1. Introduction
The transcription factor p53 is a key player in the cellular stresses response and is activated by DNA damage, and by oncogenic, oxidative and metabolic stress [
1]. p53 activation triggers cell cycle arrest and apoptosis, but is also involved in cellular survival programs through the induction of DNA damage repair and metabolic regulation. Collectively, these programs contribute to the maintenance of genome integrity and protect the organism from over-proliferation of cells that carry oncogenic mutations. p53 stabilization and function are controlled by post-translational modifications (PTMs) such as phosphorylation, acetylation, ubiquitination and methylation that may vary depending on the type of stress [
2].
Reactive oxygen species (ROS), mainly in the form of hydrogen peroxide (H
2O
2), act as a second messenger in so-called redox signaling, which involves oxidative modification of cysteine thiol side chains to regulate the function of target proteins [
3,
4]. Oxidation of thiols to form sulfenic acid (S-OH) or disulfide (S-S-) is reversible by the cellular antioxidant system, enabling to switch the redox signal on and off. The extent of cysteine oxidation in the cellular proteome therefore depends on the rates of production and clearance of ROS and the rates of oxidation and reduction in thiols. A particular attractive mode of redox modification is the formation of intermolecular disulfides, because it can stabilize otherwise weak protein–protein interactions. In this way, protein function can be modified to an extent which correlates with the local redox environment. Intermolecular disulfide formation has been shown to play roles in signaling in species from yeast to human [
5].
p53 has also been found to be oxidized on multiple cysteines upon oxidant treatment, both in vitro and in live cells. In vitro, C182 and C277 were identified to be reactive to the alkylating agent N-ethylmaleimide [
6]. C182 can also form an intramolecular disulfide bond with one of the three Zinc-binding cysteines (C176, 238 and 242), resulting in the loss of Zinc and protein unfolding [
7]. Consistently, Held et al. quantified the extent of site-specific reversible cysteine oxidation in endogenous p53 and found that both C182 and C277 were sensitive to the thiol oxidant diamide [
8], but the exact type of reversible cysteine oxidation remained unknown. Here, we set out to study whether p53 forms disulfide-dependent intermolecular interactions upon oxidation. To this end we combined immunoprecipitation and quantitative Mass spectrometry on wild type and p53 cysteine mutants expressed in HEK293T cells to identify redox-dependent interaction partners of p53. Intriguingly, in line with the observations by Held et al., diamide but not H
2O
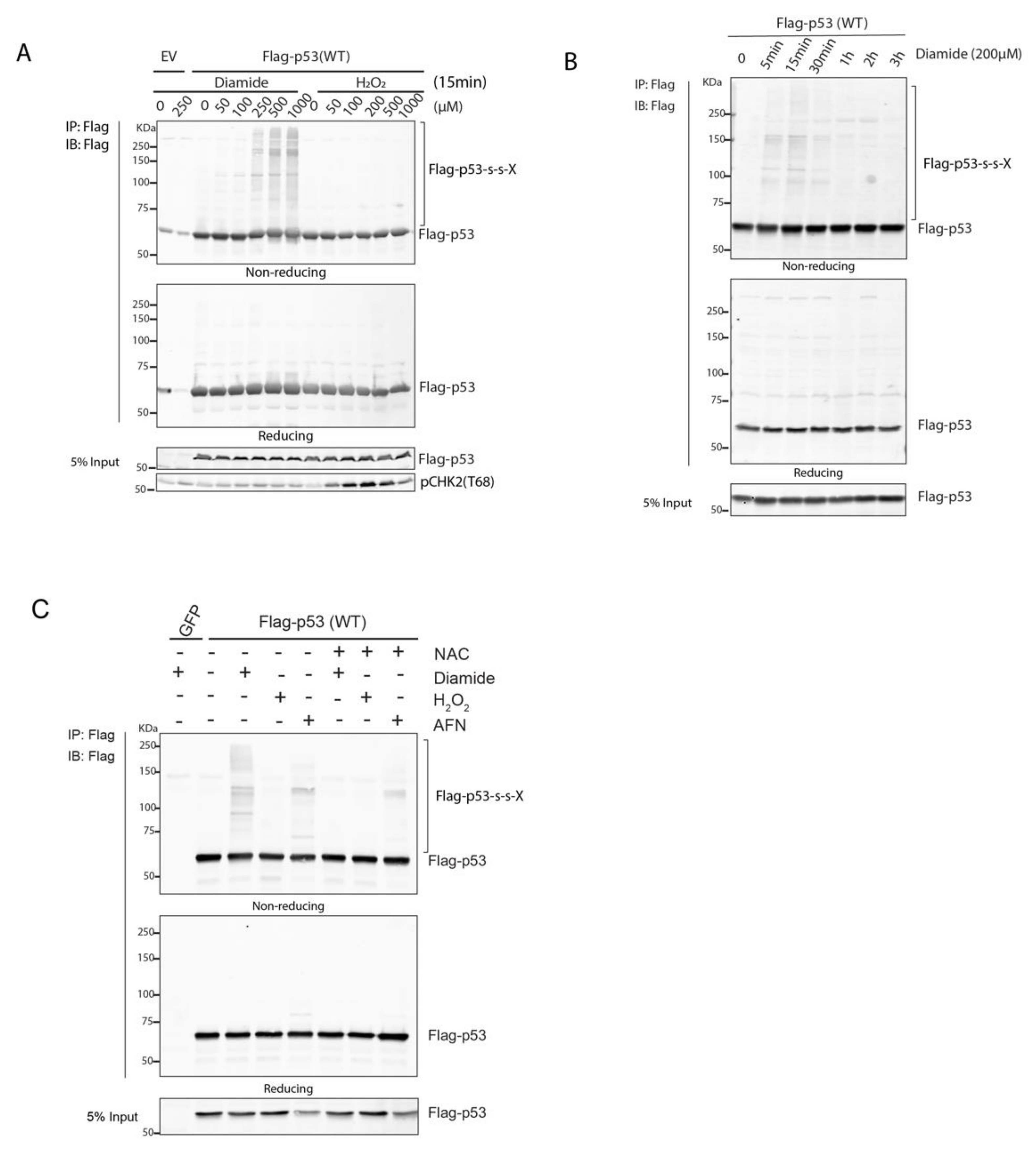
2 induced oxidative stress, which stimulated the formation of disulfide-dependent complexes with p53. Some well-known p53 regulators were among the identified disulfide-dependent binders, including 14-3-3θ and 53BP1, and these depended on the presence of C277. Nevertheless, the p53 277S mutant was still activated by Nutlin-3 treatment, oxidative signaling and DNA damage, suggesting that the identified disulfide-dependent interactors are not critical for p53 function per se. We propose that the observed covalent interactions with p53 could be involved in fine tuning the spatiotemporal p53 response by stabilization otherwise weak protein–protein interactions under oxidizing conditions.
2. Materials and Methods
2.1. Constructs, Reagents and Antibodies
The pDONR223-p53 WT plasmid was a gift from Jesse Boehm, William Hahn and David Root (Addgene plasmid # 81754 [
9]). pDON223-p53 Cysteine mutants (Cys to Ser or Ala) were generated by site-directed mutagenesis PCR using pDONR223-p53 WT as the template. The primers used for mutagenesis PCR are shown in
Table S1. N-terminally tagged Flag- and HA-p53 expression as well as doxycycline-inducible Flag-p53 WT and -C277S constructs were obtained by a Gateway cloning with pcDNA3 or pInducer20 backbones (pInducer20 was a gift from Stephen Elledge (Addgene plasmid # 44012) [
10]).
Diamide(D3648), hydrogen peroxide solution 30% (7722-84-1), Neocarzinostatin (NCS)(N9162), Auranofin (AFN) (A6733) and N-Ethylmaleimide (NEM)(E3876) were from Sigma. Nutlin-3(10004372) was from Sanbio. The 14-3-3θ siRNA(sc-29586) was from Santa Cruz Biotechnology (Dallas, TX, USA).
Anti-Flag®M2 affinity gel (A220), anti-HA agarose (A2095), Anti-FLAG®(rabbit)(F7425) and anti-FlagM2 antibody (F1804) were from Sigma-Aldrich (Burlington, MA, USA). Antibodies against p53(DO-1), p21(M-19), 53BP1(H-300) and 14-3-3θ (5J20) were from Santa Cruz Technology (Dallas, TX, USA). Anti-pCHK2(Thr68) (CS2661) antibody was from Cell Signaling Technology (Danvers, MA, USA). Anti-GAPDH (MAB374) antibody was from EMD Millipore (Burlington, MA, USA). Anti-HA (12CA5) antibody was prepared in-house from hybridoma cell lines. Goat anti-mouse IgG-HPR (170-6516) and Goat anti-rabbit IgG-HRP (170-6515) were form Bio-Rad (Hercules, CA, USA). Fluorescence-conjugated secondary antibodies: IRDye 680RD goat anti-mouse IgG (925-68070), IRDye 800CW goat anti-mouse IgG (926-32210), IRDye 680 goat anti-rabbit IgG (926-32221) and IRDye 800CW goat anti-rabbit IgG (926-32211) from Li-Cor (Lincoln, NE, USA).
2.2. Cell Culture
HEK293T, non-small cell lung cancer cells (NCI-H1299) (p53-deficient) cells were cultured in DMEM high-glucose (4,5g/L) containing 10% FBS, 2mM L-glutamine and 100 Units Penicillin-Streptomycin (All from Sigma Aldrich, Burlington, MA, USA), under a 6% CO
2 atmosphere and at 37 °C. Transient transfections were performed using the polyethyleneimine (PEI) transfection reagent (Sigma Aldrich, Burlington, MA, USA). p53 KO RPE
Tert cells were a gift from René Medema [
11], and cultured in DMEM/F-12 high-glucose supplemented with 10 % FBS and 100 U Penicillin-Streptomycin (Sigma Aldrich, Burlington, MA, USA) under a 6% CO
2 atmosphere and at 37 °C. Doxycycline-inducible expression of Flag-p53 WT and C277S cells was generated by transduction with lentiviral constructs pInducer20-Flag-p53 WT and C277S in the p53-KO RPE
Tert cells, followed by the selection by Neomycin (400 µg/mL) for two weeks. The dox-inducible expression of Flag-p53 was confirmed by Western blot detection and polyclonal cells were used for subsequent experiments.
2.3. Cell Lysis, Immunoprecipitation and Western Blot
For total lysates, cells seeded in 6-well dishes were directly scraped in loading sample buffer (Tris-HCI pH 6.8, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, 0.002% bromophenol blue). For immunoprecipitation experiments, HEK293T or H1299 cells were seeded in 10 cm dishes and transiently transfected with the indicated constructs. 48 h after transfection, cells were treated with diamide or H2O2 for the indicated time, followed by incubation with 100 mM N-ethylmaleimide (NEM) in PBS at 37 °C for 5 min to prevent post-lysis oxidation and to inactivate disulfide-reducing enzymes. Cells were scraped in the same NEM buffer and collected by centrifugation at 1500 rpm for 5 min. Cell pellets were resuspended in 1mL of lysis buffer containing 50 mM Tris pH 7.5, 1% Triton, 1.5 mM MgCl2, 1 mM EDTA, 100 mM NaCl supplemented with Aprotinin, Leupeptin, NaF and 100 mM Iodoacetamide to further prevent post-lysis oxidation.
Cell lysates were subsequently centrifuged at 14,000 rpm for 10 min. 50 µL of supernatants were taken as a control (‘input’) and the rest was used for immunoprecipitation and incubated with 15 µL of anti-FlagM2 or HA Affinity beads. After 2 h of incubation at 4 °C, beads were washed with wash buffer (50 mM Tris pH 7.5, 1% Triton, 1.5 mM MgCl2, 1 mM EDTA, 1 M NaCl supplemented with Aprotinin, Leupeptin and NaF) three times to minimize non-specific binding and enrich for disulfide-dependent interactions. After washing, samples were firstly resuspended in 1× non-reducing sample buffer (without β mercaptoethanol) and boiled at 95 °C for 10 min. Half of the samples were loaded for non-reducing sample detection and the rest half was added 5× reducing buffer (with β mercaptoethanol), boiled again at 95 °C for 5 min and loaded for reducing sample detection.
For SDS-PAGE followed by Western blot, samples were run on 7.5 % or 10 % SDS-PAGE gels depending on the molecular weight of the proteins of interest. After that, proteins were transferred to a PVDF (polyvinylidene difluoride), nitrocellulose or immobilon-FL membrane (Millipore, Burlington, MA, USA) through a traditional wet transfer method. Membranes were blocked with 2% BSA in TBST for 1 h at 4 °C and then incubated with primary antibodies overnight at 4 °C, followed by washing with TBST solution before secondary antibody staining. Secondary antibody staining was performed using HRP or fluorescence-conjugated secondary antibodies for 1h at 4 °C. For imaging, membranes were washed again with TBST and subsequently analyzed on a FujiFilm LAS-3000 Luminescent Image Analyzer (for HRP) (Tokyo, Japan) or Amersham™ Typhoon™ Biomolecular Imager (GE Healthcare, Chicago, IL, USA) for fluorescence.
2.4. Sample Preparation for Mass Spectrometry
HEK293T cells were seeded in 15 cm dishes (4 replicates per condition) and transfected with 20 µg of Flag-p53, Flag-C182S,C277S or Flag-C182S DNA constructs. After 48 h, cells were treated with diamide for 15 min, followed by incubation with 100 mM NEM in PBS at 37 °C for 5 min to alkylate free thiols and prevent post-lysis oxidation. Cells were scraped in NEM buffer, and all replicates were collected in the same 15 mL tube followed by centrifugation at 1500 rpm for 5 min. Cell pellets were lysed in 8 mL of lysis buffer as describe in
Section 2.3 and 1% of supernatant was taken as input. 80 µL of FlagM2 agarose beads were taken for immunoprecipitation against Flag following the procedure as described above in
Section 2.3. After final cleaning of the beads, 1% beads solution was taken for Western blotting detection pre-MS and the rest was used for MS experiment. Proteins on beads were incubated with reduction and alkylation buffer (1 M Ammonium bicarbonate, 50 mM Acetonitrile, 10 mM TCEP, 40 mM CAA and 8M urea) at room temperature for 30 min, and then digested with 250 ng of trypsin overnight at 37 °C on a shaker. Peptides were then loaded on C18 StageTips and washed twice with 0.1% formic acid solution (diluted in water). Peptides on C18 StageTips are stable and can be stored at 4 °C up to one month.
2.5. Mass Spectrometry
Mass spectrometry was performed as previously described [
12]. Briefly, peptides were separated on a 30 cm pico-tip column (75 µm ID, New Objective) and were packed in-house with 3 µm aquapur gold C-18 material (Dr. Maisch) using a 140 min gradient (7–80%ACN, 0.1% FA), delivered by an easy-nLC 1000 (LC 120, Waltham, MA, USA, Thermo Scientific) and electro-sprayed directly into an Orbitrap Fusion Tribrid Mass Spectrometer (LC 120, Waltham, MA, USA, Thermo Scientific). This was then run in data-dependent mode with the resolution of the full scan set at 240,000, after which the top N peaks were selected for HCD fragmentation (30% collision energy) using the top speed option with a cycle time of 1 second with a target intensity of 1E4. The mass spectrometry proteomics data were submitted to ProteomeXchange via the PRIDE database with identifier PXD026893 [
13].
2.6. Mass Spectrometry Data Analysis
The raw mass spectrometry files were processed using Maxquant software (version 1.5.2.8). The human protein database of UniProt was searched with both proteins and peptides (false discovery rate set to 1%). Data analysis regarding the identified proteins was further analyzed in R (version 3.6.1). Proteins were filtered for reverse hits and standard contaminants. Proteins for which less than 2 peptides were identified were also removed. Label-Free-Quantification (LFQ) values were log
2-transformed, and the
proDA (inference of protein differential abundance by probabilistic dropout analysis) model was used to impute missing values following data analysis [
14]. Significant hits between conditions (e.g., CTRL vs. Diamide) were judged by at least 2-fold change in protein abundance with an adjusted
p-value (Benjamini-Hochberg) smaller than 0.01. The
ggplot2 package was used to plot the data. The R scripts, raw and processed data are deposited in
https://github.com/Taoshi2021/p53-oxidation (deposited on 17 June 2021).
2.7. Immunofluorescence Microscopy
RPETert p53 KO cells expressing Doxycycline-inducible Flag-p53 WT and C277S were grown on glass coverslips in 6-well dishes and treated with Dox for 48 h. The cells were then fixed with 3.7% Formaldehyde solution at room temperature for 15 min, followed by permeabilization using 0.1% Triton for 5 min and subsequent blocking with 2% BSA (w/v) and purified goat IgG in 1:10,000 in PBS for 45 min at room temperature. The cells were then incubated with the primary antibody DO-1 against p53 at a final 1:500 dilution overnight, followed by 1 h incubation with a secondary antibody conjugated with Alexa fluor 488 (ThermoFisher Scientific, Waltham, MA, USA) and Hoechst 33,342 (Life Technologies, Carlsbad, CA, USA) after washing twice with PBS. All antibody staining was performed at 4 °C and in the dark. Finally, the coverslips were mounted in a drop of mounting medium and saved at 4 °C in the dark for further analysis. Imaging was performed on a Zeiss confocal microscope LSM880 and images were processed in Fiji (ImageJ) software.
2.8. Ubiquitination Assay
HEK293T cells in 10 cm dishes were transiently transfected with the in the text indicated DNA constructs. After 48 h, cells were treated with H2O2 or diamide for 15 min, and then scraped in lysis buffer (100 mM NaH2PO4/Na2HPO4, 10 mM Tris, 8 M Urea, 10 mM NEM, 10 mM Imidazole and 0.2% Triton X-100, pH 8.0). Cell lysates were sonicated and then centrifuged at 10,000 rpm for 10 min. 50 µL of supernatant were taken as a control for input and the remainder was subjected to pulldown using Ni-NTA beads to enrich for His-Ubiquitin-tagged proteins. After 2 h of incubation at room temperature, beads were washed twice with wash buffer (100 mM NaH2PO4/Na2HPO4, 10 mM Tris, 8 M Urea, 10 mM Imidazole and 0.2% Triton X-100, pH 6.3), followed by one-time wash with elution buffer (100 mM NaCl, 20% glycerol, 20 mM Tris, 1 mM DTT and 10 mM Imidazole, pH 8.0). Ultimately, samples were resuspended in 1x reducing sample buffer for subsequent analysis.
2.9. RNA Isolation and qPCR
p53 target gene expression was analyzed by qPCR on RNA extracted from Dox-inducible expressing p53 WT and C277S in p53 KO RPE
Tert cells. Total RNA was isolated using a RNeasy kit (QIAGEN, Hilden, Germany). 500 ng of RNA was used for cDNA synthesis according to the manufacturer’s instructions using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). qPCR was performed with SYBR Green FastStart Master Mix in the CFX Connect Real-time PCR detection system (Bio-Rad, Hercules, CA, USA). The procedures were as follows: pre-denaturing at 95 °C for 10 min, followed by denaturing at 95 °C for 10 seconds, annealing at 58 °C for 10 s and extending at 72 °C for 30 s for 39 cycles. All the primers used for the qPCRs are shown in
Table S1.
2.10. Sequence Alignment
The p53 protein sequence of vertebrate species and its paralogs (p63 and p73) were downloaded from ENSEMBL database [
15]. Sequence alignment was performed in Jalview (version 15.0) software and was colored by the extent of conservation (threshold 15) [
16]. Cysteines were subsequently colored in orange regardless of conservation status using Adobe Illustrator.
2.11. Gene Ontology Enrichment Analysis
Gene Ontology (GO) enrichment analysis was performed using the online PANTHER Classification System (
http://pantherdb.org/) (accessed on 17 April 2020). 162 proteins that were identified to significantly bind to wild-type p53 upon diamide treatment were selected for GO analysis with the annotation sets of ‘biological process’, ‘molecular function’ and ‘cellular component’. All genes (
Homo sapiens) in the database were used as the reference list.
p value was evaluated by the classic Fisher test, and the value lower 0.001 was a cutoff of significance.
4. Discussion
Oxidation of protein cysteine thiols leads to a suite of PTMs that can reversibly alter protein structure and function. In this way, cysteine oxidation-dependent redox signaling regulates a variety of biological processes including cell proliferation, differentiation, migration and regeneration [
32,
33,
34]. The methods used to detect reversible protein oxidation are in general based on differential alkylation of cysteines prior and post reduction, and hence the type of reversible oxidation (e.g., sulfenic acid,
S-glutathionylation, sulfenamide, inter- or intramolecular disulfide) is lost in this process. Strategies using sequential reduction in specific oxidative PTMs have been used to discriminate for instance proteome-wide
S-GSHylation and
S-nitrosylation [
35] by MS/MS. However, no method exists to date to identify or distinguish intra- or intermolecular disulfides in a proteome-wide manner. The amount of theoretically possible tryptic digests containing peptides from two distinct proteins and an intact disulfide is virtually endless. Intermolecular disulfides can be identified for a protein of interest by first comparing the interactomes of the wildtype protein and a cysteine mutant and subsequently test whether the protein of interest and a cysteine-dependent interactor indeed migrate as a reduction-sensitive complex on SDS-PAGE under non-reducing conditions [
5]. The tumor suppressor p53 had already been reported to undergo reversible cysteine oxidation in response to oxidizing agents both in vitro and at endogenous levels in live cells [
8], but the nature of the reversible oxidation remained elusive in that study. In the present study, we provide evidence that cysteine oxidation of p53 leads to the formation of several intermolecular disulfide-dependent complexes, most of which depend on C277. This cysteine is also implicated in the binding to cysteine-directed covalent drugs aimed at refolding mutant p53 [
36], which means that these compounds could also likely interfere with the intermolecular disulfide-dependent, p53-containing complexes described in this study. Note that the presented MS screen compares the binding of proteins to WT p53 with and without diamide treatment to binders of the C182S and C182S,C277S mutants in the presence of diamide. No changes were observed comparing WT p53 and C182S, whereas many proteins did not bind the C182S,C277S double mutant, but based on the screen we cannot exclude that proteins can bind to either C182 or C277. It is therefore important to validate hits from the MS screen by other means using the single p53 C182S and p53 C277S mutants as well. Similarly, for practical and financial reasons the interactome of p53 cysteine mutants without diamide treatment was not assessed, and validation experiments should include both treated and untreated samples.
A number of disulfide-dependent and validated hits from our MS screen are known interactors and regulators of p53, including MDM2, 53BP1 and 14-3-3. However, the original studies describing the interactions of these proteins with p53 did not study redox or cysteine dependency [
28,
30,
37]. We show that at least for 53BP1 the interaction with p53 is indeed not strictly dependent on the disulfide (
Figure S8). This experiment shows that the disulfide stabilizes the interaction in the co-immunoprecipitation assay and makes it resistant to a stringent high-salt wash. It remains to be seen to what extent this translates in the in vivo situation, and whether this means that in cells the strength or duration of the p53-53BP1 protein–protein interaction is also significantly enhanced as compared with a purely electrostatic interaction upon disulfide formation. The observation that the C277S mutant can still interact with 53BP1 suggests that the p53-53BP1 interaction occurs prior to oxidation, and that the disulfide forms between two cysteines that are already in close proximity. This could be a general concept for the formation of intermolecular disulfides, and potentially explains why p53 does not form random intermolecular disulfides with a wide range of proteins. The disulfide could in this case either strengthen a functional protein–protein interaction or lead to a conformational change that alters the protein–protein interaction in such a way that it interferes with its function. If the latter were the case, one might predict that C277 would maybe not be conserved, similar to C182 and C229, whereas it displays strong evolutionary conservation. On the other hand, if the disulfide-dependent interaction would greatly enhance the regulatory function of an interaction partner we would expect to have observed differences in the transcriptional activity or stability in the C277S mutant, which we did not. The latter might be because multiple proteins with opposing regulatory functions for p53 seem to interact with C277. Since both the C277S and C277A mutants still have transcriptional activity, we can conclude that cysteine oxidation is not absolutely required for p53 function, but that it could maybe provide a means for fine-tuning target selection or the duration of a regulatory response. An alternative function for disulfide formation could be to prevent irreversible oxidation of cysteines in p53, although as far as we know there is no evidence that this actually occurs.
We previously showed that diamide (but not peroxide) -mediated oxidizing conditions induce p53 stabilization and activation through p38MAPK-dependent signaling, which was independent of surface-exposed p53 cysteines (including C277) [
38]. p38MAPK-dependent p53 activation under oxidizing conditions could therefore obscure the effects of disulfide-dependent binding partners under the conditions tested. Furthermore, the majority of WT p53 is still reduced upon diamide treatment, and this could conceal potential regulatory effects of the disulfide-dependent interactions which have a relatively low stoichiometry.
Interestingly, p53 C277 mutations have been identified in several human tumor tissues [
39,
40]. However, since Cys277 is in the DNA-binding domain and actually is in contact with the DNA [
41], it would be difficult to distinguish whether these mutations (the majority of which are changes to large hydrophobic residues) would contribute to oncogenic transformation because of loss of protein–protein interactions or because of altered DNA binding.
Although in the present study we aimed to identify a potential functional role for redox regulation of p53, it might also be that the functional consequence lies ‘at the other end’ of the intermolecular disulfides. The disulfide-mediated p53-53BP1 interaction may for instance alter the efficiency of 53BP1-dependent non-homologous end joining in DNA-damage repair. Likewise, locking p53 to MDM2 may interfere with the ubiquitination-dependent breakdown of MDM2 substrates other than p53. The 14-3-3 proteins also have many more binding partners besides p53, and the intermolecular disulfide-dependent interaction may alter its adaptor function towards other proteins. We have previously shown that the FOXO transcription factors do not bind 14-3-3 proteins in a cysteine-dependent manner [
42], suggesting that not all 14-3-3 interactors bind in a redox-dependent manner. To what extent covalent binding of p53 to these proteins will affect their function depends of course on the stoichiometry of the interaction.
The intrinsic sensitivity for oxidation of cysteine thiols in proteins depends on a number of variables including their pKa, solvent accessibility and local protein folding. Reactivity to H
2O
2, for instance, can vary between several orders of magnitude. It has therefore been proposed that within live cells, oxidation of most cysteines by relatively low levels of H
2O
2 probably occurs indirectly, for instance, catalyzed by peroxiredoxins [
12,
43]. In this study, we observed that disulfide-containing complexes of p53 were only detectable in response to diamide but not H
2O
2 treatment when high glucose media was used. We have previously shown that intermolecular disulfides can be detected in the human 2-Cys peroxiredoxins [
12] as well as the FOXO3 and FOXO4 transcription factors [
24,
42,
44] upon treatment of cells with H
2O
2, starting even at much lower concentrations and cultured in high glucose media. The work by Held et al. showed before that cysteines in endogenous p53 are oxidized by diamide and not by H
2O
2. Furthermore, the diamide-induced oxidation of p53 in live cells occurs at much lower concentrations as compared in vitro on recombinant p53 [
8]. These observations suggest that in live cells, p53 cysteine oxidation also does not occur directly or requires an additional factor or catalyst. Glutathione is, due to its abundance, the most likely direct target of the thiol oxidant diamide, and it might be that in cells, p53 oxidation is mediated by oxidized glutathione, but future work is needed to explore this idea. Alternatively, diamide-induced inhibition of (GSH-dependent) disulfide reduction could expose the continuous turnover of intermolecular disulfides that form between p53 and interacting proteins. The latter could be in line with the observation that under low glucose culturing conditions, or when continuously produced by the addition of glucose oxidase to the media, H
2O
2 does lead to detectable p53 containing intermolecular disulfides. Glucose, through the pentose phosphate pathway, drives production of NADPH which is required for both the GSH- and Trx-dependent disulfide reduction systems. The question remains why inhibition of the reductive system is needed to expose the formation of disulfides upon H
2O
2 treatment for some proteins (e.g., p53) but not others (e.g., FOXOs). In any case, a differential pattern of cysteine oxidation in response to different oxidants is an example of specificity in redox signaling, and it is not unthinkable that a differential cellular response is required upon oxidizing conditions induced by more oxidants (i.e., H
2O
2) or by lower reductive power (i.e., diamide or Auranofin).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}