
FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Chemistry
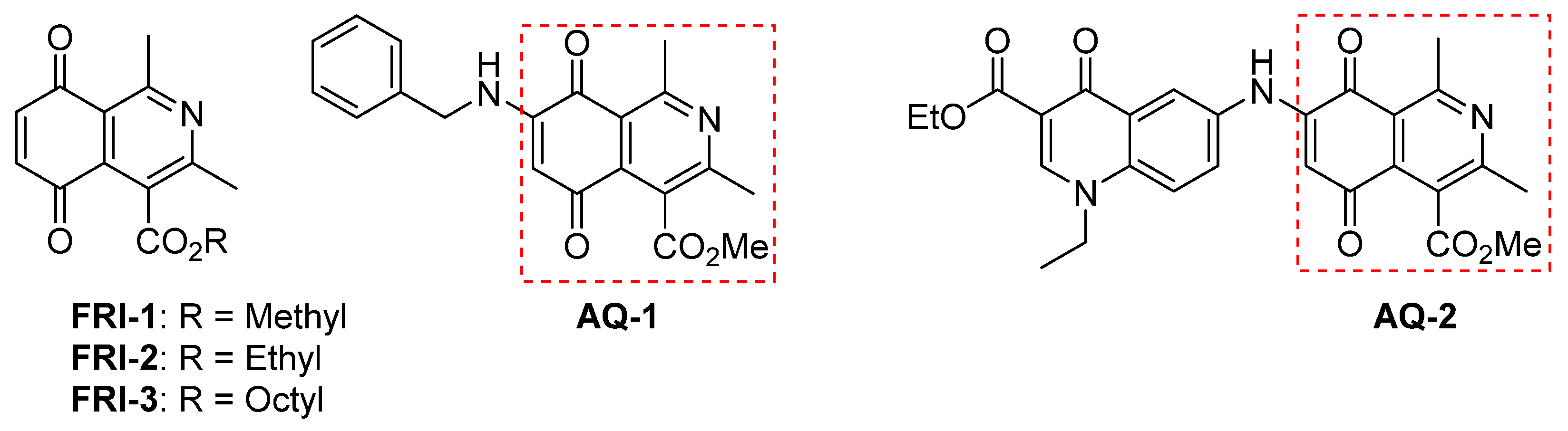
2.2.1. Synthesis of Octyl and Methyl 1,3-Dimethyl-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates (FRI-1 and FRI-3)
2.2.2. Synthesis of Octyl 1,3-Dimethyl-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylate (FRI-3)
2.3. Breast Cell Lines
2.4. Measurement of Mitochondrial Membrane Potential (Δψm)
2.5. Clonogenic Assay
2.6. Cellular Respiration in Real-Time
2.7. Respiration in Isolated Mitochondria
2.8. Mitochondrial ROS (mtROS) Levels
2.9. NAD(P)H and ATP Levels
2.10. Cell Death Assay
2.11. MTT Assay
2.12. Western Blotting
2.13. Statistical Analysis
3. Results
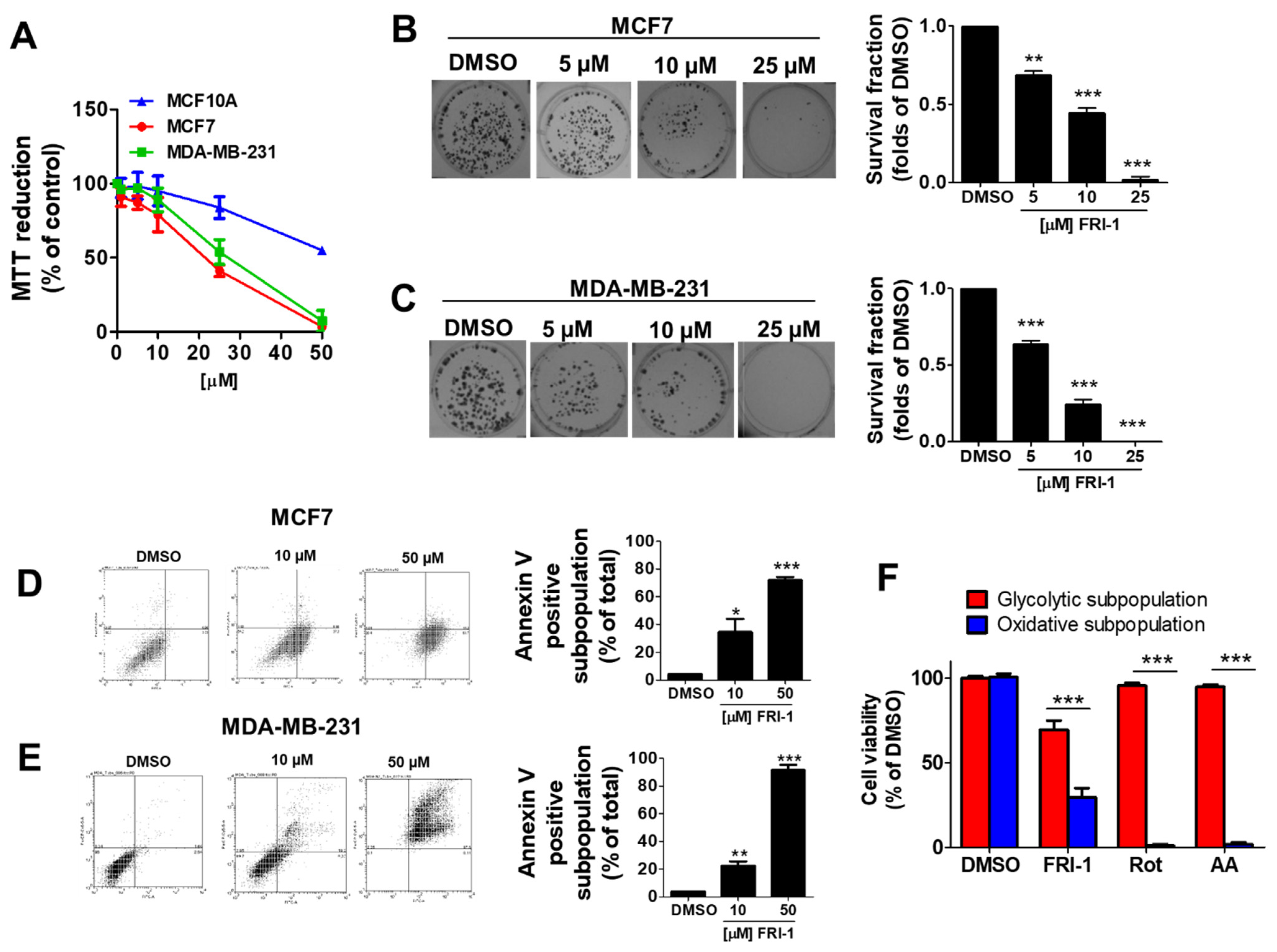
3.1. FRI-1 Inhibits Cell Proliferation, Clonogenic Capacity, and Induces Apoptosis in Breast Cancer Cells
3.2. FRI-1 Inhibits Mitochondrial Respiration and Blocks Metabolic Remodeling under Energetic Stress in Breast Cancer Cells
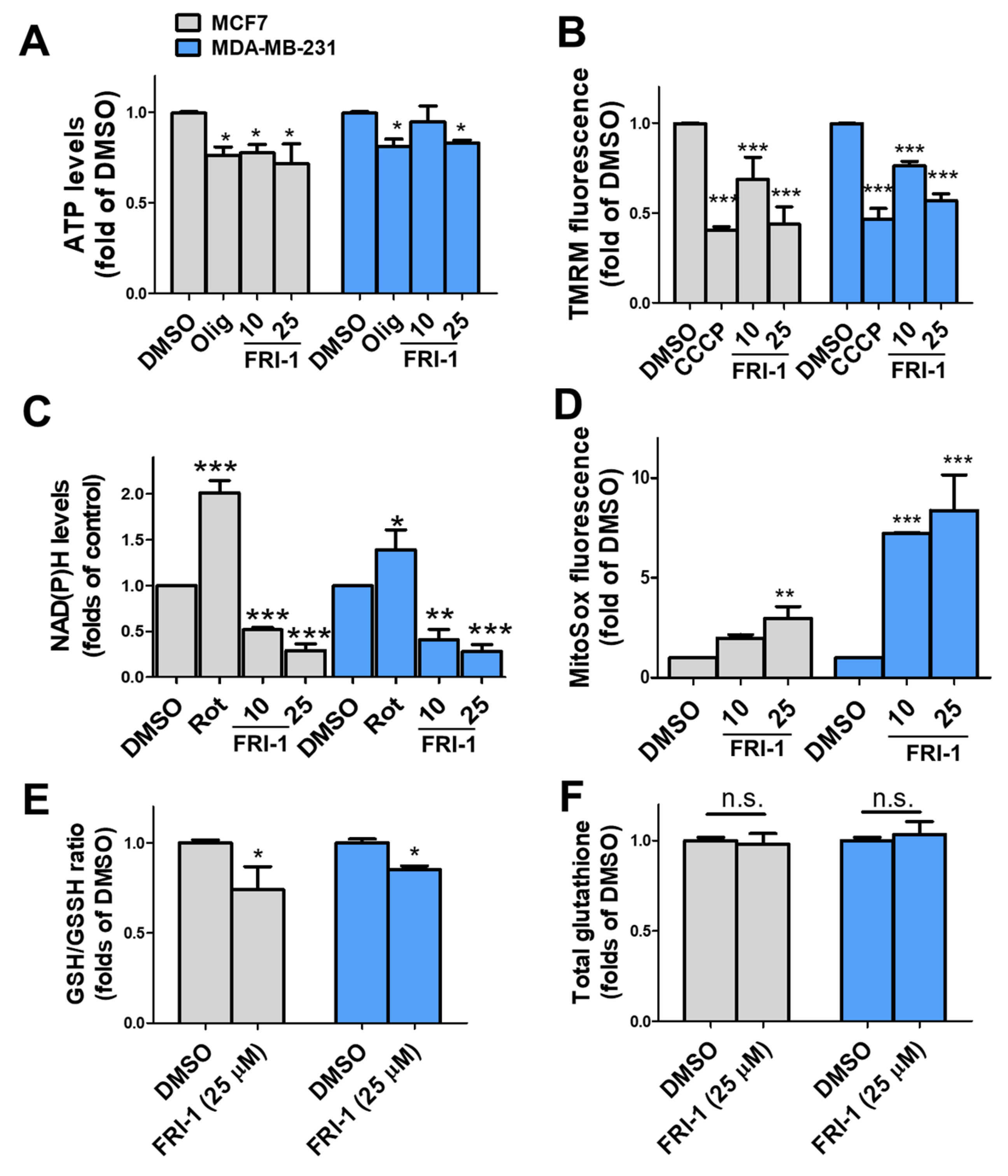
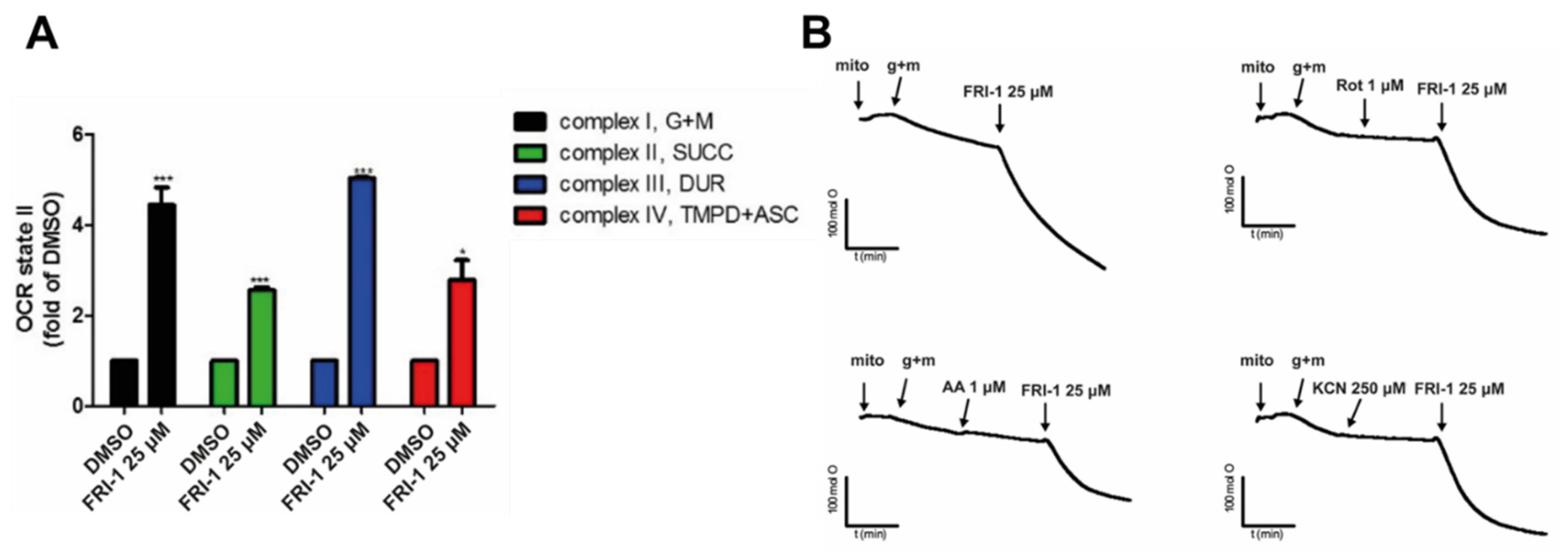
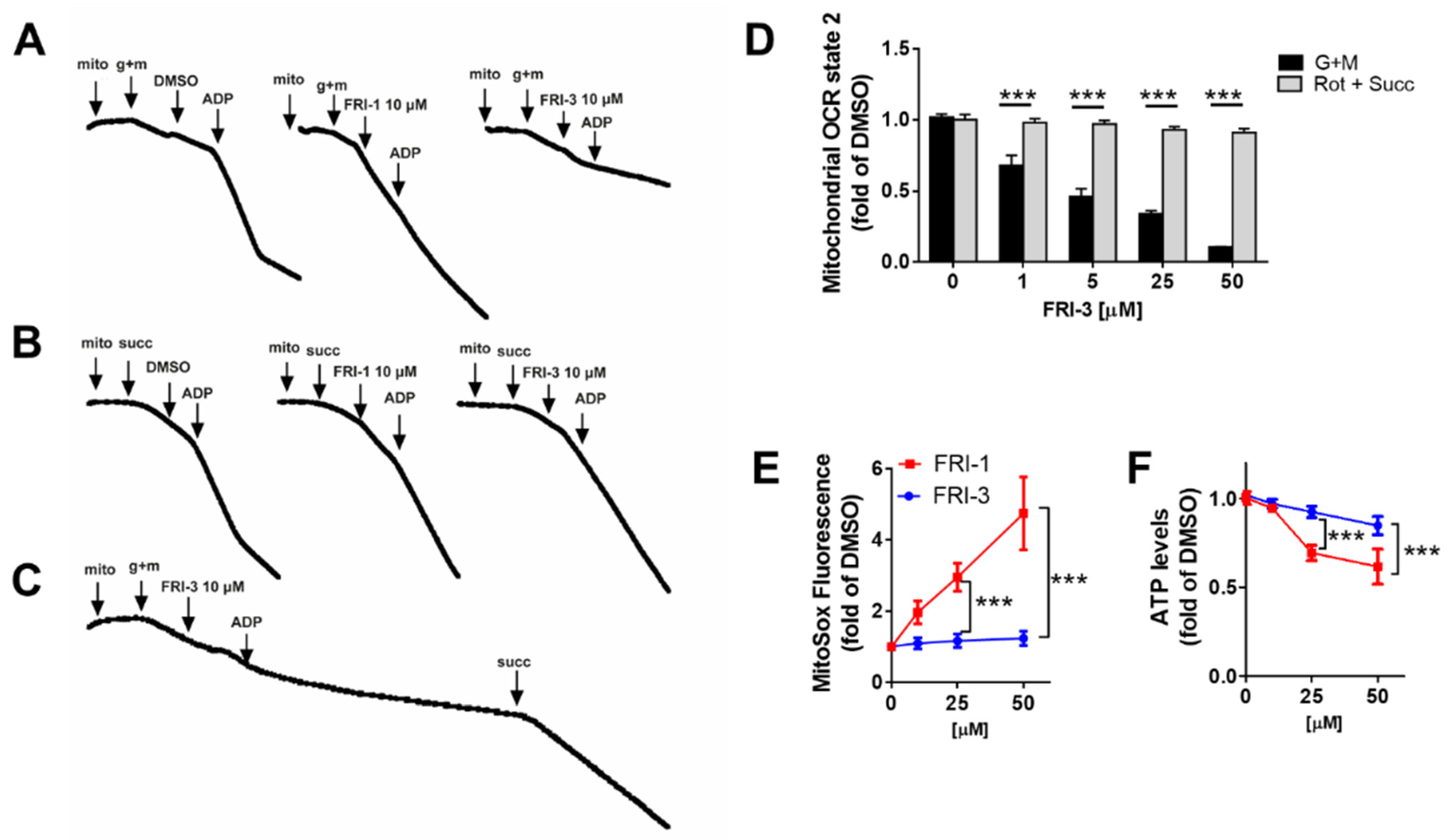
3.3. FRI-1 Produces Inhibition of Mitochondrial Bioenergetics and Redox Stress in Breast Cancer Cells
3.4. FRI-1 Increases Oxygen Consumption Rate in Tumor Mitochondria
3.5. FRI-1 Induces a Pro-Survival AMPK Signaling in BC Cells
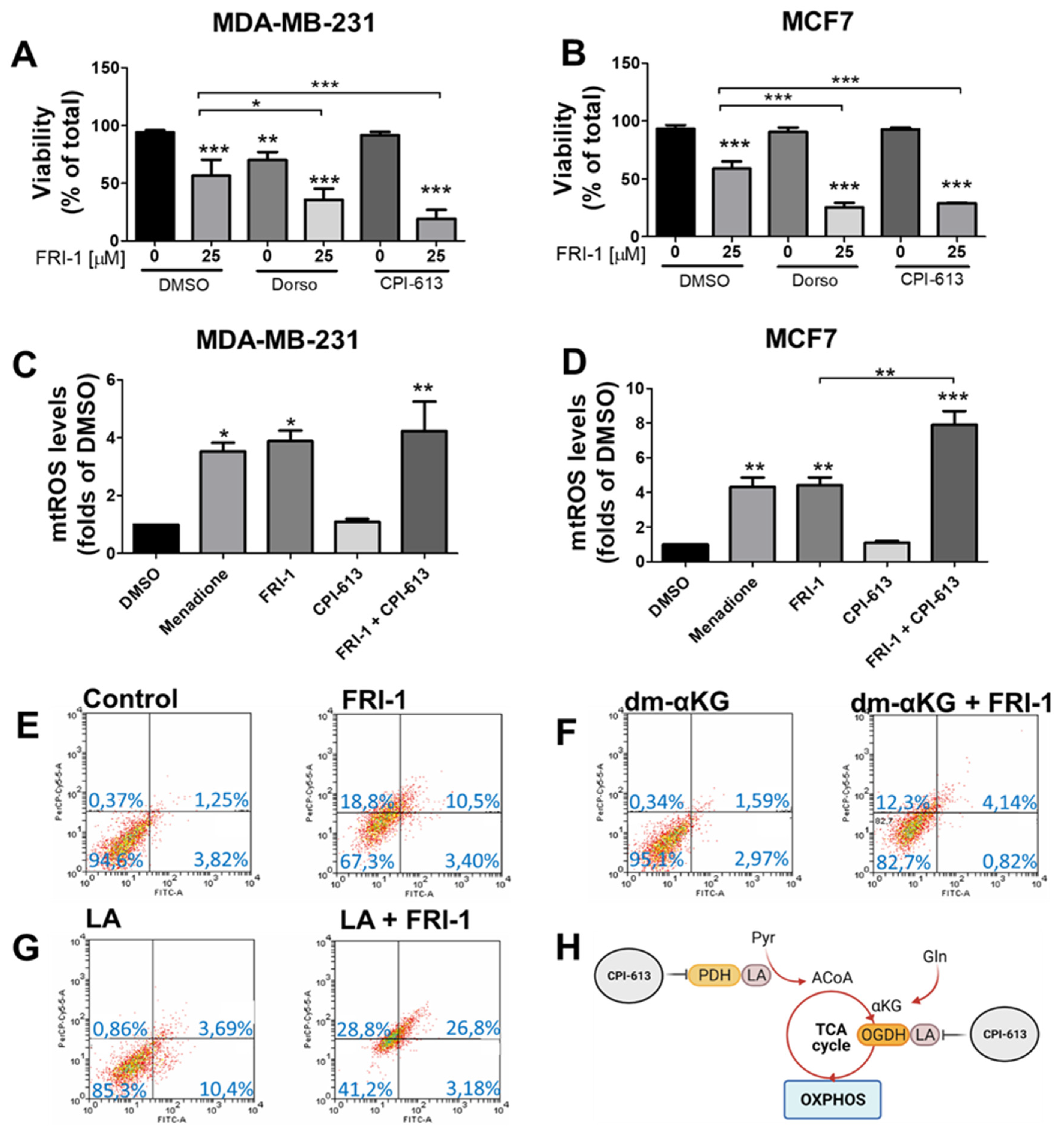
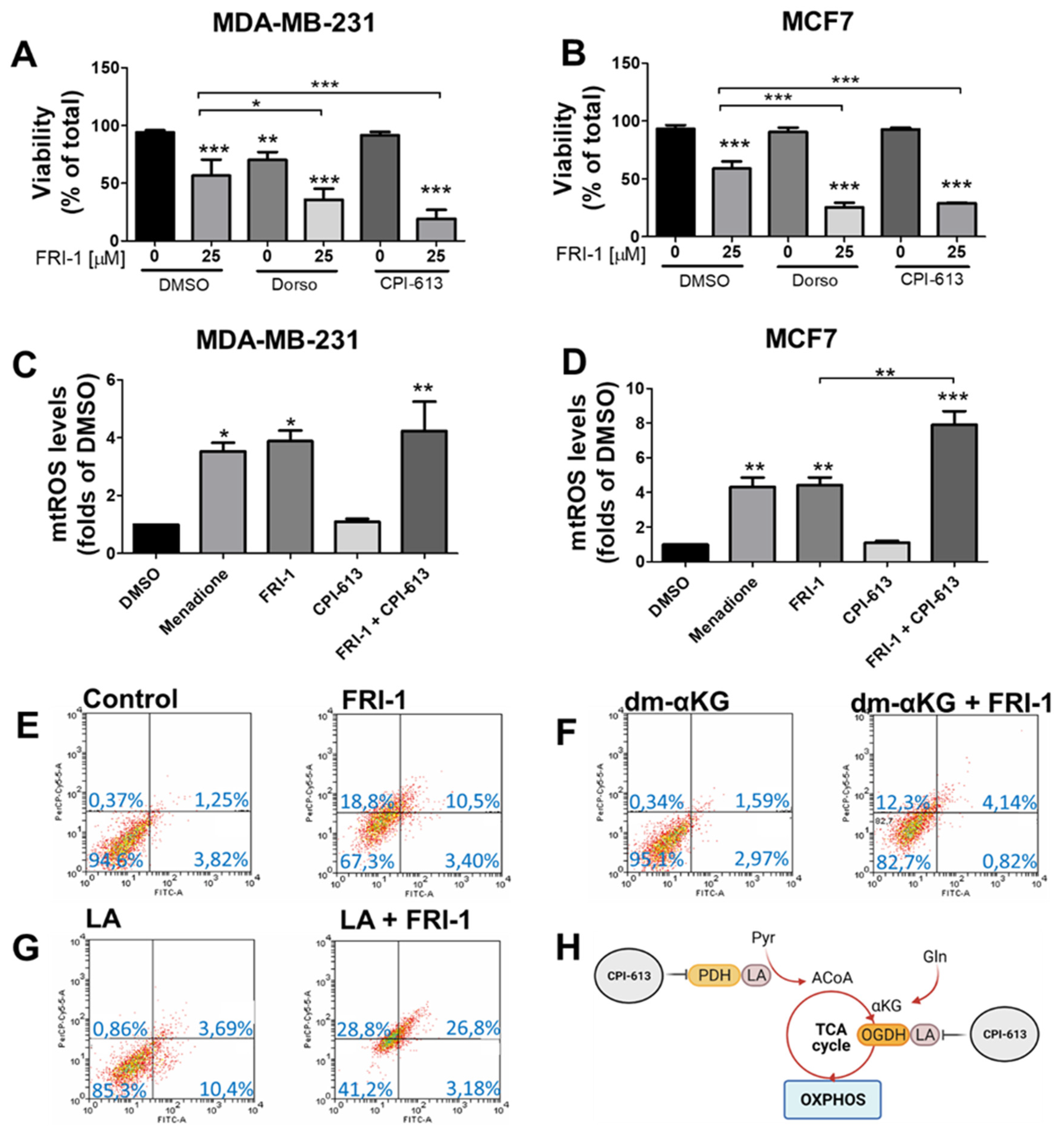
3.6. FRI-1-Induced BC Cell Death Is Prevented by Dimethyl-aKG and Sensitized by Lipoic Acid or CPI-613 Combination
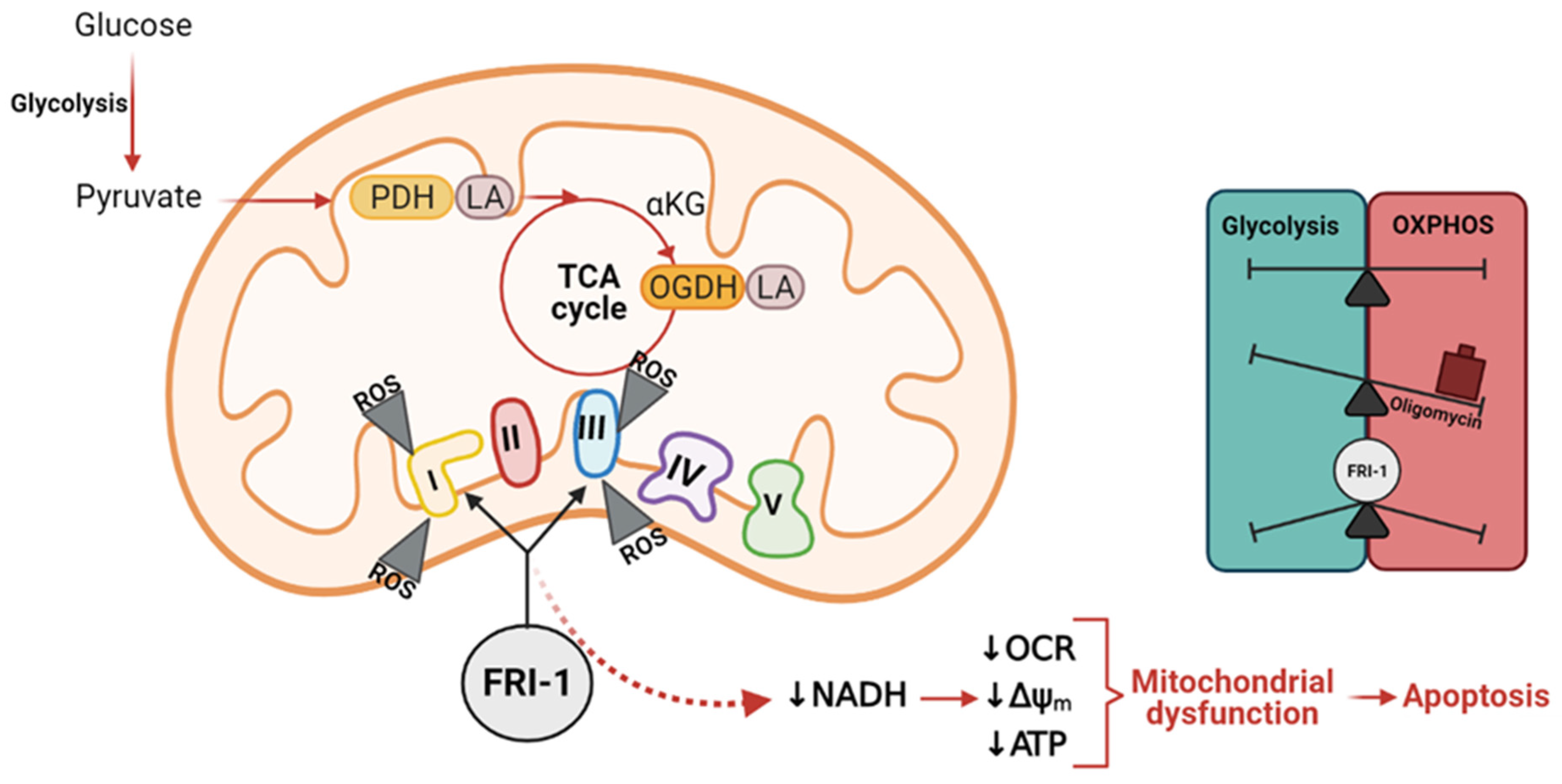
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.; Zhang, G.; Xu, S.; Song, Y. Recent advances of quinones as a privileged structure in drug discovery. Eur. J. Med. Chem. 2021, 223, 113632. [Google Scholar] [CrossRef]
- Allen, G.R.; Weiss, M.J. Behavior of 2-carbomethoxy- and 2-acetyl-1,4-benzoquinone in the Nenitzescu indole syntheses. J. Org. Chem. 1968, 33, 198–200. [Google Scholar] [CrossRef]
- Valderrama, J.A.; González, M.F.; Pessoa-Mahana, D.; Tapia, R.A.; Fillion, H.; Pautet, F.; Rodriguez, J.A.; Theoduloz, C.; Schmeda-Hirschmann, G. Studies on quinones. Part 41: Synthesis and cytotoxicity of isoquinoline-containing polycyclic quinones. Bioorganic Med. Chem. 2006, 14, 5003–5011. [Google Scholar] [CrossRef] [PubMed]
- Novais, J.S.; Campos, V.R.; Silva, A.C.J.A.; de Souza, M.C.B.V.; Ferreira, V.F.; Keller, V.G.L.; Ferreira, M.O.; Dias, F.R.F.; Vitorino, M.I.; Sathler, P.C.; et al. Synthesis and antimicrobial evaluation of promising 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates and their halogenated amino compounds for treating Gram-negative bacterial infections. RSC Adv. 2017, 7, 18311–18320. [Google Scholar] [CrossRef] [Green Version]
- Ryu, C.K.; Song, A.L.; Hong, J.A. Synthesis and antifungal evaluation of 7-arylamino-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylates. Bioorganic Med. Chem. Lett. 2013, 23, 2065–2068. [Google Scholar] [CrossRef] [PubMed]
- Kruschel, R.D.; Buzid, A.; Khandavilli, U.B.R.; Lawrence, S.E.; Glennon, J.D.; McCarthy, F.O. Isoquinolinequinone N-oxides as anticancer agents effective against drug resistant cell lines. Org. Biomol. Chem. 2020, 18, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Mello, A.L.; Sagrillo, F.S.; de Souza, A.G.; Costa, A.R.P.; Campos, V.R.; Cunha, A.C.; Imbroisi Filho, R.; da Costa Santos Boechat, F.; Sola-Penna, M.; de Souza, M.; et al. Selective AMPK activator leads to unfolded protein response downregulation and induces breast cancer cell death and autophagy. Life Sci. 2021, 276, 119470. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Saikia, R.; Joseph, J. AMPK: A key regulator of energy stress and calcium-induced autophagy. J. Mol. Med. Berl 2021. [Google Scholar] [CrossRef]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Hudis, C.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Marmé, F.; Schneeweiss, A. Targeted Therapies in Triple-Negative Breast Cancer. Breast Care 2015, 10, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.; Tabariès, S.; Annis, M.; McGuirk, S.; Northey, J.; Chénard, V.; Sriram, U.; Papadopoli, D.; et al. PGC-1α promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. Roles of mitochondria in the hallmarks of metastasis. Br. J. Cancer 2021, 124, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.; Serganova, I.; Kruchevsky, N.; Leftin, A.; Shestov, A.; Thaler, H.; Sukenick, G.; Locasale, J.; Blasberg, R.; Koutcher, J.; et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial flexibility of breast cancers: A growth advantage and a therapeutic opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.; Payen, V.; Pérez-Escuredo, J.; De Saedeleer, C.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.; O’Connell, J.; Gonzalez Herrera, K.; Wikman, H.; Pantel, K.; Haigis, M.; de Carvalho, F.; Damascena, A.; Domingos Chinen, L.; Rocha, R.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, H.H.; Cao, Y.T.; Zhang, L.L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.; Linehan, W.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.; Wheaton, W.; Jin, E.; Chen, P.; Sullivan, L.; Cheng, T.; Yang, Y.; Linehan, W.; Chandel, N.; DeBerardinis, R. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.T.; Anderson, J.L.; Denton, R.M. Studies on the regulation of the human E1 subunit of the 2-oxoglutarate dehydrogenase complex, including the identification of a novel calcium-binding site. Biochem. J. 2014, 459, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Porpaczy, Z.; Sumegi, B.; Alkonyi, I. Interaction between NAD-dependent isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase complex, and NADH:ubiquinone oxidoreductase. J. Biol. Chem. 1987, 262, 9509–9514. [Google Scholar] [CrossRef]
- Vatrinet, R.; Iommarini, L.; Kurelac, I.; De Luise, M.; Gasparre, G.; Porcelli, A. Targeting respiratory complex I to prevent the Warburg effect. Int. J. Biochem. Cell Biol. 2015, 63, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Retamal, S.; Sandoval-Acuna, C.; Peredo-Silva, L.; Guzman-Rivera, D.; Pavani, M.; Torrealba, N.; Truksa, J.; Castro-Castillo, V.; Catalan, M.; Kemmerling, U.; et al. Complex Mitochondrial Dysfunction Induced by TPP(+)-Gentisic Acid and Mitochondrial Translation Inhibition by Doxycycline Evokes Synergistic Lethality in Breast Cancer Cells. Cells 2020, 9, 407. [Google Scholar] [CrossRef] [Green Version]
- Urra, F.; Córdova-Delgado, M.; Lapier, M.; Orellana-Manzano, A.; Acevedo-Arévalo, L.; Pessoa-Mahana, H.; González-Vivanco, J.; Martínez-Cifuentes, M.; Ramírez-Rodríguez, O.; Millas-Vargas, J.; et al. Small structural changes on a hydroquinone scaffold determine the complex I inhibition or uncoupling of tumoral oxidative phosphorylation. Toxicol. Appl. Pharmacol. 2016, 291, 46–57. [Google Scholar] [CrossRef]
- Urra, F.; Muñoz, F.; Córdova-Delgado, M.; Ramírez, M.; Peña-Ahumada, B.; Rios, M.; Cruz, P.; Ahumada-Castro, U.; Bustos, G.; Silva-Pavez, E.; et al. FR58P1a; a new uncoupler of OXPHOS that inhibits migration in triple-negative breast cancer cells via Sirt1/AMPK/β1-integrin pathway. Sci. Rep. 2018, 8, 13190. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.; Weiss-López, B.; Araya-Maturana, R. Determinants of anti-cancer effect of mitochondrial electron transport chain inhibitors: Bioenergetic profile and metabolic flexibility of cancer cells. Curr. Pharm. Des. 2016, 22, 5998–6008. [Google Scholar] [CrossRef] [PubMed]
- Suárez, M.; de Armas, M.; Ramírez, O.; Alvarez, A.; Martínez-Alvarez, R.; Molero, D.; Seoane, C.; Liz, R.; de Armas, H.N.; Blaton, N.M.; et al. Synthesis and structural study of new highly lipophilic 1,4-dihydropyridines. New J. Chem. 2005, 29, 1567–1576. [Google Scholar] [CrossRef]
- Franken, N.; Rodermond, H.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Urra, F.A.; Martínez-Cifuentes, M.; Pavani, M.; Lapier, M.; Jaña-Prado, F.; Parra, E.; Maya, J.D.; Pessoa-Mahana, H.; Ferreira, J.; Araya-Maturana, R. An ortho-carbonyl substituted hydroquinone derivative is an anticancer agent that acts by inhibiting mitochondrial bioenergetics and by inducing G2/M-phase arrest in mammary adenocarcinoma TA3. Toxicol. Appl. Pharmacol. 2013, 267, 218–227. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.; Wagenaars, J.; Smeitink, J.; Willems, P.; Koopman, W. Acute stimulation of glucose influx upon mitoenergetic dysfunction requires LKB1, AMPK, Sirt2 and mTOR-RAPTOR. J. Cell Sci. 2016, 129, 4411–4423. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Donoso-Bustamante, V.; Borrego, E.A.; Schiaffino-Bustamante, Y.; Gutiérrez, D.A.; Millas-Vargas, J.P.; Fuentes-Retamal, S.; Correa, P.; Carrillo, I.; Aguilera, R.J.; Miranda, D.; et al. An acylhydroquinone derivative produces OXPHOS uncoupling and sensitization to BH3 mimetic ABT-199 (Venetoclax) in human promyelocytic leukemia cells. Bioorg. Chem. 2020, 100, 103935. [Google Scholar] [CrossRef]
- Arivazhagan, P.; Ramanathan, K.; Panneerselvam, C. Effect of DL-alpha-lipoic acid on mitochondrial enzymes in aged rats. Chem. Biol. Interact. 2001, 138, 189–198. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Urra, F.; Muñoz, F.; Lovy, A.; Cárdenas, C. The Mitochondrial Complex(I)ty of Cancer. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Urra, F.A.; Fuentes-Retamal, S.; Palominos, C.; Araya-Maturana, R. Recent advances in molecular mechanisms of anticancer natural products that target mitochondrial bioenergetics. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 71, pp. 1–41. [Google Scholar]
- Hao, W.; Chang, C.; Tsao, C.; Xu, J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J. Biol. Chem. 2010, 285, 12647–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Yuan, Q.; Cao, X.; Zhang, J.; Cao, J.; Zhang, J.; Xia, L. Isovitexin Suppresses Stemness of Lung Cancer Stem-Like Cells through Blockage of MnSOD/CaMKII/AMPK Signaling and Glycolysis Inhibition. Biomed. Res. Int. 2021, 2021, 9972057. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullarky, E.; Cantley, L.C. Diverting glycolysis to combat oxidative stress. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 3–23. [Google Scholar] [CrossRef]
- Hyun, D.H. Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts. Cancers 2020, 12, 1822. [Google Scholar] [CrossRef] [PubMed]
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1α Stability in Aerobic Conditions. Cell Metab. 2016, 24, 740–752. [Google Scholar] [CrossRef] [Green Version]
- Paredes, F.; Sheldon, K.; Lassègue, B.; Williams, H.C.; Faidley, E.A.; Benavides, G.A.; Torres, G.; Sanhueza-Olivares, F.; Yeligar, S.M.; Griendling, K.K.; et al. Poldip2 is an oxygen-sensitive protein that controls PDH and αKGDH lipoylation and activation to support metabolic adaptation in hypoxia and cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 1789–1794. [Google Scholar] [CrossRef] [Green Version]
- Atlante, S.; Visintin, A.; Marini, E.; Savoia, M.; Dianzani, C.; Giorgis, M.; Sürün, D.; Maione, F.; Schnütgen, F.; Farsetti, A.; et al. α-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis. Cell Death Dis. 2018, 9, 756. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Córdova-Delgado, M.; Fuentes-Retamal, S.; Palominos, C.; López-Torres, C.; Guzmán-Rivera, D.; Ramírez-Rodríguez, O.; Araya-Maturana, R.; Urra, F.A. FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells. Antioxidants 2021, 10, 1618. https://doi.org/10.3390/antiox10101618
Córdova-Delgado M, Fuentes-Retamal S, Palominos C, López-Torres C, Guzmán-Rivera D, Ramírez-Rodríguez O, Araya-Maturana R, Urra FA. FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells. Antioxidants. 2021; 10(10):1618. https://doi.org/10.3390/antiox10101618
Chicago/Turabian StyleCórdova-Delgado, Miguel, Sebastián Fuentes-Retamal, Charlotte Palominos, Camila López-Torres, Daniela Guzmán-Rivera, Oney Ramírez-Rodríguez, Ramiro Araya-Maturana, and Félix A. Urra. 2021. "FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells" Antioxidants 10, no. 10: 1618. https://doi.org/10.3390/antiox10101618
APA StyleCórdova-Delgado, M., Fuentes-Retamal, S., Palominos, C., López-Torres, C., Guzmán-Rivera, D., Ramírez-Rodríguez, O., Araya-Maturana, R., & Urra, F. A. (2021). FRI-1 Is an Anti-Cancer Isoquinolinequinone That Inhibits the Mitochondrial Bioenergetics and Blocks Metabolic Shifts by Redox Disruption in Breast Cancer Cells. Antioxidants, 10(10), 1618. https://doi.org/10.3390/antiox10101618