
Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses

 , , and
, , and 
Abstract
1. Introduction
2. Reactive Oxygen Species
3. Sources of Reactive Oxygen Species
4. The Essential Role of ROS in the Maintenance of Haemopoietic Stem Cells (HSCs), and Innate and Adaptive Immunity
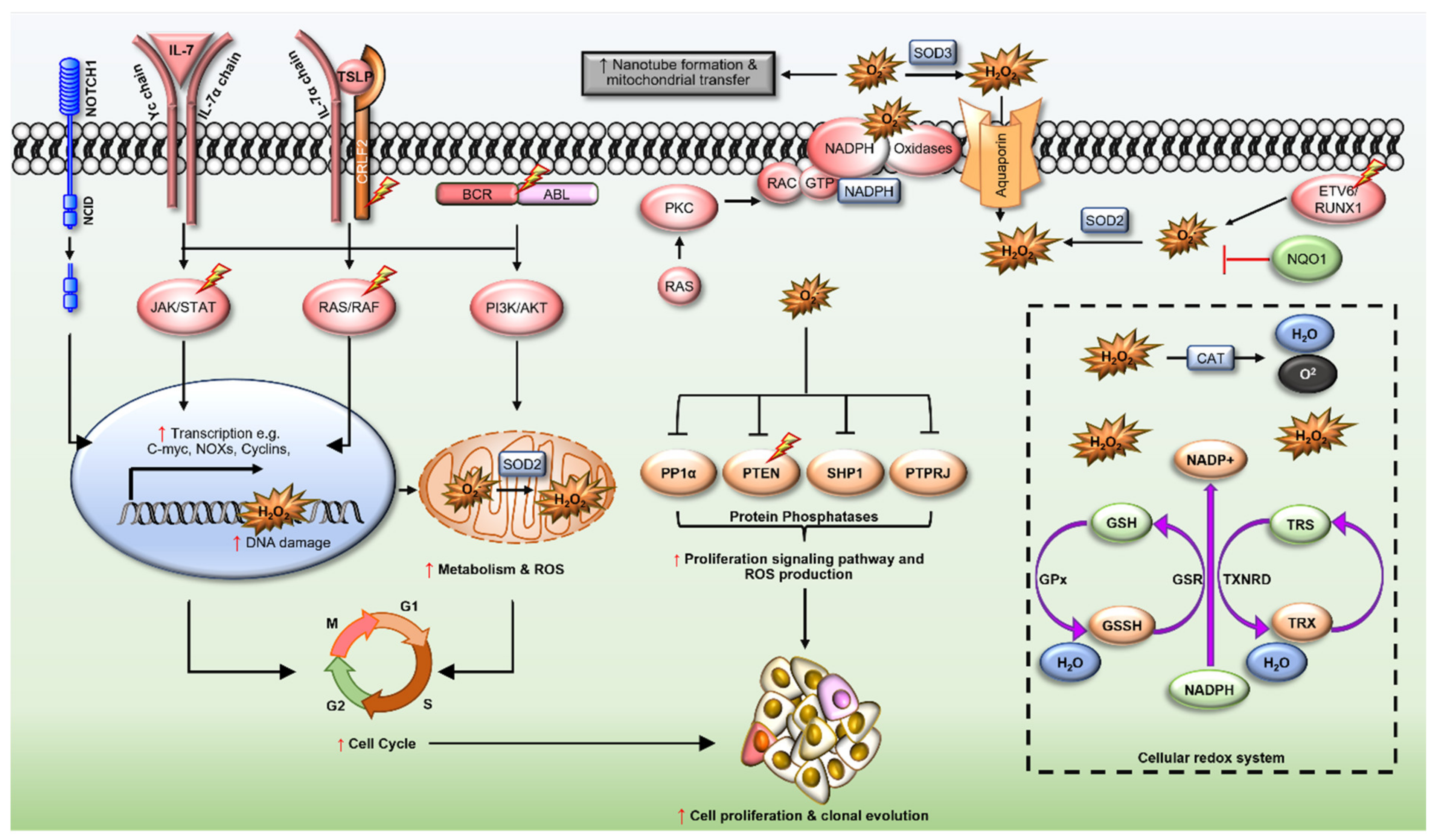
5. Redox Dysregulation in ALL
5.1. ETV6/RUNX1 Fusions
5.2. BCR/ABL Oncogene
5.3. Cytokine Receptor-like Factor 2 (CRLF2)
5.4. Interleukin-7 Receptor α (IL7R)
5.5. Transcription Factors PU.1 (SPI1) and Spi-B (SPIB)
5.6. Neurogenic Locus Notch Homolog Protein 1 (NOTCH1)
5.7. Ras GTPases (N- and KRAS)
5.8. Rho-Family GTPases
5.9. NAD(P)H Quinone Dehydrogenase 1 (NQO1)
6. Redox Homeostasis in ALL
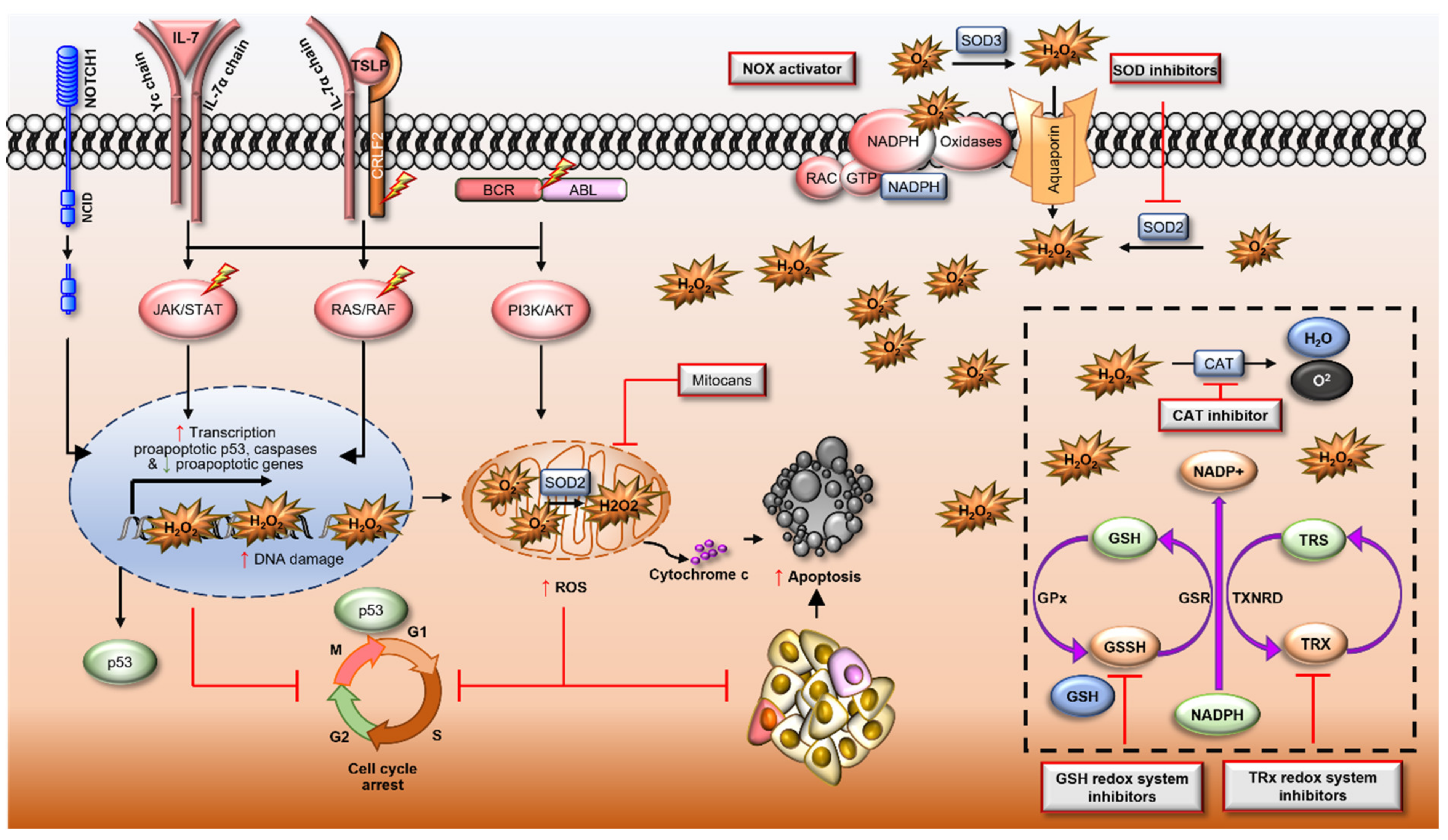
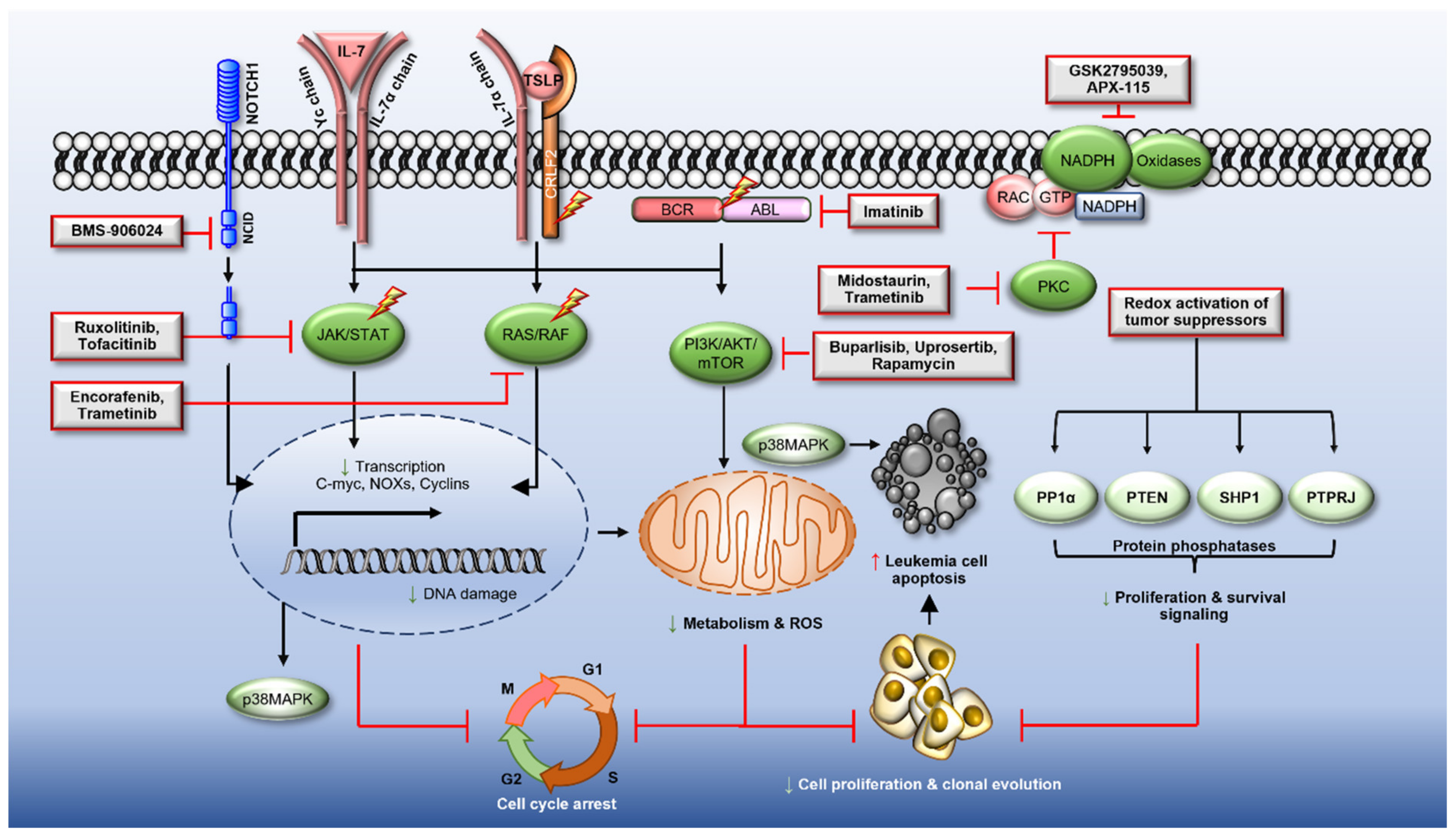
7. The Modulation of ROS to Target ALL
7.1. Pro-Oxidant Therapies

{kind=link}
{kind=link}
{kind=link}
| Drug (Status) | Signalling Pathways | Possible Mechanism of Action | References |
|---|---|---|---|
| Arsenic trioxide (FDA approved drug for AML) | Mitochondrial membrane NOX2 | ↑ ROS by inhibiting reducing GSH expression, and inhibiting GPx, GST and catalase activity, and increasing NOX2 expression and activity | [217,218] |
| Doxorubicin | Mitochondria | ↑ ROS due to topoisomerase-IIβ transcriptome mediated loss of ΔΨm | [219] |
| Tigecycline (FDA approved drug for disease other than cancer other disease) | Mitochondria | ↑ ROS by suppressing the translation of complex I and IV of mitochondrial proteins | [220] |
| NOV-002 (In Clinical trials for Myelodysplastic Syndrome (MDS) and NSLC) | Redox enzymes | ↑ ROS by acting as GSSG mimetic Also stimulates blood cell production | [221] |
| 2-methoxyestradiol (Panzem) (FDA approved drug for multiple cancers including meyloma) | Redox enzymes | ↑ ROS by inhibiting Superoxide dismutases (SODs) | [222] |
| ATN-224 (Completed phase 2 clinical trials for MM and prostate cancer) | Redox enzymes | ↑ ROS by inhibiting copper/zinc SOD activity | [223] |
| Imexon (Completed phase 2 clinical trials for multiple myeloma and lymphoma) | Redox homeostasis | ↑ ROS by reducing cysteine and GSH pool | [224] |
| PX-12 (Completed phase 2 clinical trials to treat solid tumours) | Redox enzymes | ↑ ROS by inhibiting thioredoxin enzymes system | [225,226] |
| Parthenolide and derivatives (Natural compound) (Phase I/II studies for various cancers) | Thiol inhibitors | ↑ ROS by depleting cellular thiol including GSH | [227] |
| BSO (L-buthionine-(S,R)-sulfoximine (BSO) | Redox system | ↑ ROS by depleting GSH | [228] |
| TPEN (zinc chelator) | Redox System | ↑ ROS and induces loss of ΔΨm by unknow mechanism | [229,230] |
| Cannabidiod CP55940 (Natural compound) | Redox system | ↑ ROS loss ΔΨm and Ca2+ overload | [231] |
| Curcumin (Natural compound) | Mitochondria | ↑ ROS loss of ΔΨm by unknow mechanism | [232] |
| Vinblastine | Redox system | ↑ ROS by depleting GSH levels | [233] |
| Quercetin (Natural compound) | Mitochondria | ↑ ROS by depleting GSH levels and inducing loss ΔΨm | [234] |
| Erastin (eradicator of RAS and ST) | Redox system | ↑ ROS by inhibiting and Cys2/glutamate antiporter leading glutathione depletion | [235] |
| Auranofin (FDA approved drug to treat rheumatoid arthritis) | Redox enzyme | ↑ ROS by inhibiting TXNRD | [236] |
| Adenanthin (ADE) (Natural compound) | Redox enzymes | ↑ ROS by inhibiting PRDX1/2 activities | [237] |
| RSL3 (RAS-synthetic-lethality 3) | Redox enzymes | ↑ ROS by inhibiting GPX4 and induce ferroptosis | [238] |
| Matrine (Natural compound) | Mitochondria | ↑ ROS, loss ΔΨm and mitochondrial swelling by unknow mechanism | [239] |
| NS1619 | Mitochondria | ↑ ROS production by unknow mechanism | [215] |
| Adaphostin | Mitochondria | ↑ ROS and loss of ΔΨm by inhibiting mitochondrial respiration | [240] |
| APR-246 (Phase I clinical trials) | Redox enzymes | ↑ ROS by inhibiting TRX1 and glutaredoxin | [241] |
| Sanguinarine (Natural compound) | Mitochondria | ↑ ROS and loss of ΔΨm by depleting glutathione | [242] |
| Alantolactone (Natural compound) | ↑ ROS by inhibiting glutathione reductase (GR) | [243] | |
| CB-839 | Redox enzyme | ↑ ROS by inhibiting glutaminase to suppress glutathione production | [244] |
| 6-Shogaol (Natural compound) | Unknown mechanism | ↑ ROS by reducing GSH | [245,246] |
7.2. Therapies to Reduce ROS
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| AP-1 | Activator protein 1 |
| Ara-C | Cytosine arabinoside |
| ARE | Antioxidant responsive elements |
| ATM | Ataxia-telangiectasia mutation |
| ATO | Arsenic trioxide |
| ATP | Guanosine diphosphate |
| BCR | B cell receptor |
| BCR-ABL | Breakpoint cluster-Abelson |
| CAT | Catalase |
| CDK | Cyclin dependent kinases |
| ClpP | caseinolytic protease P |
| CRLF2 | Cytokine receptor-like factor 2 |
| CREB | cAMP-responsive element binding |
| DPI | Diphenyleneiodonium |
| DS | Down syndrome |
| DSB | Double-strand break |
| DUOX | Dual oxidase |
| EPOR | Erythropoietin receptor |
| EPX | Eosinophil peroxidase |
| ETC | Electron transport chain |
| ETS | E26-transformation-specific (ETS) transcription factors |
| ETV | ETS variant transcription factor 6 |
| FLT3 | FMS-like tyrosine kinase 3 |
| FLT3-ITD | FLT3-internal tandem duplication |
| FoXO | Forkhead box class O |
| GPx | Glutathione peroxidase |
| GSH | Reduced glutathione |
| GSK-3 | Glycogen synthase kinase-3 |
| GSK-3 | Glycogen synthase kinase-3 |
| GSSG | Oxidized glutathione |
| GSR | GSSG reductase |
| GST | Glutathione S-transferases |
| GTP | Guanosine triphosphate |
| HDAC | Histone deacetylase |
| H2O2 | Hydrogen peroxide |
| HIF | Hypoxia inducible factors |
| HO− | Hydroxyl radical |
| HRR | Homologous recombinational repair |
| HSCs | Hematopoietic stem cells |
| Ikzf3 | IKAROS family zinc finger 3 |
| IL-2 | Interleukin-2 |
| IL7R | Interleukin-7 receptor |
| JAK | Janus Kinase |
| Keap1 | Helch-like ECH-associated protein |
| LICs | Leukaemic initiating cells |
| LPO | Lactoperoxidase |
| LPS | lipopolysaccharide |
| LSCs | Leukemic stem cells |
| MDV | Mitochondria-derived vesicle |
| MLL | Mixed-lineage leukemia |
| MnSOD | Manganese superoxide dismutase |
| MPO | Myeloperoxidase |
| mTOR | Mammalian target of rapamycin |
| NAC | N-acetyl-L-cysteine |
| NFAT | Nuclear factor of activated T-cells |
| NHEJ | Non-homologous end joining |
| NO− | Nitric oxide |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor (erythroid-derived)-like 2 |
| O2− | Superoxide anion |
| O3 | Ozone |
| OS | Overall survival |
| Ph | Philadelphia chromosome |
| PIKK | PI3K-like protein kinase |
| PKC | Protein kinase C |
| PRX | Peroxiredoxin |
| PTEN | Phosphatase and tensin homolog |
| PTMs | Post-translational modifications |
| RASGRP1 | RAS guanine nucleotide-releasing protein 1 |
| RAC | Ras-related C3 botulinum toxin substrate |
| Rap1 | Ras-related protein 1 |
| RASGRP | RAS guanyl nucleotide-releasing protein |
| Rb | Retinoblastoma protein |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosin kinase |
| RUNX1 | Runt-related transcription factor 1 |
| SDH | Succinate dehydrogenase |
| SOD | Superoxide dismutase |
| STAT | Signal transducer and activator of transcription |
| SYK | Spleen tyrosine kinase |
| TBB | Tetrabromobenzotriazole |
| TCR | T cell receptor |
| TLR | Toll-like receptor |
| TNTs | Tunnelling nanotubes |
| TPGS | D-α-tocopheryl polyethylene glycol 1000 succinate |
| TRX | Thioredoxin |
| TSC | Tuberous sclerosis complex |
| TSLP | thymic stromal lymphopoietin |
| TXNRD | Thioredoxin reductase |
References
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Key Statistics for Acute Lymphocytic Leukemia (ALL). Available online: https://www.cancer.org/cancer/acute-lymphocytic-leukemia/about/key-statistics.html#references (accessed on 12 October 2021).
- Hunger, S.P.; Mullighan Charles, G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, B.C.; Sensel, M.R.; Sather, H.N.; Gaynon, P.S.; La, M.K.; Johnston, K.; Erdmann, G.R.; Gold, S.; Heerema, N.A.; Hutchinson, R.J.; et al. Dexamethasone versus prednisone and daily oral versus weekly intravenous mercaptopurine for patients with standard-risk acute lymphoblastic leukemia: A report from the Children’s Cancer Group. Blood 2003, 101, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. Studies on the nature of the active serum constituent: Histological mechanism of the regression: Tests for effects of guinea pig serum on lymphoma cells in vitro: Discussion. J. Exp. Med. 1953, 98, 583–606. [Google Scholar] [CrossRef]
- Escherich, G.; Zimmermann, M.; Janka-Schaub, G.; CoALL Study Group. Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr. Blood Cancer 2013, 60, 254–257. [Google Scholar] [CrossRef]
- Nguyen, K.; Devidas, M.; Cheng, S.C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L.; et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef]
- Oriol, A.; Vives, S.; Hernández-Rivas, J.-M.; Tormo, M.; Heras, I.; Rivas, C.; Bethencourt, C.; Moscardó, F.; Bueno, J.; Grande, C.; et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica 2010, 95, 589–596. [Google Scholar] [CrossRef]
- Board, P.P.T.E. PDQ Childhood Acute Lymphoblastic Leukemia Treatment. In Bethesda; National Cancer Institute: Bethesda, MD, USA, 2021. [Google Scholar]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.H.; et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kuusanmaki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brummendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itala-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.t.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Kharas, M.G.; Janes, M.R.; Scarfone, V.M.; Lilly, M.B.; Knight, Z.A.; Shokat, K.M.; Fruman, D.A. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J. Clin. Investig. 2008, 118, 3038–3050. [Google Scholar] [CrossRef]
- Yamamoto, T.; Isomura, M.; Xu, Y.; Liang, J.; Yagasaki, H.; Kamachi, Y.; Kudo, K.; Kiyoi, H.; Naoe, T.; Kojma, S. PTPN11, RAS and FLT3 mutations in childhood acute lymphoblastic leukemia. Leuk. Res. 2006, 30, 1085–1089. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Mabon, M.E.; Silverman, L.B.; Li, A.; Gribben, J.G.; Fox, E.A.; Sallan, S.E.; Korsmeyer, S.J. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004, 103, 3544–3546. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Demeyer, S.; Govaerts, I.; Bornschein, S.; Verbeke, D.; Jacobs, K.; Binos, S.; Skerrett-Byrne, D.A.; Murray, H.C.; et al. Mutant JAK3 phosphoproteomic profiling predicts synergism between JAK3 inhibitors and MEK/BCL2 inhibitors for the treatment of T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 788–800. [Google Scholar] [CrossRef]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef]
- Paulsson, K.; Lilljebjorn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjogren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676. [Google Scholar] [CrossRef]
- Neri, A.; Knowles, D.M.; Greco, A.; McCormick, F.; Dalla-Favera, R. Analysis of RAS oncogene mutations in human lymphoid malignancies. Proc. Natl. Acad. Sci. USA 1988, 85, 9268–9272. [Google Scholar] [CrossRef]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Pearn, L.; Tonks, A.J.; James, P.E.; Burnett, A.K.; Darley, R.L.; Tonks, A. Ras-induced reactive oxygen species promote growth factor–independent proliferation in human CD34+ hematopoietic progenitor cells. Blood 2010, 115, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Adane, B.; Ye, H.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhnyy, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254. [Google Scholar] [CrossRef]
- Maraldi, T.; Angeloni, C.; Prata, C.; Hrelia, S. NADPH Oxidases: Redox Regulators of Stem Cell Fate and Function. Antioxidants 2021, 10, 973. [Google Scholar] [CrossRef]
- Chen, C.; Hao, X.; Lai, X.; Liu, L.; Zhu, J.; Shao, H.; Huang, D.; Gu, H.; Zhang, T.; Yu, Z.; et al. Oxidative phosphorylation enhances the leukemogenic capacity and resistance to chemotherapy of B cell acute lymphoblastic leukemia. Sci. Adv. 2021, 7, eabd6280. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative resistance of leukemic stem cells and oxidative damage to hematopoietic stem cells under pro-oxidative therapy. Cell Death Dis. 2020, 11, 291. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Cafe, S.L.; Nixon, B.; Dun, M.D.; Roman, S.D.; Bernstein, I.R.; Bromfield, E.G. Oxidative Stress Dysregulates Protein Homeostasis Within the Male Germ Line. Antioxid. Redox Signal. 2020, 32, 487–503. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell. Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH Oxidase and Associated Ion Fluxes During Phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14. [Google Scholar] [CrossRef]
- Ruhnau, J.; Schulze, K.; Gaida, B.; Langner, S.; Kessler, C.; Bröker, B.; Dressel, A.; Vogelgesang, A. Stroke Alters Respiratory Burst in Neutrophils and Monocytes. Stroke 2014, 45, 794–800. [Google Scholar] [CrossRef]
- Iles, K.E.; Forman, H.J. Macrophage signaling and respiratory burst. Immunol. Res. 2002, 26, 95–105. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Valente, A.J.; Zhou, Q.; Lu, Z.; He, W.; Qiang, M.; Ma, W.; Li, G.; Wang, L.; Banfi, B.; Steger, K.; et al. Regulation of NOX1 expression by GATA, HNF-1α, and Cdx transcription factors. Free. Radic. Biol. Med. 2008, 44, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, N.; Katsuyama, M.; Matsuno, K.; Urao, N.; Tabuchi, Y.; Okigaki, M.; Matsubara, H.; Yabe-Nishimura, C. Novel transcripts of Nox1 are regulated by alternative promoters and expressed under phenotypic modulation of vascular smooth muscle cells. Biochem. J. 2006, 398, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Youngson, C.; Wong, V.; Yeger, H.; Dinauer, M.C.; de Miera, E.V.-S.; Rudy, B.; Cutz, E. NADPH-oxidase and a hydrogen peroxide-sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proc. Natl. Acad. Sci. USA 1996, 93, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Hancock, J.T.; Jones, O.T.; Neubauer, A.; Topley, N. The expression of NADPH oxidase components in human glomerular mesangial cells: Detection of protein and mRNA for p47phox, p67phox, and p22phox. J. Am. Soc. Nephrol. 1995, 5, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Cao, Z.; Xu, X.; Meir, E.G.V.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Jones, S.A.; O’Donnell, V.B.; Wood, J.D.; Broughton, J.P.; Hughes, E.J.; Jones, O.T. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol.-Heart Circ. Physiol. 1996, 271, H1626–H1634. [Google Scholar] [CrossRef]
- Bánfi, B.; Molnár, G.; Maturana, A.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.-H. A Ca2+-activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes *. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef]
- Koschmann, C.; Farooqui, Z.; Kasaian, K.; Cao, X.; Zamler, D.; Stallard, S.; Venneti, S.; Hervey-Jumper, S.; Garton, H.; Muraszko, K.; et al. Multi-focal sequencing of a diffuse intrinsic pontine glioma establishes PTEN loss as an early event. npj Precision Oncology 2017, 1, 32. [Google Scholar] [CrossRef]
- Finelli, M.J. Redox Post-translational Modifications of Protein Thiols in Brain Aging and Neurodegenerative Conditions—Focus on S-Nitrosation. Front. Aging Neurosci. 2020, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem. 2017, 292, 16810–16816. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain *. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Magder, S. Reactive oxygen species: Toxic molecules or spark of life? Crit. Care 2006, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Zhao, Z. Redox Control in Acute Lymphoblastic Leukemia: From Physiology to Pathology and Therapeutic Opportunities. Cells 2021, 10, 1218. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Zhang, J.; Fang, J.; Huang, Y.; Wang, P.; Ma, J. Production of Hydroxyl Radical via the Activation of Hydrogen Peroxide by Hydroxylamine. Environ. Sci. Technol. 2015, 49, 10373–10379. [Google Scholar] [CrossRef] [PubMed]
- Murley, J.S.; Kataoka, Y.; Miller, R.C.; Li, J.J.; Woloschak, G.; Grdina, D.J. SOD2-Mediated Effects Induced by WR1065 and Low-Dose Ionizing Radiation on Micronucleus Formation in RKO Human Colon Carcinoma Cells. Radiat. Res. 2010, 175, 57–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Melov, S.; Coskun, P.; Patel, M.; Tuinstra, R.; Cottrell, B.; Jun, A.S.; Zastawny, T.H.; Dizdaroglu, M.; Goodman, S.I.; Huang, T.-T.; et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc. Natl. Acad. Sci. USA 1999, 96, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Li, H.; Pazhanisamy, S.K.; Meng, A.; Wang, Y.; Zhou, D. Reactive oxygen species and hematopoietic stem cell senescence. Int. J. Hematol. 2011, 94, 24–32. [Google Scholar] [CrossRef]
- Eliasson, P.; Rehn, M.; Hammar, P.; Larsson, P.; Sirenko, O.; Flippin, L.A.; Cammenga, J.; Jönsson, J.-I. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term–reconstituting hematopoietic stem cells during in vitro culture. Exp. Hematol. 2010, 38, 301–310. [Google Scholar] [CrossRef]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem. Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Rouault-Pierre, K.; Lopez-Onieva, L.; Foster, K.; Anjos-Afonso, F.; Lamrissi-Garcia, I.; Serrano-Sanchez, M.; Mitter, R.; Ivanovic, Z.; deVerneuil, H.; Gribben, J.; et al. HIF-2α; Protects Human Hematopoietic Stem/Progenitors and Acute Myeloid Leukemic Cells from Apoptosis Induced by Endoplasmic Reticulum Stress. Cell Stem. Cell 2013, 13, 549–563. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a Is Essential for Maintenance of the Hematopoietic Stem Cell Pool. Cell Stem. Cell 2007, 1, 101–112. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Jackson, E.R.; Alvaro, F.; Nixon, B.; Hondermarck, H.; Dun, M.D. Signal Transduction in Diffuse Intrinsic Pontine Glioma. Proteomics 2019, 19, e1800479. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Chen, Q.; Powell, D.W.; Rane, M.J.; Singh, S.; Butt, W.; Klein, J.B.; McLeish, K.R. Akt Phosphorylates p47phox and Mediates Respiratory Burst Activity in Human Neutrophils. J. Immunol. 2003, 170, 5302–5308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Juntilla, M.M.; Patil, V.D.; Calamito, M.; Joshi, R.P.; Birnbaum, M.J.; Koretzky, G.A. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030–4038. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.-L.; Liu, Y.; Zheng, P. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Mak, T.W.; Suda, T. Regulation of Oxidative Stress by ATM Is Required for the Self-Renewal of Haematopoietic Stem Cells. Blood 2004, 104, 369. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Fasouli, E.S.; Katsantoni, E. JAK-STAT in Early Hematopoiesis and Leukemia. Front. Cell Dev. Biol. 2021, 9, 669363. [Google Scholar] [CrossRef] [PubMed]
- Akada, H.; Akada, S.; Hutchison, R.E.; Sakamoto, K.; Wagner, K.-U.; Mohi, G. Critical Role of Jak2 in the Maintenance and Function of Adult Hematopoietic Stem Cells. Stem. Cells 2014, 32, 1878–1889. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Müller, J.P.; Bauer, R.; Böhmer, S.A.; Lässig, J.; Cerny-Reiterer, S.; Sperr, W.R.; Valent, P.; Maurer, B.; Moriggl, R.; et al. NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 2016, 30, 473–483. [Google Scholar] [CrossRef]
- Vilas-Zornoza, A.; Agirre, X.; Martin-Palanco, V.; Martin-Subero, J.I.; San Jose-Eneriz, E.; Garate, L.; Alvarez, S.; Miranda, E.; Rodriguez-Otero, P.; Rifon, J.; et al. Frequent and simultaneous epigenetic inactivation of TP53 pathway genes in acute lymphoblastic leukemia. PLoS ONE 2011, 6, e17012. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lee, J.; Yoon, S.R.; Lee, H.G.; Jung, H. Regulation of Hematopoietic Stem Cell Fate and Malignancy. Int. J. Mol. Sci. 2020, 21, 4780. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic granulomatous disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.-C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.e625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, L.; Chu, Y. Reactive oxygen species: The signal regulator of B cell. Free Radic. Biol. Med. 2019, 142, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Maly, F.E.; Cross, A.R.; Jones, O.T.; Wolf-Vorbeck, G.; Walker, C.; Dahinden, C.A.; De Weck, A.L. The superoxide generating system of B cell lines. Structural homology with the phagocytic oxidase and triggering via surface Ig. J. Immunol. 1988, 140, 2334–2339. [Google Scholar] [PubMed]
- Maly, F.E.; Nakamura, M.; Gauchat, J.F.; Urwyler, A.; Walker, C.; Dahinden, C.A.; Cross, A.R.; Jones, O.T.; de Weck, A.L. Superoxide-dependent nitroblue tetrazolium reduction and expression of cytochrome b-245 components by human tonsillar B lymphocytes and B cell lines. J. Immunol. 1989, 142, 1260–1267. [Google Scholar]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete Generation of Superoxide and Hydrogen Peroxide by T Cell Receptor Stimulation: Selective Regulation of Mitogen-Activated Protein Kinase Activation and Fas Ligand Expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Ferrante, A.; Arrigo, A.P.; Dayer, J.M. Neutrophil stimulation and priming by direct contact with activated human T lymphocytes. J. Immunol. 1992, 148, 177–181. [Google Scholar]
- Kwon, J.; Shatynski, K.E.; Chen, H.; Morand, S.; de Deken, X.; Miot, F.; Leto, T.L.; Williams, M.S. The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci. Signal. 2010, 3, ra59. [Google Scholar] [CrossRef]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Schenk, H.; Hexel, K.; Baeuerle, P.A.; Droge, W.; Schulze-Osthoff, K. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995, 14, 3731–3740. [Google Scholar] [CrossRef]
- Rolli, V.; Gallwitz, M.; Wossning, T.; Flemming, A.; Schamel, W.W.A.; Zürn, C.; Reth, M. Amplification of B Cell Antigen Receptor Signaling by a Syk/ITAM Positive Feedback Loop. Mol. Cell 2002, 10, 1057–1069. [Google Scholar] [CrossRef]
- Wheeler, M.L.; Defranco, A.L. Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J. Immunol. 2012, 189, 4405–4416. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Urbánek, P.; Rolink, A.; Busslinger, M. Essential functions of Pax5 (BSAP) in pro-B cell development: Difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev. 1997, 11, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, Y.; Coll, M.D.; Ortega, J.J.; Bastida, P.; Dastugue, N.; Robert, A.; Cervera, J.; Verdeguer, A.; Tasso, M.; Aventin, A.; et al. Genetic abnormalities associated with the t(12;21) and their impact in the outcome of 56 patients with B-precursor acute lymphoblastic leukemia. Cancer Genet. Cytogenet. 2005, 162, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.P.; Deltcheva, E.M.; Boiers, C.; Richardson, S.E.; Chettri, J.B.; Gagrica, S.; Guo, Y.; Illendula, A.; Martens, J.H.A.; Stunnenberg, H.G.; et al. Cell cycle corruption in a preleukemic ETV6-RUNX1 model exposes RUNX1 addiction as a therapeutic target in acute lymphoblastic leukemia. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kantner, H.-P.; Warsch, W.; Delogu, A.; Bauer, E.; Esterbauer, H.; Casanova, E.; Sexl, V.; Stoiber, D. ETV6/RUNX1 Induces Reactive Oxygen Species and Drives the Accumulation of DNA Damage in B Cells. Neoplasia 2013, 15, 1292-IN1228. [Google Scholar] [CrossRef]
- Schröder, K.; Kohnen, A.; Aicher, A.; Liehn, E.A.; Büchse, T.; Stein, S.; Weber, C.; Dimmeler, S.; Brandes, R.P. NADPH Oxidase Nox2 Is Required for Hypoxia-Induced Mobilization of Endothelial Progenitor Cells. Circ. Res. 2009, 105, 537–544. [Google Scholar] [CrossRef]
- Teachey, D.T.; Pui, C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Kuan, J.W.; Su, A.T.; Leong, C.F.; Osato, M.; Sashida, G. Systematic Review of Normal Subjects Harbouring BCR-ABL1 Fusion Gene. Acta Haematol. 2020, 143, 96–111. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. Molecular Pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL Tyrosine Kinase Induces Production of Reactive Oxygen Species in Hematopoietic Cells*. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef]
- Naughton, R.; Quiney, C.; Turner, S.D.; Cotter, T.G. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009, 23, 1432–1440. [Google Scholar] [CrossRef]
- Landry, W.D.; Woolley, J.F.; Cotter, T.G. Imatinib and Nilotinib inhibit Bcr–Abl-induced ROS through targeted degradation of the NADPH oxidase subunit p22phox. Leuk. Res. 2013, 37, 183–189. [Google Scholar] [CrossRef]
- Cho, Y.J.; Zhang, B.; Kaartinen, V.; Haataja, L.; Curtis, I.d.; Groffen, J.; Heisterkamp, N. Generation of rac3 Null Mutant Mice: Role of Rac3 in Bcr/Abl-Caused Lymphoblastic Leukemia. Mol. Cell. Biol. 2005, 25, 5777–5785. [Google Scholar] [CrossRef]
- Miyano, K.; Koga, H.; Minakami, R.; Sumimoto, H. The insert region of the Rac GTPases is dispensable for activation of superoxide-producing NADPH oxidases. Biochem. J. 2009, 422, 373–382. [Google Scholar] [CrossRef]
- Koptyra, M.; Cramer, K.; Slupianek, A.; Richardson, C.; Skorski, T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia 2008, 22, 1969–1972. [Google Scholar] [CrossRef]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.O.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nowicki, M.O.; Koptyra, M.; Skorski, T. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA Repair 2006, 5, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL Regulates Mammalian RecA Homologs, Resulting in Drug Resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int. J. Mol. Sci. 2018, 19, 3198. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.C.; Enjeti, A.K.; Kahl, R.G.S.; Flanagan, H.M.; Sillar, J.; Skerrett-Byrne, D.A.; Al Mazi, J.G.; Au, G.G.; de Bock, C.E.; Evans, K.; et al. Quantitative phosphoproteomics uncovers synergy between DNA-PK and FLT3 inhibitors in acute myeloid leukaemia. Leukemia 2021, 35, 1782–1787. [Google Scholar] [CrossRef]
- Al-Shami, A.; Spolski, R.; Kelly, J.; Fry, T.; Schwartzberg, P.L.; Pandey, A.; Mackall, C.L.; Leonard, W.J. A role for thymic stromal lymphopoietin in CD4(+) T cell development. J. Exp. Med. 2004, 200, 159–168. [Google Scholar] [CrossRef]
- Russell, L.J.; Capasso, M.; Vater, I.; Akasaka, T.; Bernard, O.A.; Calasanz, M.J.; Chandrasekaran, T.; Chapiro, E.; Gesk, S.; Griffiths, M.; et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 2009, 114, 2688–2698. [Google Scholar] [CrossRef]
- Jain, N.; Roberts, K.G.; Jabbour, E.; Patel, K.; Eterovic, A.K.; Chen, K.; Zweidler-McKay, P.; Lu, X.; Fawcett, G.; Wang, S.A.; et al. Ph-like acute lymphoblastic leukemia: A high-risk subtype in adults. Blood 2017, 129, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Pei, D.; Campana, D.; Payne-Turner, D.; Li, Y.; Cheng, C.; Sandlund, J.T.; Jeha, S.; Easton, J.; Becksfort, J.; et al. Outcomes of children with BCR-ABL1-like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J. Clin. Oncol. 2014, 32, 3012–3020. [Google Scholar] [CrossRef]
- Tasian, S.K.; Doral, M.Y.; Borowitz, M.J.; Wood, B.L.; Chen, I.M.; Harvey, R.C.; Gastier-Foster, J.M.; Willman, C.L.; Hunger, S.P.; Mullighan, C.G.; et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012, 120, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, L.; Vendramini, E.; Ganmore, I.; Cazzaniga, G.; Schmitz, M.; Chalker, J.; Shiloh, R.; Iacobucci, I.; Shochat, C.; Zeligson, S.; et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: A report from the International BFM Study Group. Blood 2010, 115, 1006–1017. [Google Scholar] [CrossRef]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematology 2012, 2012, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Yoda, A.; Yoda, Y.; Chiaretti, S.; Bar-Natan, M.; Mani, K.; Rodig, S.J.; West, N.; Xiao, Y.; Brown, J.R.; Mitsiades, C.; et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 252–257. [Google Scholar] [CrossRef]
- Shochat, C.; Tal, N.; Bandapalli, O.R.; Palmi, C.; Ganmore, I.; te Kronnie, G.; Cario, G.; Cazzaniga, G.; Kulozik, A.E.; Stanulla, M.; et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011, 208, 901–908. [Google Scholar] [CrossRef]
- Rochman, Y.; Kashyap, M.; Robinson, G.W.; Sakamoto, K.; Gomez-Rodriguez, J.; Wagner, K.-U.; Leonard, W.J. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7–induced signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 19455–19460. [Google Scholar] [CrossRef]
- Silva, A.; Gírio, A.; Cebola, I.; Santos, C.I.; Antunes, F.; Barata, J.T. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 2011, 25, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zwet, J.C.G.; Buijs-Gladdines, J.G.C.A.M.; Cordo, V.; Debets, D.; Smits, W.K.; Chen, Z.; Dylus, J.; Zaman, G.; Altelaar, M.; Oshima, K.; et al. The Central Role of MAPK-ERK Signaling in IL7-Dependent and IL7-Independent Steroid Resistance Reveals a Broad Application of MEK-Inhibitors Compared to JAK1/2-Inhibition in T-ALL. Blood 2020, 136, 20. [Google Scholar] [CrossRef]
- Polli, M.; Dakic, A.; Light, A.; Wu, L.; Tarlinton, D.M.; Nutt, S.L. The development of Funct. B lymphocytes in conditional PU.1 knock-out mice. Blood 2005, 106, 2083–2090. [Google Scholar] [CrossRef]
- Su, G.H.; Chen, H.-M.; Muthusamy, N.; Garrett-Sinha, L.A.; Baunoch, D.; Tenen, D.G.; Simon, M.C. Defective B cell receptor-mediated responses in mice lacking the Ets protein, Spi-B. EMBO J. 1997, 16, 7118–7129. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Lim, M.; Batista, C.R.; Oliveira, B.R.d.; Creighton, R.; Ferguson, J.; Clemmer, K.; Knight, D.; Iansavitchous, J.; Mahmood, D.; Avino, M.; et al. Janus Kinase Mutations in Mice Lacking PU.1 and Spi-B Drive B Cell Leukemia through Reactive Oxygen Species-Induced DNA Damage. Mol. Cell. Biol. 2020, 40, e00189-20. [Google Scholar] [CrossRef]
- Staber, P.B.; Zhang, P.; Ye, M.; Welner, R.S.; Nombela-Arrieta, C.; Bach, C.; Kerenyi, M.; Bartholdy, B.A.; Zhang, H.; Alberich-Jordà, M.; et al. Sustained PU.1 Levels Balance Cell-Cycle Regulators to Prevent Exhaustion of Adult Hematopoietic Stem Cells. Mol. Cell 2013, 49, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Manea, S.A.; Gafencu, A.V.; Raicu, M.; Simionescu, M. AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: Role of p22phox subunit. Arter. Thromb. Vasc. Biol. 2008, 28, 878–885. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate p53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, H.; Qian, X.; Gao, S.; Xia, J.; Liu, J.; Cheng, Y.; Man, J.; Zhai, X. Genetic mutational analysis of pediatric acute lymphoblastic leukemia from a single center in China using exon sequencing. BMC Cancer 2020, 20, 211. [Google Scholar] [CrossRef] [PubMed]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; van de Ven, C.; de Groot-Kruseman, H.A.; de Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 931–940. [Google Scholar] [CrossRef]
- Tang, C.; Li, M.-H.; Chen, Y.-L.; Sun, H.-Y.; Liu, S.-L.; Zheng, W.-W.; Zhang, M.-Y.; Li, H.; Fu, W.; Zhang, W.-J.; et al. Chemotherapy-induced niche perturbs hematopoietic reconstitution in B-cell acute lymphoblastic leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 204. [Google Scholar] [CrossRef]
- Irving, J.; Matheson, E.; Minto, L.; Blair, H.; Case, M.; Halsey, C.; Swidenbank, I.; Ponthan, F.; Kirschner-Schwabe, R.; Groeneveld-Krentz, S.; et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014, 124, 3420–3430. [Google Scholar] [CrossRef]
- Ksionda, O.; Melton, A.A.; Bache, J.; Tenhagen, M.; Bakker, J.; Harvey, R.; Winter, S.S.; Rubio, I.; Roose, J.P. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene 2016, 35, 3658–3668. [Google Scholar] [CrossRef]
- Mues, M.; Roose, J.P. Distinct oncogenic Ras signals characterized by profound differences in flux through the RasGDP/RasGTP cycle. Small GTPases 2017, 8, 20–25. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Kerstjens, M.; Driessen, E.M.C.; Willekes, M.; Pinhanços, S.S.; Schneider, P.; Pieters, R.; Stam, R.W. MEK inhibition is a promising therapeutic strategy for MLL-rearranged infant acute lymphoblastic leukemia patients carrying RAS mutations. Oncotarget 2017, 8, 14835–14846. [Google Scholar] [CrossRef]
- Holland, M.; Castro, F.V.; Alexander, S.; Smith, D.; Liu, J.; Walker, M.; Bitton, D.; Mulryan, K.; Ashton, G.; Blaylock, M.; et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood 2011, 118, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Kondoh, K.; Fukushima, S.; Hashiguchi, A.; Du, W.; Hayashi, M.; Fujimoto, J.-i.; Hata, J.-i.; Yamada, T. Mutated D4-guanine diphosphate–dissociation inhibitor is found in human leukemic cells and promotes leukemic cell invasion. Exp. Hematol. 2008, 36, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Freret, M.; Gouel, F.; Buquet, C.; Legrand, E.; Vannier, J.-P.; Vasse, M.; Dubus, I. Rac-1 GTPase controls the capacity of human leukaemic lymphoblasts to migrate on fibronectin in response to SDF-1α (CXCL12). Leuk. Res. 2011, 35, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.-P.; Vallée, A.; Robert, G.; Bonneau, J.; Leroy, C.; Varin-Blank, N.; Rio, A.-G.; Troadec, M.-B.; Galibert, M.-D.; Gandemer, V. CD9, a key actor in the dissemination of lymphoblastic leukemia, modulating CXCR4-mediated migration via RAC1 signaling. Blood 2015, 126, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Geiszt, M.; Leto, T.L. Involvement of Rac1 in Activation of Multicomponent Nox1- and Nox3-Based NADPH Oxidases. Mol. Cell. Biol. 2006, 26, 2160–2174. [Google Scholar] [CrossRef]
- Madajewski, B.; Boatman, M.A.; Chakrabarti, G.; Boothman, D.A.; Bey, E.A. Depleting Tumor-NQO1 Potentiates Anoikis and Inhibits Growth of NSCLC. Mol. Cancer Res. 2016, 14, 14–25. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Diao, J.; Bao, J.; Peng, J.; Mo, J.; Ye, Q.; He, J. Correlation between NAD(P)H: Quinone oxidoreductase 1 C609T polymorphism and increased risk of esophageal cancer: Evidence from a meta-analysis. Ther. Adv. Med Oncol. 2017, 9, 13–21. [Google Scholar] [CrossRef]
- Smith, M.T.; Wang, Y.; Skibola, C.F.; Slater, D.J.; Nigro, L.L.; Nowell, P.C.; Lange, B.J.; Felix, C.A. Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood 2002, 100, 4590–4593. [Google Scholar] [CrossRef]
- Freriksen, J.J.M.; Salomon, J.; Roelofs, H.M.J.; te Morsche, R.H.M.; van der Stappen, J.W.J.; Dura, P.; Witteman, B.J.M.; Lacko, M.; Peters, W.H.M. Genetic polymorphism 609C>T in NAD(P)H:quinone oxidoreductase 1 enhances the risk of proximal colon cancer. J. Hum. Genet. 2014, 59, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Pagnamenta, A.; Taylor, G.M.; Eden, O.B.; Alexander, F.E.; Greaves, M.F. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res. 1999, 59, 4095–4099. [Google Scholar] [PubMed]
- Manesia, J.K.; Xu, Z.; Broekaert, D.; Boon, R.; van Vliet, A.; Eelen, G.; Vanwelden, T.; Stegen, S.; Van Gastel, N.; Pascual-Montano, A.; et al. Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem. Cell Res. 2015, 15, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kong, J.; Yan, G.; Ren, X.; Jin, D.; Jin, T.; Lin, L.; Lin, Z. NQO1 overexpression is associated with poor prognosis in squamous cell carcinoma of the uterine cervix. BMC Cancer 2014, 14, 414. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Jin, T.; Men, J.; Lin, Z.; Qi, P.; Piao, Y.; Yan, G. NQO1 protein expression predicts poor prognosis of non-small cell lung cancers. BMC Cancer 2015, 15, 207. [Google Scholar] [CrossRef]
- Motea, E.A.; Huang, X.; Singh, N.; Kilgore, J.A.; Williams, N.S.; Xie, X.J.; Gerber, D.E.; Beg, M.S.; Bey, E.A.; Boothman, D.A. NQO1-dependent, Tumor-selective Radiosensitization of Non-small Cell Lung Cancers. Clin. Cancer Res. 2019, 25, 2601–2609. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Et. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Fidyt, K.; Pastorczak, A.; Goral, A.; Szczygiel, K.; Fendler, W.; Muchowicz, A.; Bartlomiejczyk, M.A.; Madzio, J.; Cyran, J.; Graczyk-Jarzynka, A.; et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol. Oncol. 2019, 13, 1180–1195. [Google Scholar] [CrossRef]
- Inoue, H.; Takemura, H.; Kawai, Y.; Yoshida, A.; Ueda, T.; Miyashita, T. Dexamethasone-resistant human Pre-B leukemia 697 cell line evolving elevation of intracellular glutathione level: An additional resistance mechanism. Jpn. J Cancer Res. 2002, 93, 582–590. [Google Scholar] [CrossRef]
- Tome, M.E.; Baker, A.F.; Powis, G.; Payne, C.M.; Briehl, M.M. Catalase-overexpressing thymocytes are resistant to glucocorticoid-induced apoptosis and exhibit increased net tumor growth. Cancer Res. 2001, 61, 2766–2773. [Google Scholar]
- Jaramillo, M.C.; Frye, J.B.; Crapo, J.D.; Briehl, M.M.; Tome, M.E. Increased manganese superoxide dismutase expression or treatment with manganese porphyrin potentiates dexamethasone-induced apoptosis in lymphoma cells. Cancer Res. 2009, 69, 5450–5457. [Google Scholar] [CrossRef]
- Kearns, P.R.; Pieters, R.; Rottier, M.M.A.; Pearson, A.D.J.; Hall, A.G. Raised blast glutathione levels are associated with an increased risk of relapse in childhood acute lymphocytic leukemia. Blood 2001, 97, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Xu, N.; Ding, Q.; Huang, S.; Zha, Y.; Zhu, H. LncRNA VPS9D1-AS1 promotes cell proliferation in acute lymphoblastic leukemia through modulating GPX1 expression by miR-491-5p and miR-214-3p evasion. Biosci. Rep. 2020, 40, BSR20193461. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-kappaB activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef]
- Vasudevan, K.M.; Gurumurthy, S.; Rangnekar, V.M. Suppression of PTEN expression by NF-kappa B prevents apoptosis. Mol. Cell Biol. 2004, 24, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. Loss of PTEN Expression Induces NF-kB Via PI3K/Akt Pathway Involving Resistance to Chemotherapy in Acute Lymphoblastic Leukemia Cell Lines. Blood 2004. [Google Scholar] [CrossRef]
- Katerndahl, C.D.S.; Heltemes-Harris, L.M.; Willette, M.J.L.; Henzler, C.M.; Frietze, S.; Yang, R.; Schjerven, H.; Silverstein, K.A.T.; Ramsey, L.B.; Hubbard, G.; et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 2017, 18, 694–704. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Schwaller, J.; Parganas, E.; Wang, D.; Cain, D.; Aster, J.C.; Williams, I.R.; Lee, C.K.; Gerthner, R.; Kitamura, T.; Frantsve, J.; et al. Stat5 is essential for the myelo- and lymphoproliferative disease induced by TEL/JAK2. Mol. Cell 2000, 6, 693–704. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100. [Google Scholar] [CrossRef]
- Wang, J.; Rao, Q.; Wang, M.; Wei, H.; Xing, H.; Liu, H.; Wang, Y.; Tang, K.; Peng, L.; Tian, Z.; et al. Overexpression of Rac1 in leukemia patients and its role in leukemia cell migration and growth. Biochem. Biophys Res. Commun. 2009, 386, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Echeverria, E.; Lenicov, F.R.; Cardama, G.; Gonzalez, N.; Davio, C.; Fernandez, N.; Menna, P.L. Pharmacological Rac1 inhibitors with selective apoptotic activity in human acute leukemic cell lines. Oncotarget 2017, 8, 98509–98523. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Akin-Bali, D.F.; Aktas, S.H.; Unal, M.A.; Kankilic, T. Identification of novel Nrf2/Keap1 pathway mutations in pediatric acute lymphoblastic leukemia. Pediatr Hematol. Oncol. 2020, 37, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Tucci, J.; Parmentier, J.-H.; Ji, L.; Behan, J.W.; Heisterkamp, N.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to daunorubicin via oxidative stress response. Oncotarget 2016, 7, 73147. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, C.E.; Rafal, R.A.; John, P.M.; Paraskevi, D.; Allison, B. Investigating chemoresistance to improve sensitivity of childhood T-cell acute lymphoblastic leukemia to parthenolide. Haematologica 2018, 103, 1493–1501. [Google Scholar] [CrossRef]
- Drab, M.; Stopar, D.; Kralj-Iglič, V.; Iglič, A. Inception Mechanisms of Tunneling Nanotubes. Cells 2019, 8, 626. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef]
- Fahy, L.; Calvo, J.; Chabi, S.; Renou, L.; Le Maout, C.; Poglio, S.; Leblanc, T.; Petit, A.; Baruchel, A.; Ballerini, P.; et al. Hypoxia favors chemoresistance in T-ALL through an HIF1α-mediated mTORC1 inhibition loop. Blood Adv. 2021, 5, 513–526. [Google Scholar] [CrossRef]
- Takahashi, H.; Inoue, J.; Sakaguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef]
- Wu, X.; Feng, X.; Zhao, X.; Ma, F.; Liu, N.; Guo, H.; Li, C.; Du, H.; Zhang, B. Role of Beclin-1-Mediated Autophagy in the Survival of Pediatric Leukemia Cells. Cell. Physiol. Biochem. 2016, 39, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Jing, B.; Jin, J.; Xiang, R.; Liu, M.; Yang, L.; Tong, Y.; Xiao, X.; Lei, H.; Liu, W.; Xu, H.; et al. Vorinostat and quinacrine have synergistic effects in T-cell acute lymphoblastic leukemia through reactive oxygen species increase and mitophagy inhibition. Cell Death Dis. 2018, 9, 589. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.C.; Woollard, K.J.; Griffiths, H.R. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br. J. Pharmacol. 2003, 138, 501–511. [Google Scholar] [CrossRef]
- Al-Aamri, H.M.; Ku, H.; Irving, H.R.; Tucci, J.; Meehan-Andrews, T.; Bradley, C. Time dependent response of daunorubicin on cytotoxicity, cell cycle and DNA repair in acute lymphoblastic leukaemia. BMC Cancer 2019, 19, 179. [Google Scholar] [CrossRef]
- Groninger, E.; Meeuwsen-De Boer, G.J.; De Graaf, S.S.N.; Kamps, W.A.; De Bont, E.S.J.M. Vincristine induced apoptosis in acute lymphoblastic leukaemia cells: A mitochondrial controlled pathway regulated by reactive oxygen species? Int. J. Oncol. 2002, 21, 1339–1345. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid Med. Cell Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Gidron, Y.; Russ, K.; Tissarchondou, H.; Warner, J. The relation between psychological factors and DNA-damage: A critical review. Biol. Psychol. 2006, 72, 291–304. [Google Scholar] [CrossRef]
- Liu, W.; Zhao, Z.; Na, Y.; Meng, C.; Wang, J.; Bai, R. Dexamethasone-induced production of reactive oxygen species promotes apoptosis via endoplasmic reticulum stress and autophagy in MC3T3-E1 cells. Int. J. Mol. Med. 2018, 41, 2028–2036. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 inhibitor venetoclax inhibits Nrf2 antioxidant pathway activation induced by hypomethylating agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef]
- Petruccelli, L.A.; Dupéré-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS ONE 2011, 6, e20987. [Google Scholar] [CrossRef]
- Scotland, S.; Saland, E.; Skuli, N.; de Toni, F.; Boutzen, H.; Micklow, E.; Sénégas, I.; Peyraud, R.; Peyriga, L.; Théodoro, F.; et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013, 27, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Silic-Benussi, M.; Scattolin, G.; Cavallari, I.; Minuzzo, S.; del Bianco, P.; Francescato, S.; Basso, G.; Indraccolo, S.; D’Agostino, D.M.; Ciminale, V. Selective killing of human T-ALL cells: An integrated approach targeting redox homeostasis and the OMA1/OPA1 axis. Cell Death Dis. 2018, 9, 822. [Google Scholar] [CrossRef] [PubMed]
- Haß, C.; Belz, K.; Schoeneberger, H.; Fulda, S. Sensitization of acute lymphoblastic leukemia cells for LCL161-induced cell death by targeting redox homeostasis. Biochem. Pharmacol. 2016, 105, 14–22. [Google Scholar] [CrossRef]
- Chou, W.-C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4578–4583. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.V.; Manju, A.; Abhilash, M.; Paul, M.V.S.; Abhilash, S.; Nair, R.H. Oxidative stress induced by the chemotherapeutic agent arsenic trioxide. 3 Biotech 2014, 4, 425–430. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef]
- Townsend, D.M.; Pazoles, C.J.; Tew, K.D. NOV-002, a mimetic of glutathione disulfide. Expert. Opin. Investig. Drugs 2008, 17, 1075–1083. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef]
- Doñate, F.; Juarez, J.C.; Burnett, M.E.; Manuia, M.M.; Guan, X.; Shaw, D.E.; Smith, E.L.P.; Timucin, C.; Braunstein, M.J.; Batuman, O.A.; et al. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224). Br. J. Cancer 2008, 98, 776–783. [Google Scholar] [CrossRef]
- Dvorakova, K.; Payne, C.M.; Tome, M.E.; Briehl, M.M.; McClure, T.; Dorr, R.T. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem. Pharmacol. 2000, 60, 749–758. [Google Scholar] [CrossRef]
- Ehrenfeld, V.; Fulda, S. Thioredoxin inhibitor PX-12 induces mitochondria-mediated apoptosis in acute lymphoblastic leukemia cells. Biol. Chem. 2020, 401, 273–283. [Google Scholar] [CrossRef]
- Kirkpatrick, D.L.; Ehrmantraut, G.; Stettner, S.; Kunkel, M.; Powis, G. Redox active disulfides: The thioredoxin system as a drug target. Oncol. Res. 1997, 9, 351–356. [Google Scholar] [PubMed]
- Sztiller-Sikorska, M.; Czyz, M. Parthenolide as Cooperating Agent for Anti-Cancer Treatment of Various Malignancies. Pharmaceuticals 2020, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [CrossRef]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. TPEN Induces Apoptosis Independently of Zinc Chelator Activity in a Model of Acute Lymphoblastic Leukemia and Ex Vivo Acute Leukemia Cells through Oxidative Stress and Mitochondria Caspase-3- and AIF-Dependent Pathways. Oxid Med. Cell Longev. 2012, 2012, 313275. [Google Scholar] [CrossRef]
- Hashemi, M.; Ghavami, S.; Eshraghi, M.; Booy, E.P.; Los, M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. Eur. J. Pharmacol. 2007, 557, 9–19. [Google Scholar] [CrossRef]
- Afrin, F.; Chi, M.; Eamens, A.L.; Duchatel, R.J.; Douglas, A.M.; Schneider, J.; Gedye, C.; Woldu, A.S.; Dun, M.D. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers 2020, 12, 1033. [Google Scholar] [CrossRef]
- Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Khan, A.Q.; Ahmed, E.I.; Akhtar, S.; Ali, T.A.; Merhi, M.; Dermime, S.; Steinhoff, M.; et al. Curcumin Induces Apoptotic Cell Death via Inhibition of PI3-Kinase/AKT Pathway in B-Precursor Acute Lymphoblastic Leukemia. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Yamada, T.; Egashira, N.; Imuta, M.; Yano, T.; Yamauchi, Y.; Watanabe, H.; Oishi, R. Role of oxidative stress in vinorelbine-induced vascular endothelial cell injury. Free. Radic. Biol. Med. 2010, 48, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers 2010, 2, 1288–1311. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free. Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Liu, C.-X.; Yin, Q.-Q.; Zhou, H.-C.; Wu, Y.-L.; Pu, J.-X.; Xia, L.; Liu, W.; Huang, X.; Jiang, T.; Wu, M.-X.; et al. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 2012, 8, 486–493. [Google Scholar] [CrossRef]
- Dächert, J.; Schoeneberger, H.; Rohde, K.; Fulda, S. RSL3 and Erastin differentially regulate redox signaling to promote Smac mimetic-induced cell death. Oncotarget 2016, 7, 63779–63792. [Google Scholar] [CrossRef]
- Aghvami, M.; Ebrahimi, F.; Zarei, M.H.; Salimi, A.; Pourahmad Jaktaji, R.; Pourahmad, J. Matrine Induction of ROS Mediated Apoptosis in Human ALL B-lymphocytes Via Mitochondrial Targeting. Asian Pac. J. Cancer Prev. 2018, 19, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Orsolic, N.; Golemovic, M.; Quintás-Cardama, A.; Scappini, B.; Manshouri, T.; Chandra, J.; Basic, I.; Giles, F.; Kantarjian, H.; Verstovsek, S. Adaphostin has significant and selective activity against chronic and acute myeloid leukemia cells. Cancer Sci. 2006, 97, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Khan, A.Q.; Akhtar, S.; Mateo, J.M.; Merhi, M.; Taha, R.; Omri, H.E.; Mraiche, F.; et al. Sanguinarine suppresses growth and induces apoptosis in childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2019, 60, 782–794. [Google Scholar] [CrossRef]
- Xu, X.; Huang, L.; Zhang, Z.; Tong, J.; Mi, J.; Wu, Y.; Zhang, C.; Yan, H. Targeting non-oncogene ROS pathway by alantolactone in B cell acute lymphoblastic leukemia cells. Life Sci. 2019, 227, 153–165. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [PubMed]
- Najafi Dorcheh, S.; Rahgozar, S.; Talei, D. 6-Shogaol induces apoptosis in acute lymphoblastic leukaemia cells by targeting p53 signalling pathway and generation of reactive oxygen species. J. Cell. Mol. Med. 2021, 25, 6148–6160. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.-H.; Ni, Z.-J.; Zhang, F.; Zhang, Y.-Y.; Liu, M.-M.; Thakur, K.; Zhang, J.-G.; Wang, S.; Wei, Z.-J. 6-Shogaol mediated ROS production and apoptosis via endoplasmic reticulum and mitochondrial pathways in human endometrial carcinoma Ishikawa cells. J. Funct. Foods 2020, 74, 104178. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Fu, X.; Liu, W.; Huang, Q.; Wang, Y.; Li, H.; Xiong, Y. Targeting mitochondrial respiration selectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am. J. Cancer Res. 2017, 7, 2395–2405. [Google Scholar]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737. [Google Scholar] [CrossRef]
- Dong, L.F.; Low, P.; Dyason, J.C.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef]
- Ruiz-Moreno, C.; Jimenez-Del-Rio, M.; Sierra-Garcia, L.; Lopez-Osorio, B.; Velez-Pardo, C. Vitamin E synthetic derivate—TPGS—selectively induces apoptosis in jurkat t cells via oxidative stress signaling pathways: Implications for acute lymphoblastic leukemia. Apoptosis 2016, 21, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, D.; Zhelev, Z.; Semkova, S.; Aoki, I.; Bakalova, R. Resveratrol Modulates the Redox-status and Cytotoxicity of Anticancer Drugs by Sensitizing Leukemic Lymphocytes and Protecting Normal Lymphocytes. Anticancer. Res. 2019, 39, 3745–3755. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Gerauer, H.; Wachter, Y.; Zunino, S.J. Resveratrol Induces Extensive Apoptosis by Depolarizing Mitochondrial Membranes and Activating Caspase-9 in Acute Lymphoblastic Leukemia Cells. Cancer Res. 2001, 61, 4731–4739. [Google Scholar] [PubMed]
- Larasati, Y.A.; Yoneda-Kato, N.; Nakamae, I.; Yokoyama, T.; Meiyanto, E.; Kato, J.-y. Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci. Rep. 2018, 8, 2039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, Z.; Jin, Y.; Chen, Y.; Yang, T.; Yang, Q.; Wu, B.; Shang, Y.; Liu, X.; Wei, Y.; et al. Azelaic Acid Exerts Antileukemia Effects against Acute Myeloid Leukemia by Regulating the Prdxs/ROS Signaling Pathway. Oxid Med. Cell Longev. 2020, 2020, 1295984. [Google Scholar] [CrossRef]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wu, Q.; Wang, J.; Yao, B.; Ma, L.; Yang, Z.; Li, J.; Liu, B. NOX4 supports glycolysis and promotes glutamine metabolism in non-small cell lung cancer cells. Free. Radic. Biol. Med. 2016, 101, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Muecke, C.; Valasarajan, C.; Schmoranzer, M.; Wietelmann, A.; Semenza, G.L.; Meister, M.; Muley, T.; Seeger-Nukpezah, T.; Samakovlis, C.; et al. A RASSF1A-HIF1α loop drives Warburg effect in cancer and pulmonary hypertension. Nat. Commun. 2019, 10, 2130. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Horiuchi, K.; Aoki, Y.; Yamada, Y.; Minamoto, T.; Oshima, H.; Oshima, M. NF-κB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene 2019, 38, 4250–4263. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wu, Q.; Chen, Y.; Deng, Y.; Yang, Z.; Zhang, L.; Liu, B. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. 2019, 22, 101116. [Google Scholar] [CrossRef]
- Ford, K.; Hanley, C.J.; Mellone, M.; Szyndralewiez, C.; Heitz, F.; Wiesel, P.; Wood, O.; Machado, M.; Lopez, M.-A.; Ganesan, A.-P.; et al. NOX4 Inhibition Potentiates Immunotherapy by Overcoming Cancer-Associated Fibroblast-Mediated CD8 T-cell Exclusion from Tumors. Cancer Res. 2020, 80, 1846–1860. [Google Scholar] [CrossRef]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. JNCI: J. Natl. Cancer Inst. 2017, 110, 109–120. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, D.P.; Bhatt, L.; Woolley, J.F.; Gough, D.R.; Wang, J.H.; Cotter, T.G.; Redmond, H.P. TLR-4 Signalling Accelerates Colon Cancer Cell Adhesion via NF-κB Mediated Transcriptional Up-Regulation of Nox-1. PLoS ONE 2012, 7, e44176. [Google Scholar] [CrossRef]
- Raad, H.; Serrano-Sanchez, M.; Harfouche, G.; Mahfouf, W.; Bortolotto, D.; Bergeron, V.; Kasraian, Z.; Dousset, L.; Hosseini, M.; Taieb, A.; et al. NADPH Oxidase-1 Plays a Key Role in Keratinocyte Responses to UV Radiation and UVB-Induced Skin Carcinogenesis. J. Investig. Dermatol. 2017, 137, 1311–1321. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.-Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e1156. [Google Scholar] [CrossRef]
- Buck, T.; Hack, C.T.; Berg, D.; Berg, U.; Kunz, L.; Mayerhofer, A. The NADPH oxidase 4 is a major source of hydrogen peroxide in human granulosa-lutein and granulosa tumor cells. Sci. Rep. 2019, 9, 3585. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Elbatreek, M.H.; Mucke, H.; Schmidt, H. NOX Inhibitors: From Bench to Naxibs to Bedside. Handb. Exp. Pharm. 2021, 264, 145–168. [Google Scholar] [CrossRef]
- Reis, J.; Massari, M.; Marchese, S.; Ceccon, M.; Aalbers, F.S.; Corana, F.; Valente, S.; Mai, A.; Magnani, F.; Mattevi, A. A closer look into NADPH oxidase inhibitors: Validation and insight into their mechanism of action. Redox Biol. 2020, 32, 101466. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Chung, C.H.; Huh, J.H.; Lee, S.H. 2033-P: APX-115, a Pan-NADPH Oxidase (NOX) Inhibitor, Ameliorates Diabetic Nephropathy in NOX5 Transgenic Mice. Diabetes 2019, 68, 2033-P. [Google Scholar] [CrossRef]
- Urner, S.; Ho, F.; Jha, J.C.; Ziegler, D.; Jandeleit-Dahm, K. NADPH Oxidase Inhibition: Preclinical and Clinical Studies in Diabetic Complications. Antioxid. Redox Signal. 2020, 33, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.L.; Xiang, X.H.; Chen, K.; Xu, W.; Xia, S.H. Potential role of NADPH oxidase in pathogenesis of pancreatitis. World J. Gastrointest Pathophysiol. 2014, 5, 169–177. [Google Scholar] [CrossRef]
- Hirano, K.; Chen, W.S.; Chueng, A.L.; Dunne, A.A.; Seredenina, T.; Filippova, A.; Ramachandran, S.; Bridges, A.; Chaudry, L.; Pettman, G.; et al. Discovery of GSK2795039, a Novel Small Molecule NADPH Oxidase 2 Inhibitor. Antioxid. Redox Signal. 2015, 23, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.-Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 Inhibitor GKT137831 Attenuates Hypoxia-Induced Pulmonary Vascular Cell Proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef]
- Gorin, Y.; Cavaglieri, R.C.; Khazim, K.; Lee, D.-Y.; Bruno, F.; Thakur, S.; Fanti, P.; Szyndralewiez, C.; Barnes, J.L.; Block, K.; et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Ren. Physiol. 2015, 308, F1276–F1287. [Google Scholar] [CrossRef]
- Ostrowski, R.P.; Tang, J.; Zhang, J.H. Hyperbaric Oxygen Suppresses NADPH Oxidase in a Rat Subarachnoid Hemorrhage Model. Stroke 2006, 37, 1314–1318. [Google Scholar] [CrossRef]
- Liu, W.; Sood, R.; Chen, Q.; Sakoglu, U.; Hendren, J.; Çetin, Ö.; Miyake, M.; Liu, K.J. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J. Neurochem. 2008, 107, 1196–1205. [Google Scholar] [CrossRef]
- Kochanski, R.; Peng, C.; Higashida, T.; Geng, X.; Hüttemann, M.; Guthikonda, M.; Ding, Y. Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: An inhibitory effect on hyperglycolysis and NADPH oxidase activation. J. Neurochem. 2013, 126, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.-C.; Fontayne, A.; Hakim, J.; El Benna, J.; Périanin, A. Protein Kinase C ζ Phosphorylates a Subset of Selective Sites of the NADPH Oxidase Component p47phox and Participates in Formyl Peptide-Mediated Neutrophil Respiratory Burst. J. Immunol. 2001, 166, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H.; Gaur, S.; Markel, S.; Lu, J.; van Balgooy, J.; Synold, T.W.; Xi, B.; Wu, X.; Juhasz, A. Effects of iodonium-class flavin dehydrogenase inhibitors on growth, reactive oxygen production, cell cycle progression, NADPH oxidase 1 levels, and gene expression in human colon cancer cells and xenografts. Free. Radic. Biol. Med. 2013, 57, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Stolk, J.; Hiltermann, T.J.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am. J. Respir. Cell Mol. Biol. 1994, 11, 95–102. [Google Scholar] [CrossRef]
- Liu, Z.; Tuo, Y.-H.; Chen, J.-W.; Wang, Q.-Y.; Li, S.; Li, M.-C.; Dai, G.; Wang, J.-S.; Zhang, Y.-L.; Feng, L.; et al. NADPH oxidase inhibitor regulates microRNAs with improved outcome after mechanical reperfusion. J. NeuroInterventional Surg. 2017, 9, 702–706. [Google Scholar] [CrossRef]
- Jaquet, V.; Marcoux, J.; Forest, E.; Leidal, K.G.; McCormick, S.; Westermaier, Y.; Perozzo, R.; Plastre, O.; Fioraso-Cartier, L.; Diebold, B.; et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011, 164, 507–520. [Google Scholar] [CrossRef]
- Cifuentes-Pagano, E.; Csanyi, G.; Pagano, P.J. NADPH oxidase inhibitors: A decade of discovery from Nox2ds to HTS. Cell Mol. Life Sci. 2012, 69, 2315–2325. [Google Scholar] [CrossRef]
- Smith, S.M.E.; Min, J.; Ganesh, T.; Diebold, B.; Kawahara, T.; Zhu, Y.; McCoy, J.; Sun, A.; Snyder, J.P.; Fu, H.; et al. Ebselen and Congeners Inhibit NADPH Oxidase 2-Dependent Superoxide Generation by Interrupting the Binding of Regulatory Subunits. Chem. Biol. 2012, 19, 752–763. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.-H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, J.; Zhang, S.; Wu, W.; Qi, R. Aspirin Inhibits the Production of Reactive Oxygen Species by Downregulating Nox4 and Inducible Nitric Oxide Synthase in Human Endothelial Cells Exposed to Oxidized Low-density Lipoprotein. J. Cardiovasc. Pharmacol. 2012, 59, 405–412. [Google Scholar] [CrossRef]



| Drug | Possible Mechanism of Action | Impact on NOX | Experimental Model | References |
|---|---|---|---|---|
| Normobaric oxygen (NBO) and Hyperbaric oxygen (HBO) | Unknown | NOX2 levels and activity downregulation | Male Sprague–Dawley rats | [281,282] |
| Ethanol | Unknown | Reduced activity enzyme and gp91 expression | Rat model of ischemia | [283] |
| PKC inhibitors (Calphostin C, Chelerythrine and Ruboxistaurin mesylate) | Inhibiting PKC mediated p47phox phosphorylation | Reduced activity of NOX | Human polymorphonuclear leukocytes | [284] |
| Diphenylene iodonium (DPI) | Forms redox adduct with the NOX catalytic core | Reduced NOX 1 expression | Human colon carcinoma cells in vivo and in vitro | [285] |
| Apocynin (4-hydroxy-3methoxy-acetophenone), natural compound) | Blocks p47phox cell membrane migration and NOX assembly | Inhibit assembly and activity of NOX1 and NOX2 | Human neutrophils | [286] |
| VAS2870 (Vasopharm), | Upregulation of NOX2 and 4 targeting microRNA | Reduced expression of NOX2 and Nox 4 | Rat model of ischemia | [287] |
| Celastrol | Disrupt NOX enzyme assembly | Binds to p47phox and disrupted p22phox | Human neutrophils, HEK293 and CHO in vitro and cell free assays | [288] |
| NOX peptide inhibitors (e.g gp91ds-tat) | Disrupt NOX enzyme assembly | Bind with NOX subunits disrupting NOX assembly | Cell free and cell-based assays | [289] |
| GSK2795039 | Compete for NADPH binding site | Specifically inhibit NOX2 by excluding NADPH binding | Cell based, cell-free assays and animal model of acute pancreatitis | [277] |
| Ebselen and analogue | Blocks p47phox cell membrane migration and NOX assembly | Inhibits NOX1 and NOX2 assembly | Human neutrophil and cell-free assays | [290] |
| GKT137831 (Phase 2 clinical trials for diabetic complications) | Direct interaction with NOX complex | Inhibits NOX1/4 activity | Cell-free assays and animal models | [268,291] |
| APX115 (Phase 1 clinical trials) (Phase 2 clinical trials for Covid-19) | Unknown | Decreases the expression of NOX1-3 proteins | Diabetic mouse model | [278] |
| Aspirin (FDA Approved drug) | Unknown | Lowers Nox 4 enzyme | Human endothelial cells | [292] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannan, A.; Germon, Z.P.; Chamberlain, J.; Sillar, J.R.; Nixon, B.; Dun, M.D. Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants 2021, 10, 1616. https://doi.org/10.3390/antiox10101616
Mannan A, Germon ZP, Chamberlain J, Sillar JR, Nixon B, Dun MD. Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants. 2021; 10(10):1616. https://doi.org/10.3390/antiox10101616
Chicago/Turabian StyleMannan, Abdul, Zacary P. Germon, Janis Chamberlain, Jonathan R. Sillar, Brett Nixon, and Matthew D. Dun. 2021. "Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses" Antioxidants 10, no. 10: 1616. https://doi.org/10.3390/antiox10101616
APA StyleMannan, A., Germon, Z. P., Chamberlain, J., Sillar, J. R., Nixon, B., & Dun, M. D. (2021). Reactive Oxygen Species in Acute Lymphoblastic Leukaemia: Reducing Radicals to Refine Responses. Antioxidants, 10(10), 1616. https://doi.org/10.3390/antiox10101616