenhoff, U.; Webert, H.; Lill, R.; Fontecave, M.; Pierrel, F. Coenzyme Q biosynthesis: Coq6 is required for the C5-hydroxylation reaction and substrate analogs rescue Coq6 deficiency. Chem. Biol. 2011, 18, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.; Clarke, C.F. Isolation and functional expression of human COQ3, a gene encoding a methyltransferase required for ubiquinone biosynthesis. J. Biol. Chem. 2000, 275, 12381–12387. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Casarin, A.; Desbats, M.A.; Doimo, M.; Trevisson, E.; Santos-Ocaña, C.; Navas, P.; Clarke, C.F.; Salviati, L. Molecular characterization of the human COQ5 C-methyltransferase in coenzyme Q10 biosynthesis. Biochim. Biophys. Acta 2014, 1841, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Marbois, B.N.; Clarke, C.F. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.; Stark, J.L.; Stefely, J.; Reddy, T.; Hebert, A.S.; et al. Conserved lipid and small-molecule modulation of COQ8 reveals regulation of the ancient kinase-like UbiB family. Cell Chem. Biol. 2018, 25, 154–165.e11. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Aydin, D.; Von Bank, H.; Smith, R.W.; Linke, V.; Weisenhorn, E.; McDevitt, M.T.; Hutchins, P.; Wilkerson, E.M.; Wancewicz, B.; et al. An isoprene lipid-binding protein promotes eukaryotic coenzyme Q biosynthesis. Mol. Cell 2019, 73, 763–774.e10. [Google Scholar] [CrossRef] [PubMed]
- He, C.H.; Black, D.S.; Nguyen, T.P.; Wang, C.; Srinivasan, C.; Clarke, C.F. Yeast Coq9 controls deamination of coenzyme Q intermediates that derive from para-aminobenzoic acid. Biochim. Biophys. Acta 2015, 1851, 1227–1239. [Google Scholar] [CrossRef] [PubMed][Green Version]
- García-Corzo, L.; Sánchez, M.L.; Doerrier, C.; García, J.A.; Guarás, A.; Acin-Perez, R.; Bullejos-Peregrín, J.; López, A.; Escames, G.; Enríquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby, C.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Marbois, B.; Gin, P.; Gulmezian, M.; Clarke, C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta 2009, 1791, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Alcázar-Fabra, M.; Rodríguez-Sánchez, F.; Trevisson, E.; Brea-Calvo, G. Primary coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of coenzyme Q10 deficiency. Mol. Syndr. 2014, 5, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Cascajo, M.V.; Alcázar-Fabra, M.; Hernández-Camacho, J.D.; Sánchez-Cuesta, A.; Rodríguez, A.B.C.; Ballesteros-Simarro, M.; Arroyo-Luque, A.; Rodríguez-Aguilera, J.C.; Fernández-Ayala, D.J.M.; et al. Secondary CoQ10 deficiency, bioenergetics unbalance in disease and aging. Biofactors 2021, 47, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; De Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; DiMauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Diomedi-Camassei, F.; Di Giandomenico, S.; Santorelli, F.M.; Caridi, G.; Piemonte, F.; Montini, G.; Ghiggeri, G.M.; Murer, L.; Barisoni, L.; Pastore, A.; et al. COQ2 Nephropathy: A newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007, 18, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Rodríguez-Hernández; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency ofCOQ4causes coenzyme Q10deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Malicdan, M.C.V.; Vilboux, T.; Ben-Zeev, B.; Guo, J.; Eliyahu, A.; Pode-Shakked, B.; Dori, A.; Kakani, S.; Chandrasekharappa, S.C.; Ferreira, C.R.; et al. A novel inborn error of the coenzyme Q10 biosynthesis pathway: Cerebellar ataxia and static encephalomyopathy due to COQ5 C-methyltransferase deficiency. Hum. Mutat. 2017, 39, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Vie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell. Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Tazir, M.; López, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; De Lonlay, P.; De Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef]
- Feng, C.; Wang, Q.; Wang, J.; Liu, F.; Shen, H.; Fu, H.; Mao, J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: Case reports and literature review. Medicine 2017, 96, e8880. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.; Hardy, J.; et al. A Nonsense Mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Danhauser, K.; Herebian, D.; Haack, T.B.; Rodenburg, R.J.; Strom, T.M.; Meitinger, T.; Klee, D.; Mayatepek, E.; Prokisch, H.; Distelmaier, F. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss-of-function variant in COQ9. Eur. J. Hum. Genet. 2015, 24, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, O.; Naini, A.; Slonim, A.E.; Skavin, N.; Hadjigeorgiou, G.L.; Krawiecki, N.; Weissman, B.M.; Tsao, C.-Y.; Mendell, J.R.; Shanske, S.; et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001, 56, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; Dubourg, O.; Benoist, J.F.; Jardel, C.; Mochel, F.; Koenig, M.; Brice, A.; Lombea, A.; Durr, A. Muscle coenzyme Q10 deficiencies in ataxia with oculomotor apraxia 1. Neurology 2007, 68, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Aeby, A.; Sznajer, Y.; Cavé, H.; Rebuffat, E.; Van Coster, R.; Rigal, O.; Van Bogaert, P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007, 30, 827. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Martín, M.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Aguilera, J.C.R.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]
- Montero, R.; Sánchez-Alcázar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martín, D.; Rivera, H.; Martín, M.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J. Inherit. Metab. Dis. 2014, 38, 145. [Google Scholar] [CrossRef] [PubMed]
- Kühl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.-G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Chaudhuri, R.; Yang, P.; Maghzal, G.J.; Thomas, K.C.; Krycer, J.R.; Humphrey, S.J.; Parker, B.L.; Fisher-Wellman, K.H.; Meoli, C.C.; et al. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 2018, 7, e32111. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Hirano, M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010, 16, 183–188. [Google Scholar] [CrossRef]
- Hargreaves, I.; Heaton, R.A.; Mantle, D. Disorders of human coenzyme Q10 metabolism: An Overview. Int. J. Mol. Sci. 2020, 21, 6695. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.; Saldanha, J.W.; Gould, A.P. A Drosophila model for primary coenzyme Q deficiency and dietary rescue in the developing nervous system. Dis. Model. Mech. 2010, 3, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, Q.; He, D.; Ma, T.; Du, L.; Dui, W.; Guo, X.; Jiao, R. Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J. Genet. Genom. 2011, 38, 225–234. [Google Scholar] [CrossRef]
- Cheng, W.; Song, C.; Anjum, K.M.; Chen, M.; Li, D.; Zhou, H.; Wang, W.; Chen, J. Coenzyme Q plays opposing roles on bacteria/fungi and viruses in Drosophila innate immunity. Int. J. Immunogenet. 2011, 38, 331–337. [Google Scholar] [CrossRef]
- Hermle, T.; Braun, D.A.; Helmstädter, M.; Huber, T.B.; Hildebrandt, F. Modeling monogenic human nephrotic syndrome in the drosophila garland cell nephrocyte. J. Am. Soc. Nephrol. 2016, 28, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ayala, D.J.; Guerra, I.; Sanz, A.; Navas, P. coq7 (cg14437) interference courses a primary coenzyme Q deficiency in Drosophila. FEBS J. 2012, 279, 13–123. [Google Scholar]
- Guerra, I.; Fernandez-Ayala, D.; Daniel, J.M.; Navas, P. RNA interference (RNAi) of genes involved in Coenzyme Q biosynthesis in Drosophila melanogaster models Coenzyme Q deficiency in humans. In Proceedings of the 22nd IUBMB and 37th FEBS Congress, Seville, Spain, 4–9 September 2012. [Google Scholar]
- Vasta, V.; Sedensky, M.; Morgan, P.; Hahn, S. Altered redox status of coenzyme Q9 reflects mitochondrial electron transport chain deficiencies in Caenorhabditis elegans. Mitochondrion 2011, 11, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Takahashi, K.; Tomita, S.; Keino, T.; Honda, S.; Yoshino, K.; Suzuki, K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat. Res. 1990, 237, 165–171. [Google Scholar] [CrossRef]
- Ishii, N.; Senoo-Matsuda, N.; Miyake, K.; Yasuda, K.; Ishii, T.; Hartman, P.S.; Furukawa, S. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mech. Ageing Dev. 2004, 125, 41–46. [Google Scholar] [CrossRef]
- Jonassen, T.; Larsen, P.L.; Clarke, C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 2001, 98, 421–426. [Google Scholar] [PubMed]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G.; Hoppel, C.L. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J. Biol. Chem. 2004, 279, 54479–54486. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Gangoiti, J.A.; Sedensky, M.M.; Morgan, P.G. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 370–376. [Google Scholar] [CrossRef]
- Yang, Y.-Y.; Vasta, V.; Hahn, S.; Gangoiti, J.A.; Opheim, E.; Sedensky, M.M.; Morgan, P.G. The role of DMQ9 in the long-lived mutant clk-1. Mech. Ageing Dev. 2011, 132, 331–339. [Google Scholar] [CrossRef][Green Version]
- Takahashi, M.; Ogawara, M.; Shimizu, T.; Shirasawa, T. Restoration of the behavioral rates and lifespan in clk-1 mutant nematodes in response to exogenous coenzyme Q10. Exp. Gerontol. 2012, 47, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Saiki, R.; Chin, R.; Srinivasan, C.; Clarke, C.F. Restoring de novo coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous coenzyme Q supplementation. Gene 2012, 506, 106–116. [Google Scholar] [CrossRef]
- Gavilan, A.; Asencio, C.; Cabello, J.; Rodriguez-Aguilera, J.C.; Schnabel, R.; Navas, P. C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors 2005, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Earls, L.R.; Hacker, M.L.; Watson, J.D.; Miller, D.M. Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 14460–14465. [Google Scholar] [CrossRef] [PubMed]
- Hihi, A.K.; Gao, Y.; Hekimi, S. Ubiquinone is necessary for caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 2002, 277, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Navas, P.; Cabello, J.; Schnabel, R.; Cypser, J.R.; Johnson, T.E.; Rodríguez-Aguilera, J.C. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Mugoni, V.; Postel, R.; Catanzaro, V.; De Luca, E.; Turco, E.; Digilio, G.; Silengo, L.; Murphy, M.; Medana, C.; Stainier, D.; et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell 2013, 152, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Garone, C.; Emmanuele, V.; Tadesse, S.; Krishna, S.; Dorado, B.; Hirano, M. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 2013, 27, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Gutiérrez, A.H.; Kleiner, G.; Lopez, L.C. The role of sulfide oxidation impairment in the pathogenesis of primary CoQ deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Gutiérrez, A.H.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ10 supplementation rescues nephrotic syndrome through normalization of H2S oxidation pathway. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Falk, M.J.; Polyak, E.; Zhang, Z.; Peng, M.; King, R.; Maltzman, J.S.; Okwuego, E.; Horyn, O.; Nakamaru-Ogiso, E.; Ostrovsky, J.; et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol. Med. 2011, 3, 410–427. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Licata, J.; Tsukikawa, M.; Marty, E.; Thomas, J.; Felix, C.A.; Xiao, R.; et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015, 24, 4829–4847. [Google Scholar] [CrossRef]
- Sidhom, E.H.; Kim, C.; Kost-Alimova, M.; Ting, M.T.; Keller, K.; Avila-Pacheco, J.; Watts, A.J.B.; Vernon, K.A.; Marshall, J.L.; Reyes-Bricio, E.; et al. Targeting a Braf/Mapk pathway rescues podocyte lipid peroxidation in CoQ-deficiency kidney disease. J. Clin. Investig. 2021, 131, e141380. [Google Scholar] [CrossRef]
- Peng, M.; Falk, M.J.; Haase, V.H.; King, R.; Polyak, E.; Selak, M.; Yudkoff, M.; Hancock, W.W.; Meade, R.; Saiki, R.; et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008, 4, e1000061. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Lu, L.-Y.; Liu, M.-F.; Yuan, Q.-J.; Sham, M.-H.; Guan, X.-Y.; Huang, J.-D. Cerebellar defects in Pdss2 conditional knockout mice during embryonic development and in adulthood. Neurobiol. Dis. 2011, 45, 219–233. [Google Scholar] [CrossRef]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 deficiency destabilizes the coenzyme Q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Mitochondrial respiration without ubiquinone biosynthesis. Hum. Mol. Genet. 2013, 22, 4768–4783. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Oxer, D.; Hekimi, S. Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat. Commun. 2015, 6, 6393. [Google Scholar] [CrossRef]
- Lapointe, J.; Wang, Y.; Bigras, E.; Hekimi, S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/− mice. J. Cell Biol. 2012, 199, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Levavasseur, F.; Miyadera, H.; Sirois, J.; Tremblay, M.L.; Kita, K.; Shoubridge, E.; Hekimi, S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001, 276, 46160–46164. [Google Scholar] [CrossRef] [PubMed]
- Nakai, D.; Yuasa, S.; Takahashi, M.; Shimizu, T.; Asaumi, S.; Isono, K.; Tako, T.; Suzuki, Y.-S.; Kuroyanagi, K.; Hirokawa, K.; et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001, 289, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef]
- Stefely, J.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.; et al. Cerebellar ataxia and coenzyme Q deficiency through loss of unorthodox kinase activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Casado, M.E.D.; Barca, E.; Tejada, M.; Montilla-García, A.; Cobos, E.J.; Escames, G.; Acuña-Castroviejo, D.; Quinzii, C.M.; López, L.C. The clinical heterogeneity of coenzyme Q 10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef]
- Rodríguez-Hidalgo, M.; Luna-Sánchez, M.; Gutiérrez, A.H.; Barriocanal-Casado, E.; Mascaraque, C.; Acuña-Castroviejo, D.; Rivera, M.; Escames, G.; López, L.C. Reduction in the levels of CoQ biosynthetic proteins is related to an increase in lifespan without evidence of hepatic mitohormesis. Sci. Rep. 2018, 8, 14013. [Google Scholar] [CrossRef]
- Luna-Sánchez, M.; Hidalgo-Gutiérrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, Á.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2016, 9, 78–95. [Google Scholar] [CrossRef] [PubMed]
- González-García, P.; Hidalgo-Gutiérrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sánchez-Hernández, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- García-Corzo, L.; Luna-Sánchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuña-Castroviejo, D.; López, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutierrez, A.; Barriocanal-Casado, E.; Bakkali, M.; Diaz-Casado, M.E.; Sanchez-Maldonado, L.; Romero, M.; Sayed, R.K.; Prehn, C.; Escames, G.; Duarte, J.; et al. beta-RA reduces DMQ/CoQ ratio and rescues the encephalopathic phenotype in Coq9 (R239X) mice. EMBO Mol. Med. 2019, 11, e9466. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Cueto-Ureña, C.; Benabdellah, K.; Gutiérrez-Guerrero, A.; Cobo, M.; Gutiérrez, A.H.; Rodríguez-Sevilla, J.J.; Martín, F.; Lopez, L.C. Gene therapy corrects mitochondrial dysfunction in hematopoietic progenitor cells and fibroblasts from Coq9R239X mice. PLoS ONE 2016, 11, e0158344. [Google Scholar] [CrossRef]
- Vázquez-Fonseca, L.; Schäefer, J.; Navas-Enamorado, I.; Santos-Ocaña, C.; Hernández-Camacho, J.D.; Guerra, I.; Cascajo, M.V.; Sánchez-Cuesta, A.; Horvath, Z.; Siendones, E.; et al. ADCK2 haploinsufficiency reduces mitochondrial lipid oxidation and causes myopathy associated with CoQ Deficiency. J. Clin. Med. 2019, 8, 1374. [Google Scholar] [CrossRef]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA 2018, 116, 277–286. [Google Scholar] [CrossRef]
- Licitra, F.; Puccio, H. An Overview of Current Mouse Models Recapitulating Coenzyme Q10 Deficiency Syndrome. Mol. Syndr. 2014, 5, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ayala, D.J.M.; Gancedo, S.J.; Guerra, I.; Navas, P. Invertebrate Models for Coenzyme Q10 Deficiency. Mol. Syndr. 2014, 5, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Jeibmann, A.; Paulus, W. Drosophila melanogaster as a Model Organism of Brain Diseases. Int. J. Mol. Sci. 2009, 10, 407–440. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta 2014, 1842, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Cristina, D.; Cary, M.; Lunceford, A.; Clarke, C.; Kenyon, C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, A.; Santos-Ocaña, C.; Ruiz-Ferrer, M.; Padilla, S.; Gavilán, A.; Rodríguez-Aguilera, J.C.; Navas, P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane of Caenorhabditis elegans. FEBS Lett. 2006, 580, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Currie, P. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Sawada, N.; Hirota, Y.; Uchino, Y.; Suhara, Y.; Hasegawa, T.; Amizuka, N.; Okamoto, T.; Tsugawa, N.; Kamao, M.; et al. Vitamin K2 biosynthetic enzyme, UBIAD1 is essential for embryonic development of mice. PLoS ONE 2014, 9, e104078. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, J.M.; Yang, H.; Chi, N.C. UBIAD1-mediated vitamin K2 synthesis is required for vascular endothelial cell survival and development. Development 2013, 140, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Tran, U.C.; Clarke, C.F. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion 2007, 7, S62–S71. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F.; Hulse, E.V. An inherited kidney disease of mice resembling human nephronophthisis. J. Med. Genet. 1971, 8, 41–48. [Google Scholar] [CrossRef][Green Version]
- Neilson, E.G.; McCafferty, E.; Feldman, A.; Clayman, M.D.; Zakheim, B.; Korngold, R. Spontaneous interstitial nephritis in kdkd mice. I. An experimental model of autoimmune renal disease. J. Immunol. 1984, 133, 2560–2565. [Google Scholar] [PubMed]
- Kelly, C.J.; Korngold, R.; Mann, R.; Clayman, M.; Haverty, T.; Neilson, E.G. Spontaneous interstitial nephritis in kdkd mice. II. Characterization of a tubular antigen-specific, H-2K-restricted Lyt-2+ effector T cell that mediates destructive tubulointerstitial injury. J Immunol. 1986, 136, 526–531. [Google Scholar] [PubMed]
- Hancock, W.W.; Tsai, T.-L.; Madaio, M.P.; Gasser, D.L. Cutting Edge: Multiple autoimmune pathways in kd/kd mice. J. Immunol. 2003, 171, 2778–2781. [Google Scholar] [CrossRef]
- Madaio, M.P.; Ahima, R.S.; Meade, R.; Rader, D.J.; Mendoza, A.; Peng, M.; Tomaszewski, J.E.; Hancock, W.W.; Gasser, D.L. Glomerular and tubular epithelial defects in kd/kd mice lead to progressive renal failure. Am. J. Nephrol. 2005, 25, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Jarett, L.; Meade, R.; Madaio, M.P.; Hancock, W.W.; George, A.L.; Neilson, E.G.; Gasser, D.L. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 2004, 66, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Rötig, A.; Mollet, J.; Rio, M.; Munnich, A. Infantile and pediatric quinone deficiency diseases. Mitochondrion 2007, 7, S112–S121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-M.; Ng, A.H.-L.; Tanner, J.A.; Wu, W.-T.; Copeland, N.G.; Jenkins, N.A.; Huang, J.-D. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis 2004, 40, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.Y.; Poon, W.; Shepherd, J.A.; Myles, D.C.; Clarke, C.F. Complementation of coq3 mutant yeast by mitochondrial targeting of the escherichia coli UbiG polypeptide: Evidence that UbiG catalyzes both O-methylation steps in ubiquinone biosynthesis. Biochemistry 1996, 35, 9797–9806. [Google Scholar] [CrossRef]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Stepanyan, Z.; Bigras, E.; Hekimi, S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J. Biol. Chem. 2009, 284, 20364–20374. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Malo, D.; Hekimi, S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1alpha in long-lived Mclk1+/− mouse mutants. J. Immunol. 2010, 184, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Lapointe, J.; Hekimi, S. Lifelong protection from global cerebral ischemia and reperfusion in long-lived Mclk1(+/) (−) mutants. Exp. Neurol. 2010, 223, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Zhang, F.; Chen, G.; Han, C.; Liu, J.; Ren, Z.; Zhu, Y.; Waddington, J.L.; Zheng, L.T.; Zhen, X. Clk1 deficiency promotes neuroinflammation and subsequent dopaminergic cell death through regulation of microglial metabolic reprogramming. Brain Behav. Immun. 2017, 60, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. Determination of Life-Span in Caenorhabditis elegans by Four Clock Genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Gutiérrez, A.; Barriocanal-Casado, E.; Díaz-Casado, M.E.; González-García, P.; Chiozzi, R.Z.; Acuña-Castroviejo, D.; López, L.C. β-RA Targets mitochondrial metabolism and adipogenesis, leading to therapeutic benefits against CoQ deficiency and age-related overweight. Biomedicines 2021, 9, 1457. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Hidalgo-Gutiérrez, A.; Raimundo, N.; González-García, P.; Acuña-Castroviejo, D.; Escames, G.; López, L.C. Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine 2019, 42, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Düsterhöft, S.; Künzel, U.; Freeman, M. Rhomboid proteases in human disease: Mechanisms and future prospects. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 2200–2209. [Google Scholar] [CrossRef]
- Herebian, D.; Seibt, A.; Smits, S.H.J.; Rodenburg, R.J.; Mayatepek, E.; Distelmaier, F. 4-Hydroxybenzoic acid restores CoQ10 biosynthesis in human COQ2 deficiency. Ann. Clin. Transl. Neurol. 2017, 4, 902–908. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).