
Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19?

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. SARS-CoV-2 Infection and Multiorgan Damage
3. Ferroptosis Signature in SARS-CoV-2 Infection and Molecular Mechanisms of Ferroptosis
4. Proposed Mechanisms of SARS-CoV-2 Infection and Ferroptosis Induction
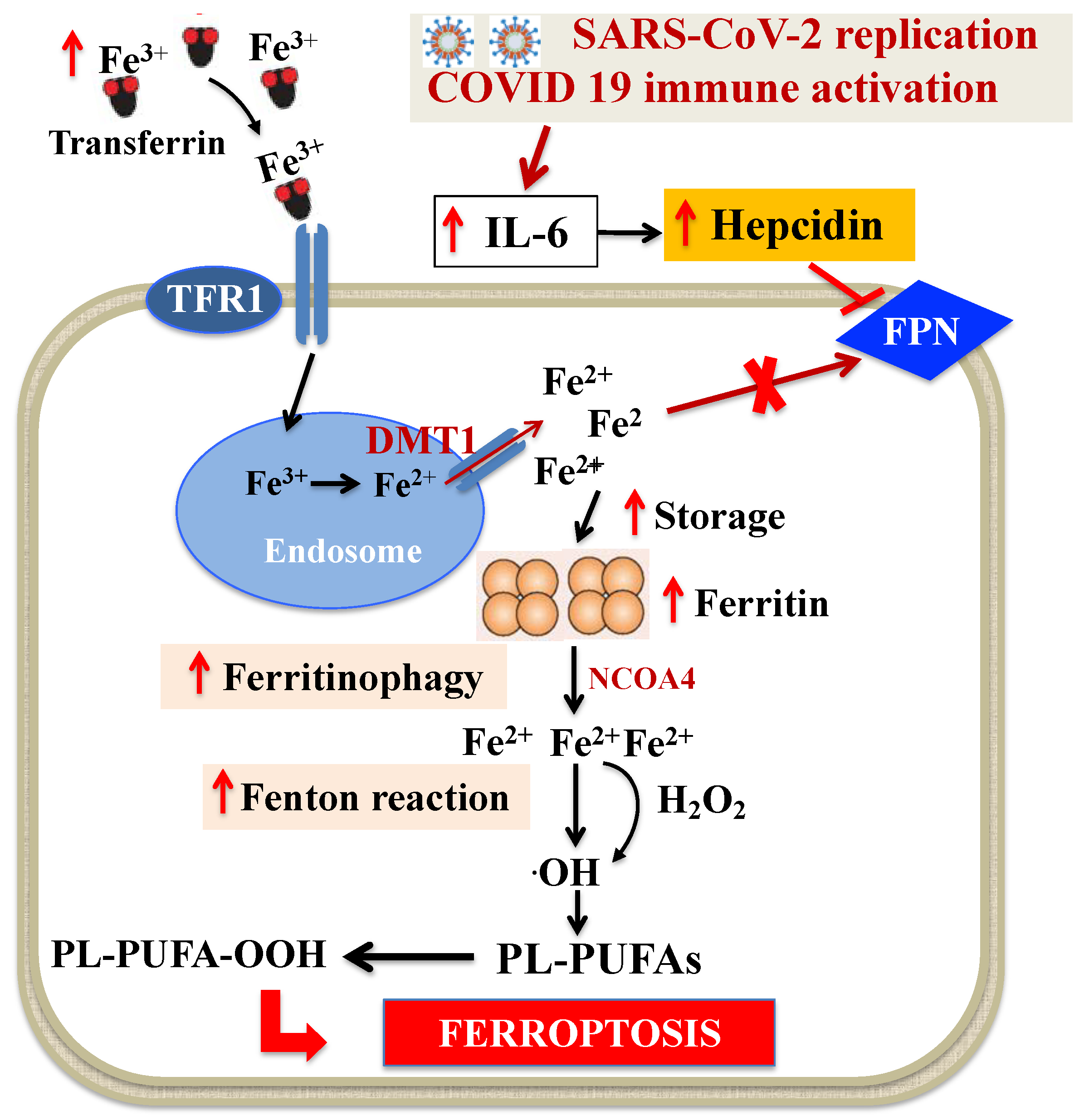
4.1. Dysregulation of Iron Metabolism in SARS-CoV-2 Infection
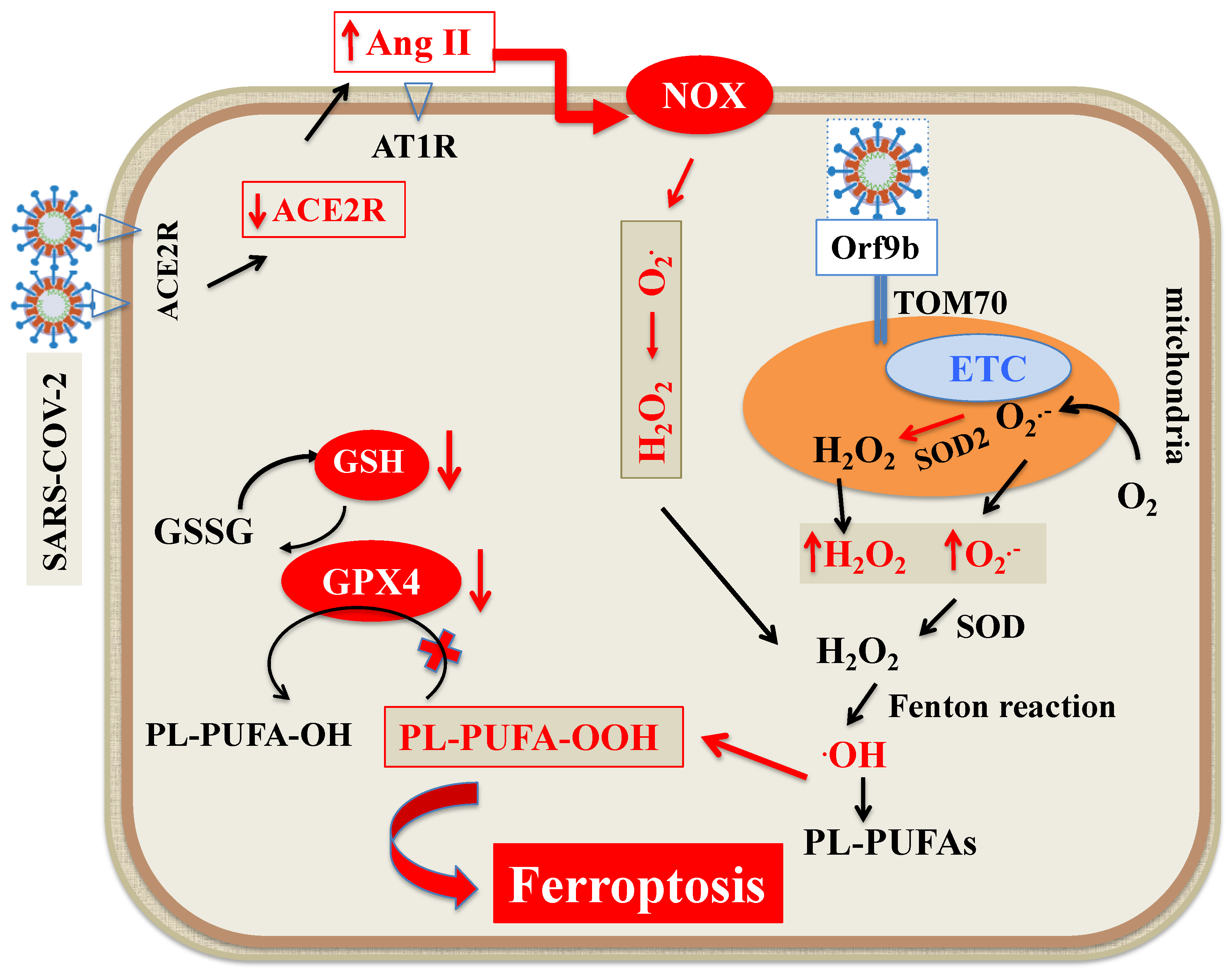
4.2. GSH-GPX4 Axis in SARS-CoV-2 Infection
4.3. ROS Generation during SARS-CoV-2 Infection
5. Potential Therapeutic Interventions
5.1. Compounds That Strengthen the GPX4-GSH-Cysteine Axis
5.2. Compounds That Induce RTA Activity and Inhibit LOXs and ACSL4 Activity
5.3. Compounds That Induce Cell Iron Depletion
5.4. Compounds That Operate through other Potential Targets
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | acyls arachidonoyl |
| AdA | adrenoyl moieties |
| ACE2R | angiotensin converting enzyme 2 receptor |
| ACLF | acute-on-chronic liver failure |
| ACSL4 | acyl-coenzyme A synthetase long-chain family member 4 |
| AKI | acute kidney injury |
| Ang II | angiotensin II |
| α-TOH | α-tocopherol |
| AT1R | angiotensin 1 receptor |
| BH4 | tetrahydrobiopterin |
| CKD | chronic kidney disease |
| CoQ10 | coenzyme Q10 |
| CLD | chronic liver disease |
| COVID-19 | coronavirus disease 2019 |
| CV | cardiovascular |
| DFO | deferoxamine |
| DHFR | dihydrofolate reductase |
| DMT1 | divalent metal transporter 1 |
| ETC | electron transport chain |
| Fer-1 | ferrostatin-1 |
| FSP1 | ferroptosis suppressor protein |
| FPN | ferroportin |
| GCH1 | GTP cyclohydrolase 1 |
| GPX | glutathione peroxidase |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| HNE | 4-hydroxynonenal |
| HO | heme-oxygenase |
| H2O2 | hydrogen peroxide |
| Keap-1 | Kelch-like enoyl-CoA hydratase-associated protein1 |
| ICU | intensive care unit |
| IL-6 | interleukin-6 |
| IFN | interferon |
| Lip-1 | liproxstatin-1 |
| LOOH | peroxides |
| LOXs | lipoxygenases |
| NAC | N-acetylcysteine |
| NCOA4 | nuclear receptor coactivator 4 |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| NRF2 | nuclear factor erythroid 2 p45-related factor2 |
| O2●− | superoxide |
| ●OH | hydroxyl radical |
| Orf9B | open reading frame-9b |
| PEs | phosphatidylethanolamines |
| PPAR γ | peroxisome proliferator-activated receptor γ |
| PUFAs | polyunsaturated fatty acids |
| PL-PUFAs | phospholipid polyunsaturated fatty acids |
| PUFA-OH | phospholipid polyunsaturated fatty acid alcohols |
| PL-PUFA-OOH | phospholipid polyunsaturated fatty acid peroxides |
| ROS | reactive oxygen species |
| ROSI | rosiglitazone |
| RRT | renal replacing therapy |
| RSL3 | RAS-selective lethal 3 |
| RTA | radical trapping antioxidant |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| Se | selenium |
| SOD | superoxide dismutase |
| TOM70 | translocase of outer membrane 70 |
| TF | transferrin |
| TFR1 | transferrin receptor 1 |
| TZDs | thiazolidinediones |
References
- WHO. Coronavirus Disease (COVID-19) Pandemic. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 15 September 2021).
- CDC. 2019 Novel Coronavirus, Wuhan, China. CDC. Available online: https://www.cdc.gov/coronavirus/2019-ncov/about/index.html (accessed on 15 September 2021).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Mahase, E. COVID-19: WHO declares pandemic because of “alarming levels” of spread, severity, and inaction. BMJ 2020, 368, m1036. [Google Scholar] [CrossRef] [Green Version]
- Do Nascimento, I.J.B.; Cacic, N.; Abdulazeem, H.M.; Von Groote, T.C.; Jayarajah, U.; Weerasekara, I.; Esfahani, M.A.; Civile, V.T.; Marusic, A.; Jerončić, A.; et al. Novel Coronavirus Infection (COVID-19) in Humans: A Scoping Review and Meta-Analysis. J. Clin. Med. 2020, 9, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, O.P.; Bhandari, P.; Raut, A.; Kacimi, S.E.O.; Huy, N.T. Coronavirus Disease (COVID-19): Comprehensive Review of Clinical Presentation. Front. Public Heal. 2021, 8, 582932. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 Expression in Human Heart Indicates New Potential Mechanism of Heart Injury Among Patients Infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Chieffo, A.; Stefanini, G.G.; Price, S.; Barbato, E.; Tarantini, G.; Karam, N.; Moreno, R.; Buchanan, G.L.; Gilard, M.; Halvorsen, S.; et al. EAPCI Position Statement on Invasive Management of Acute Coronary Syndromes during the COVID-19 pandemic. Eur. Hear. J. 2020, 41, 1839–1851. [Google Scholar] [CrossRef]
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T., Jr. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef]
- Dalen, H.; Holte, E.; Guldal, A.U.; Hegvik, J.-A.; Stensaeth, K.H.; Braaten, A.T.; Mjølstad, O.C.; Rossvoll, O.; Wiseth, R. Acute perimyocarditis with cardiac tamponade in COVID-19 infection without respiratory disease. BMJ Case Rep. 2020, 13, e236218. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Liu, J.; Bao, J.; Li, R.; Xu, Y.; Guo, W.; Hu, Y.; Gao, Z. Acute Myocardical Injuryat Hospital Admission Is Associated with All-Cause Mortality in COVID-19. J. Am. Coll. Cardiol. 2020, 76, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Korompoki, E.; Fotiou, D.; Ntanasis-Stathopoulos, I.; Psaltopoulou, T.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Organ-specific manifestations of COVID-19 infection. Clin. Exp. Med. 2020, 20, 493–506. [Google Scholar] [CrossRef]
- Amann, K.; Boor, P.; Wiech, T.; Singh, J.; Vonbrunn, E.; Knöll, A.; Hermann, M.; Büttner-Herold, M.; Daniel, C.; Hartmann, A. COVID-19 effects on the kidney. Der Pathol. 2021, 1, 1–5. [Google Scholar] [CrossRef]
- Rubin, S.; Orieux, A.; Prevel, R.; Garric, A.; Bats, M.-L.; Dabernat, S.; Camou, F.; Guisset, O.; Issa, N.; Mourissoux, G.; et al. Characterization of acute kidney injury in critically ill patients with severe coronavirus disease 2019. Clin. Kidney J. 2020, 13, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Northwell COVID-19 Research Consortium. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized with COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Han, B.; Wang, J. COVID-19: Gastrointestinal Manifestations and Potential Fecal–Oral Transmission. Gastroenterology 2020, 158, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, L.; Wang, F.-S. Liver injury in COVID-19: Management and challenges. Lancet Gastroenterol. Hepatol. 2020, 5, 428–430. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 1–7. [Google Scholar] [CrossRef]
- Zhao, B.; Ni, C.; Gao, R.; Wang, Y.; Yang, L.; Wei, J.; Lv, T.; Liang, J.; Zhang, Q.; Xu, W.; et al. Recapitulation of SARS-CoV-2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell. 2020, 11, 771–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, N.C.; Kim, A.; Vitkovski, T.; Xia, J.; Ramirez, G.; Bernstein, D.; Crawford, J.M. Post–COVID-19 Cholangiopathy: A Novel Entity. Am. J. Gastroenterol. 2021, 116, 1077–1082. [Google Scholar] [CrossRef]
- Bangash, M.N.; Patel, J.; Parekh, D. COVID-19 and the liver: Little cause for concern. Lancet Gastroenterol. Hepatol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.Y.; Chen, L.; Wang, M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [Green Version]
- Boettler, T.; Newsome, P.; Mondelli, M.U.; Maticic, M.; Cordero, E.; Cornberg, M.; Berg, T. Care of patients with liver disease during the COVID-19 pandemic: EASL-ESCMID position paper. JHEP Rep. 2020, 2, 100113. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Roth, N.C.; Kvasnovsky, C.; Hirsch, J.S.; Trindade, A.J.; Molmenti, E.; Barish, M.; Hirschwerk, D.; Da, B.L.; Bernstein, D. Risk factors and outcomes for acute-on-chronic liver failure in COVID-19: A large multi-center observational cohort study. Hepatol. Int. 2021, 7, 1–14. [Google Scholar] [CrossRef]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Li, S.; Fang, M.; Hu, M.; Bian, Y.; Ling, J.; Yu, S.; Jing, L.; Li, D.; Huang, J. Acute Physiology and Chronic Health Evaluation II Score as a Predictor of Hospital Mortality in Patients of Coronavirus Disease 2019. Crit. Care Med. 2020, 48, e657–e665. [Google Scholar] [CrossRef]
- Jacobs, W.; Lammens, M.; Kerckhofs, A.; Voets, E.; Van San, E.; Van Coillie, S.; Peleman, C.; Mergeay, M.; Sirimsi, S.; Matheeussen, V.; et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): Autopsy reveals a ferroptosis signature. ESC Hear. Fail. 2020, 7, 3772–3781. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Neely, G.; Yaghubian-Malhami, R.; Perkmann, T.; Van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Edeas, M.; Saleh, J.; Peyssonnaux, C. Iron: Innocent bystander or vicious culprit in COVID-19 pathogenesis? Int. J. Infect. Dis. 2020, 97, 303–305. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, D.M.; Anderson, G. The regulation of iron transport. BioFactors 2013, 40, 206–214. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- Ganz, T. Cellular iron: Ferroportin is the only way out. Cell Metab. 2005, 1, 155–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomme, P.T.; McCann, K.B.; Bertolini, J. Transferrin: Structure, function and potential therapeutic actions. Drug Discov. Today 2005, 10, 267–273. [Google Scholar] [CrossRef]
- Wang, Z.; Ding, Y.; Wang, X.; Lu, S.; Wang, C.; He, C.; Wang, L.; Piao, M.; Chi, G.; Luo, Y.; et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018, 428, 21–33. [Google Scholar] [CrossRef]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Philpott, C.C. The flux of iron through ferritin in erythrocyte development. Curr. Opin. Hematol. 2018, 25, 183–188. [Google Scholar] [CrossRef]
- Codina, N.S.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.; Miyamoto, M.; Sastre, A.; Schnaar, R.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Gill, I.; Valivety, R. Polyunsaturated fatty acids, part 1: Occurrence, biological activities and applications. Trends Biotechnol. 1997, 15, 401–409. [Google Scholar] [CrossRef]
- Porter, N.A.; Wolf, R.A.; Yarbro, E.M.; Weenen, H. The autoxidation of arachidonic acid: Formation of the proposed SRS-A intermediate. Biochem. Biophys. Res. Commun. 1979, 89, 1058–1064. [Google Scholar] [CrossRef]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Bellmann-Weiler, R.; Lanser, L.; Barket, R.; Rangger, L.; Schapfl, A.; Schaber, M.; Fritsche, G.; Wöll, E.; Weiss, G. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J. Clin. Med. 2020, 9, 2429. [Google Scholar] [CrossRef]
- Zhao, K.; Huang, J.; Dai, D.; Feng, Y.; Liu, L.; Nie, S. Serum iron level as a potential predictor of coronavirus disease 2019 severity and mortality: A retrospective study. Open Forum Infect. Dis. 2020, 7, 250. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Taneri, P.E.; Gómez-Ochoa, S.A.; Llanaj, E.; Raguindin, P.F.; Rojas, L.Z.; Roa-Díaz, Z.M.; Salvador, D., Jr.; Groothof, D.; Minder, B.; Kopp-Heim, D.; et al. Anemia and iron metabolism in COVID-19: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 763–773. [Google Scholar] [CrossRef]
- Hippchen, T.; Altamura, S.; Muckenthaler, M.U.; Merle, U. Hypoferremia is Associated with Increased Hospitalization and Oxygen Demand in COVID-19 Patients. HemaSphere 2020, 4, e492. [Google Scholar] [CrossRef]
- Shah, A.; Frost, J.N.; Aaron, L.; Donovan, K.; Drakesmith, H.; McKechnie, S.R.; Stanworth, S.J. Collaborators Systemic hypoferremia and severity of hypoxemic respiratory failure in COVID-19. Crit. Care 2020, 24, 1–4. [Google Scholar] [CrossRef]
- Girelli, D.; Marchi, G.; Busti, F.; Vianello, A. Iron metabolism in infections: Focus on COVID-19. Semin. Hematol. 2021, 58, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobune, M.; Kohgo, Y.; Kato, J.; Miyazaki, E.; Niitsu, Y. Interleukin-6 enhances hepatic transferrin uptake and ferritin expression in rat. Hepatology 1994, 19, 1468–1475. [Google Scholar] [CrossRef]
- Buszko, M.; Nita-Lazar, A.; Park, J.-H.; Schwartzberg, P.L.; Verthelyi, D.; Young, H.A.; Rosenberg, A.S. Lessons learned: New insights on the role of cytokines in COVID-19. Nat. Immunol. 2021, 22, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, Y.; Ji, Y.; He, X.; Xue, D. Increased Serum Levels of Hepcidin and Ferritin Are Associated with Severity of COVID-19. Med. Sci. Monit. 2020, 26, 926178. [Google Scholar] [CrossRef] [PubMed]
- Banchini, F.; Vallisa, D.; Maniscalco, P.; Capelli, P. Iron overload and Hepcidin overexpression could play a key role in COVID infection, and may explain vulnerability in elderly, diabetics, and obese patients. Acta Biomed. 2020, 91, e2020013. [Google Scholar] [CrossRef]
- Nai, A.; Lorè, N.I.; Pagani, A.; De Lorenzo, R.; Di Modica, S.; Saliu, F.; Cirillo, D.M.; Rovere-Querini, P.; Manfredi, A.A.; Silvestri, L. Hepcidin levels predict COVID-19 severity and mortality in a cohort of hospitalized Italian patients. Am. J. Hematol. 2021, 96, 32. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nat. Cell Biol. 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.W. The oxidative stress-induced niacin sink (OSINS) model for HIV pathogenesis. Toxicology 2010, 278, 124–130. [Google Scholar] [CrossRef]
- Polonikov, A. Endogenous Deficiency of Glutathione as the Most Likely Cause of Serious Manifestations and Death in COVID-19 Patients. ACS Infect. Dis. 2020, 6, 1558–1562. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, R.I.; Freeman, P.R.; Bruzzese, J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: A report of 2 cases. Respir. Med. Case Rep. 2020, 30, 101063. [Google Scholar] [CrossRef]
- Guloyan, V.; Oganesian, B.; Baghdasaryan, N.; Yeh, C.; Singh, M.; Guilford, F.; Ting, Y.-S.; Venketaraman, V. Glutathione Supplementation as an Adjunctive Therapy in COVID-19. Antioxidants 2020, 9, 914. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Sun, Y.; Stubbs, D.; He, J.; Li, W.; Wang, F.; Liu, Z.; Ruzicka, J.A.; Taylor, E.W.; et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem. Toxicol. 2021, 153, 112286. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Treberg, J.R. Review: Using isolated mitochondria to investigate mitochondrial hydrogen peroxide metabolism. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2021, 256, 110614. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Anand, S.K.; Tikoo, S.K. Viruses as Modulators of Mitochondrial Functions. Adv. Virol. 2013, 2013, 738794. [Google Scholar] [CrossRef]
- De las Heras, N.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef]
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the Cross fire of SARS-CoV-2 and immunity. Science 2020, 23, 101631. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- McBride, R.; Fielding, B.C. The Role of Severe Acute Respiratory Syndrome (SARS)-Coronavirus Accessory Proteins in Virus Pathogenesis. Viruses 2012, 4, 2902–2923. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Danielle, L. A SARS-CoV-2 protein interaction map reveals targets for drug-repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, H.; Meng, Q.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Leal, N.J.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol. 2018, 28, 369–382.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Zablocki, D.; Sadoshima, J. Angiotensin II and Oxidative Stress in the Failing Heart. Antioxidants Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxidants Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zou, T.; Tuo, Q.; Shuo, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct Target Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Yuan, H.; Pratte, J.; Giardina, C. Ferroptosis and its potential as a therapeutic target. Biochem. Pharmacol. 2021, 186, 114486. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279.e25. [Google Scholar] [CrossRef] [Green Version]
- Guillin, O.M.; Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium, Selenoproteins and Viral Infection. Nutrients 2019, 11, 2101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Taylor, E.W.; Bennett, K.; Saad, R.; Rayman, M.P. Association between regional selenium status and reported outcome of COVID-19 cases in China. Am. J. Clin. Nutr. 2020, 111, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, A.; Heller, R.; Sun, Q.; Seelig, J.; Cherkezov, A.; Seibert, L.; Hackler, J.; Seemann, P.; Diegmann, J.; Pilz, M.; et al. Selenium Deficiency Is Associated with Mortality Risk from COVID-19. Nutrients 2020, 12, 2098. [Google Scholar] [CrossRef]
- Im, J.H.; Je, Y.S.; Baek, J.; Chung, M.-H.; Kwon, H.Y.; Lee, J.-S. Nutritional status of patients with COVID-19. Int. J. Infect. Dis. 2020, 100, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Fakhrolmobasheri, M.; Nasr-Esfahany, Z.; Khanahmad, H.; Zeinalian, M. Selenium supplementation can relieve the clinical complications of COVID-19 and other similar viral infections. Int. J. Vitam. Nutr. Res. 2021, 91, 197–199. [Google Scholar] [CrossRef]
- Kieliszek, M.; Lipinski, B. Selenium supplementation in the prevention of coronavirus infections (COVID-19). Med. Hypotheses 2020, 143, 109878. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Bermano, G.; Méplan, C.; Mercer, D.K.; Hesketh, J.E. Selenium and viral infection: Are there lessons for COVID-19? Br. J. Nutr. 2021, 125, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Puyo, C.A. N-Acetylcysteine to Combat COVID-19: An Evidence Review. Ther. Clin. Risk Manag. 2020, 16, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Codenotti, S.; Poli, M.; Asperti, M.; Zizioli, D.; Marampon, F.; Fanzani, A. Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1717–1730. [Google Scholar] [CrossRef]
- Han, F.; Li, S.; Yang, Y.; Bai, Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered 2021, 12, 5279–5288. [Google Scholar] [CrossRef]
- Gupta, G.; Gliga, A.; Hedberg, J.; Serra, A.; Greco, D.; Wallinder, I.O.; Fadeel, B. Cobalt nanoparticles trigger ferroptosis-like cell death (oxytosis) in neuronal cells: Potential implications for neurodegenerative disease. FASEB J. 2020, 34, 5262–5281. [Google Scholar] [CrossRef] [Green Version]
- Fratta Pasini, A.M.; Stranieri, C.; Cominacini, L.; Mozzini, C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-CoV-2 Complications. Antioxidants 2021, 10, 272. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef]
- Ingold, K.U.; Pratt, D.A. Advances in Radical-Trapping Antioxidant Chemistry in the 21st Century: A Kinetics and Mechanisms Perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.; Tyurin, V.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Skouta, R.; Himmerkus, N.; Mulay, S.R.; Dewitz, C.; De Zen, F.; Prokai, A.; Zuchtriegel, G.; Krombach, F.; Welz, P.S.; et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16836–16841. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Banthiya, S.; Van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 308–330. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.; Shchepinov, M.S.; Derek, A.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhuang, Y.; Liu, M.; Che, J.; Dong, X. Inhibiting ferroptosis: A novel approach for stroke therapeutics. Drug Discov. Today 2021, 26, 916–930. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.E.; Tyurina, Y.; Zhao, J.; Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.; Anthonymuthu, T.S.; Kapralov, O.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lewin, T.M.; Coleman, R.A. Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J. Biol. Chem. 2001, 276, 24667–24673. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, X.; Cheng, Y.; Yang, M.; Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. 2020, 34, 16262–16275. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, S.; Nekhai, S.; Liu, S. Depriving Iron Supply to the Virus Represents a Promising Adjuvant Therapeutic Against Viral Survival. Curr. Clin. Microbiol. Rep. 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Perricone, C.; Bartoloni, E.; Bursi, R.; Cafaro, G.; Guidelli, G.M.; Shoenfeld, Y.; Gerli, R. COVID-19 as part of the hyperferritinemic syndromes: The role of iron depletion therapy. Immunol. Res. 2020, 68, 213–224. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Temraz, S.; Santini, V.; Musallam, K.; Taher, A. Iron overload and chelation therapy in myelodysplastic syndromes. Crit. Rev. Oncol. 2014, 91, 64–73. [Google Scholar] [CrossRef]
- Imoto, S.; Kono, M.; Suzuki, T.; Shibuya, Y.; Sawamura, T.; Mizokoshi, Y.; Sawada, H.; Ohbuchi, A.; Saigo, K. Haemin-induced cell death in human monocytic cells is consistent with ferroptosis. Transfus. Apher. Sci. 2018, 57, 524–531. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Laloux, C.; Moreau, C.; Bordet, R.; Devos, D.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’sdisease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Song, X.; Sun, X.; Huang, J.; Zhong, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016, 473, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-C.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.N.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Central Sci. 2019, 6, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soula, M.; Weber, R.A.; Zilka, O.; Alwaseem, H.; La, K.; Yen, F.; Molina, H.; Garcia-Bermudez, J.; Pratt, D.A.; Birsoy, K. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 2020, 16, 1351–1360. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [Green Version]
- Protein-Protein Interaction Networks Functional Enrichment Analysis. Available online: https://string-db.org/cgi/input.pl (accessed on 21 September 2021).
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Cadanet, J.B.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. Identification of SARS-CoV-2-mediated suppression of NRF2 signaling reveals a potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Cuadrado, A.; Pajares, M.; Benito, C.; Jiménez-Villegas, J.; Escoll, M.; Fernández-Ginés, R.; Garcia Yague, A.J.; Lastra, D.; Manda, G.; Ana, I.; et al. Can activation of NRF2 be a strategy against COVID-19? Trends Pharmacol. Sci. 2020, 41, 598–610. [Google Scholar] [CrossRef]
- Zheng, K.; Dong, Y.; Yang, R.; Liang, Y.; Wu, H.; He, Z. Regulation of ferroptosis by bioactive phytochemicals: Implications for medical nutritional therapy. Pharmacol. Res. 2021, 168, 105580. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fratta Pasini, A.M.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants 2021, 10, 1677. https://doi.org/10.3390/antiox10111677
Fratta Pasini AM, Stranieri C, Girelli D, Busti F, Cominacini L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants. 2021; 10(11):1677. https://doi.org/10.3390/antiox10111677
Chicago/Turabian StyleFratta Pasini, Anna Maria, Chiara Stranieri, Domenico Girelli, Fabiana Busti, and Luciano Cominacini. 2021. "Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19?" Antioxidants 10, no. 11: 1677. https://doi.org/10.3390/antiox10111677
APA StyleFratta Pasini, A. M., Stranieri, C., Girelli, D., Busti, F., & Cominacini, L. (2021). Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants, 10(11), 1677. https://doi.org/10.3390/antiox10111677