
Cystine and Methionine Deficiency Promotes Ferroptosis by Inducing B-Cell Translocation Gene 1

 , ,
, ,  ,
,
Abstract
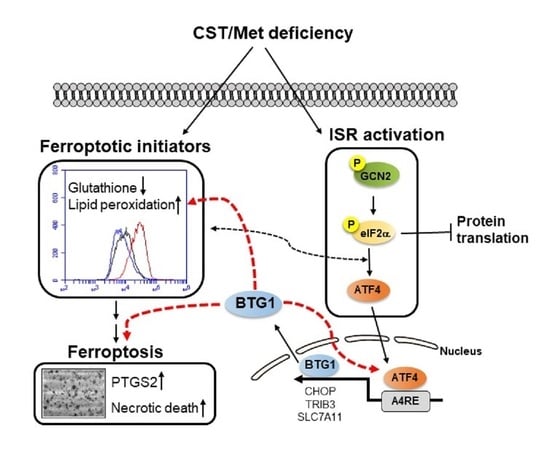
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Treatment
2.3. Measurement of Glutathione
2.4. MTT Assay
2.5. Measurement of Necrotic Cell Death
2.6. Quantitative Polymerase Chain Reaction Analysis
2.7. Measurement of Reactive Oxygen Species and Lipid Peroxidation
2.8. Immunoblot Analysis
2.9. Plasmid Construction, Transient Transfection, and Reporter Gene Assay
2.10. In Silico Analysis of BTG1 Promoter
2.11. Statistical Analysis
3. Results
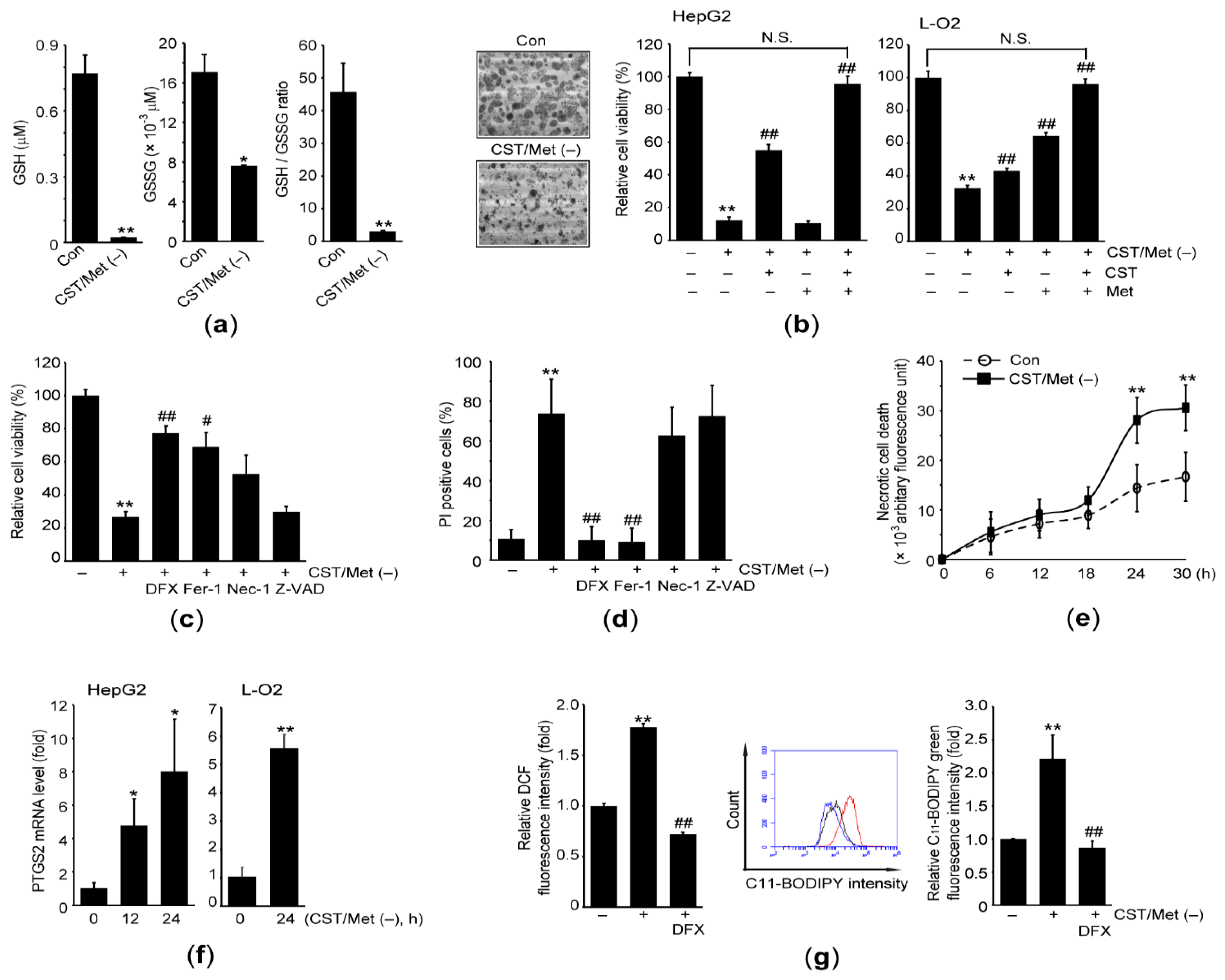
3.1. Cystine and Methionine Deficiency Promotes Ferroptosis in Hepatocyte-Derived Cells
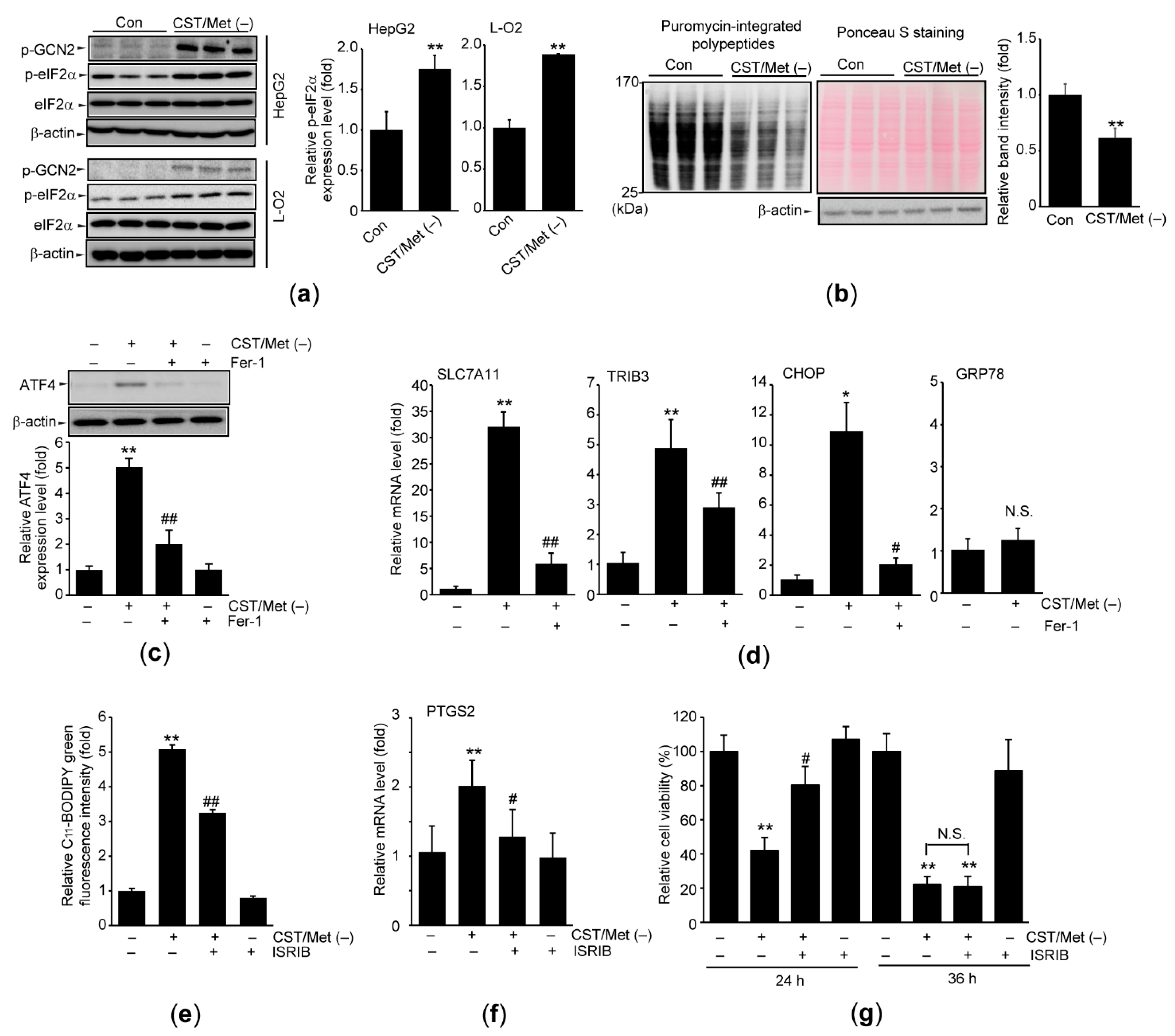
3.2. Cystine and Methionine Deficiency Activates ISR in Hepatocyte-Derived Cells
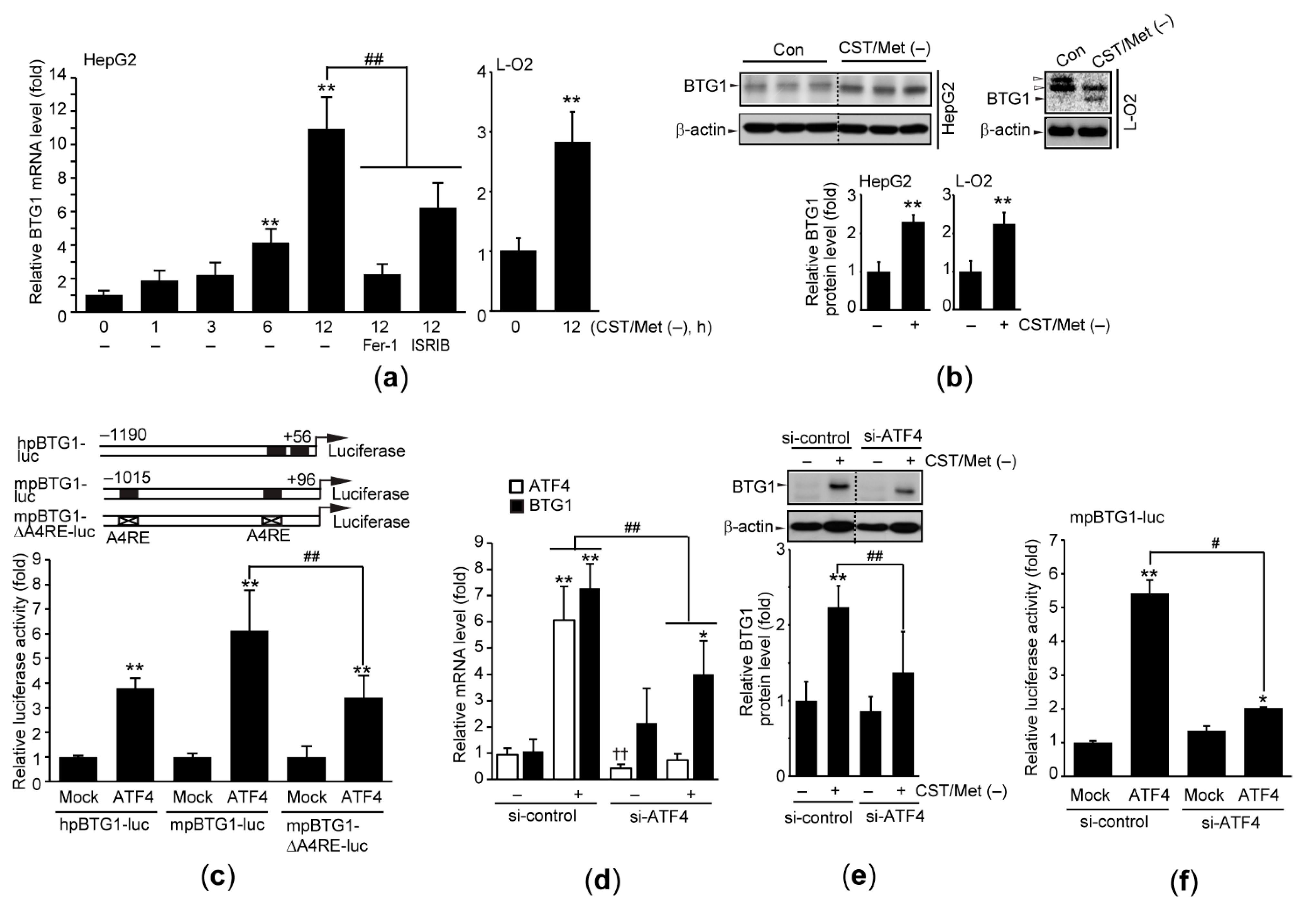
3.3. Cystine and Methionine Deficiency Induces BTG1 in an ATF4-Dependent Manner
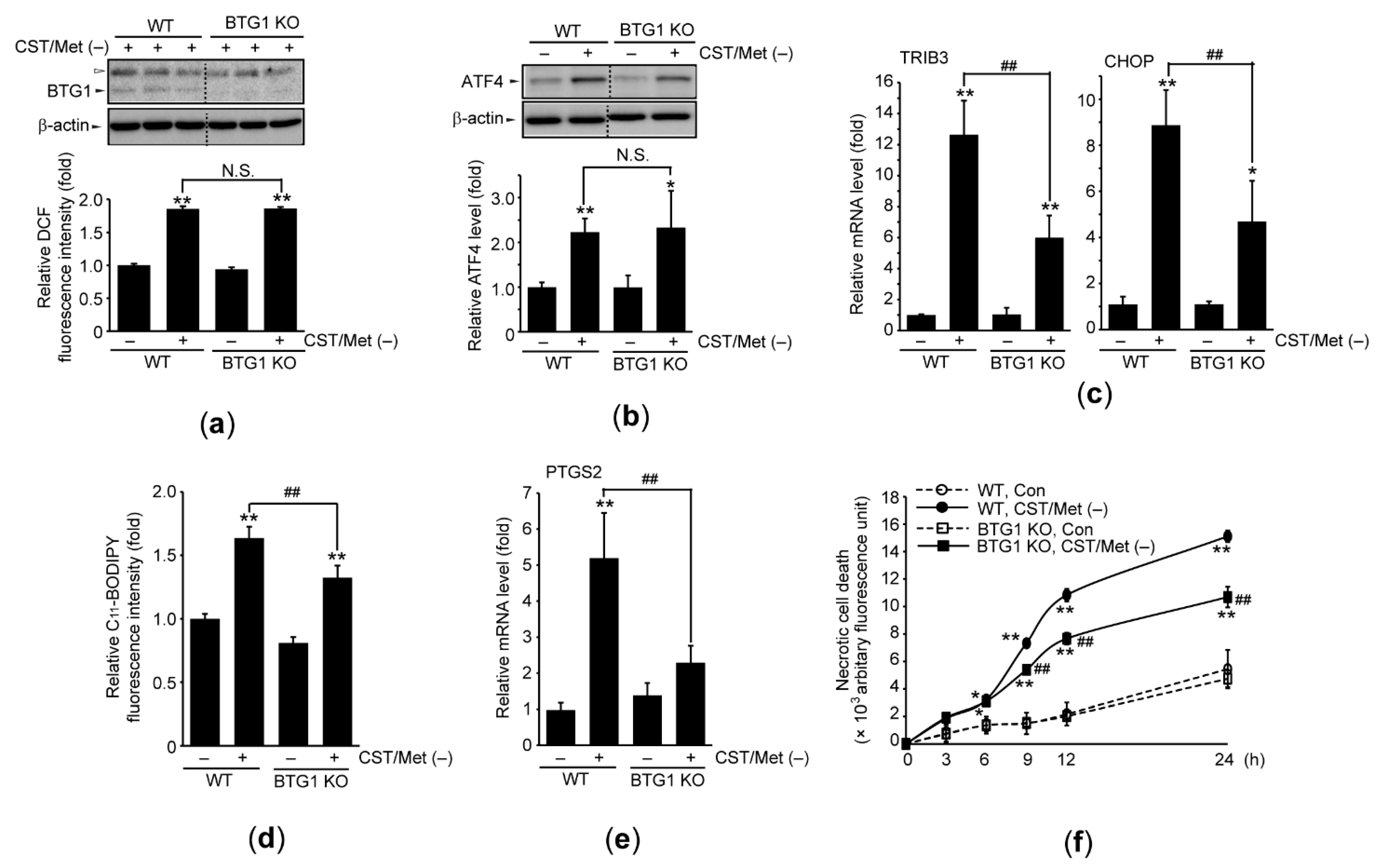
3.4. BTG1 KO Prevents Ferroptosis Induced by Cystine and Methionine Deficiency
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, A.K.; Kang, K.W.; Kim, Y.G.; Cho, M.K.; Lee, M.G.; Shim, C.K.; Chung, S.J.; Kim, S.G. Identification of genes enhanced by protein-calorie malnutrition by differential display polymerase chain reaction (expression of fibrinogen B beta chain, B cell translocation gene 1 and thyroid hormone responsive protein genes). Mol. Cell. Biochem. 2002, 231, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.K.; Kim, Y.G.; Lee, M.G.; Kim, S.G. The effect of cysteine on the altered expression of class alpha and mu glutathione S-transferase genes in the rat liver during protein-calorie malnutrition. Biochim. Biophys. Acta 2000, 1502, 235–246. [Google Scholar] [CrossRef]
- Cho, M.K.; Kim, Y.G.; Lee, M.G.; Kim, S.G. Suppression of rat hepatic cytochrome P450s by protein-calorie malnutrition: Complete or partial restoration by cysteine or methionine supplementation. Arch. Biochem. Biophys. 1999, 372, 150–158. [Google Scholar] [CrossRef]
- Dröge, W. Oxidative stress and ageing: Is ageing a cysteine deficiency syndrome? Philos. Trans. R. Soc. Lond. B 2005, 360, 2355–2372. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Wu, J.; Xu, Y.; Ding, C.C.; Mestre, A.A.; Lin, C.C.; Yang, W.H.; Chi, J.T. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 2021, 12, 198. [Google Scholar] [CrossRef]
- Lin, C.C.; Ding, C.C.; Sun, T.; Wu, J.; Chen, K.Y.; Zhou, P.; Chi, J.T. The regulation of ferroptosis by MESH1 through the activation of the integrative stress response. Cell Death Dis. 2021, 12, 727. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Z.; Tsai, H.I.; Liu, Y.; Gao, J.; Wang, M.; Song, L.; Cao, X.; Xu, Z.; Chen, H.; et al. Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells. Cell Death Differ. 2021, 28, 1222–1236. [Google Scholar] [CrossRef]
- Kim, K.M.; Cho, S.S.; Ki, S.H. Emerging roles of ferroptosis in liver pathophysiology. Arch. Pharm. Res. 2020, 43, 985–996. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef]
- Wang, S.F.; Wung, C.H.; Chen, M.S.; Chen, C.F.; Yin, P.H.; Yeh, T.S.; Chang, Y.L.; Chou, Y.C.; Hung, H.H.; Lee, H.C. Activated Integrated Stress Response Induced by Salubrinal Promotes Cisplatin Resistance in Human Gastric Cancer Cells via Enhanced xCT Expression and Glutathione Biosynthesis. Int. J. Mol. Sci. 2018, 19, 3389. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef]
- Poursaitidis, I.; Wang, X.; Crighton, T.; Labuschagne, C.; Mason, D.; Cramer, S.L.; Triplett, K.; Roy, R.; Pardo, O.E.; Seckl, M.J.; et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep. 2017, 18, 2547–2556. [Google Scholar] [CrossRef]
- Rimokh, R.; Rouault, J.P.; Wahbi, K.; Gadoux, M.; Lafage, M.; Archimbaud, E.; Charrin, C.; Gentilhomme, O.; Germain, D.; Samarut, J.; et al. A chromosome 12 coding region is juxtaposed to the MYC protooncogene locus in a t(8;12)(q24;q22) translocation in a case of B-cell chronic lymphocytic leukemia. Genes Chromosomes Cancer 1991, 3, 24–36. [Google Scholar] [CrossRef]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell. Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [PubMed]
- Yuniati, L.; van der Meer, L.T.; Tijchon, E.; van Ingen Schenau, D.; van Emst, L.; Levers, M.; Palit, S.A.; Rodenbach, C.; Poelmans, G.; Hoogerbrugge, P.M.; et al. Tumor suppressor BTG1 promotes PRMT1-mediated ATF4 function in response to cellular stress. Oncotarget 2016, 7, 3128–3143. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Deng, J.; Guo, Y.; Niu, Y.; Yuan, F.; Yu, J.; Chen, S.; Guo, F. BTG1 ameliorates liver steatosis by decreasing stearoyl-CoA desaturase 1 (SCD1) abundance and altering hepatic lipid metabolism. Sci. Signal. 2016, 9, ra50. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Deng, J.; Yu, J.; Guo, Y.; Chen, S.; Guo, F. A novel function of B-cell translocation gene 1 (BTG1) in the regulation of hepatic insulin sensitivity in mice via c-Jun. FASEB J. 2016, 30, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Busson, M.; Carazo, A.; Seyer, P.; Grandemange, S.; Casas, F.; Pessemesse, L.; Rouault, J.P.; Wrutniak-Cabello, C.; Cabello, G. Coactivation of nuclear receptors and myogenic factors induces the major BTG1 influence on muscle differentiation. Oncogene 2005, 24, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Prévôt, D.; Voeltzel, T.; Birot, A.M.; Morel, A.P.; Rostan, M.C.; Magaud, J.P.; Corbo, L. The leukemia-associated protein Btg1 and the p53-regulated protein Btg2 interact with the homeoprotein Hoxb9 and enhance its transcriptional activation. J. Biol. Chem. 2000, 275, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Rouault, J.P.; Prévôt, D.; Berthet, C.; Birot, A.M.; Billaud, M.; Magaud, J.P.; Corbo, L. Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcriptional regulatory complex. J. Biol. Chem. 1998, 273, 22563–22569. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Sugimoto, H.; Nomoto, S.; Oya, H.; Hibino, S.; Shimizu, D.; Takami, H.; Hashimoto, R.; Okamura, Y.; Yamada, S.; et al. B-cell translocation gene 1 serves as a novel prognostic indicator of hepatocellular carcinoma. Int. J. Oncol. 2015, 46, 641–648. [Google Scholar] [CrossRef][Green Version]
- Lu, S.C.; Huang, H.Y. Comparison of sulfur amino acid utilization for GSH synthesis between HepG2 cells and cultured rat hepatocytes. Biochem. Pharmacol. 1994, 47, 859–869. [Google Scholar] [CrossRef]
- Cho, I.J.; Kim, J.K.; Kim, E.O.; Park, S.M.; Kim, S.C.; Ki, S.H.; Ku, S.K. Hemistepsin A induces apoptosis of hepatocellular carcinoma cells by downregulating STAT3. Int. J. Mol. Sci. 2021, 22, 4743. [Google Scholar] [CrossRef]
- Bakker, W.J.; Blázquez-Domingo, M.; Kolbus, A.; Besooyen, J.; Steinlein, P.; Beug, H.; Coffer, P.J.; Löwenberg, B.; von Lindern, M.; van Dijk, T.B. FoxO3a regulates erythroid differentiation and induces BTG1, an activator of protein arginine methyl transferase 1. J. Cell Biol. 2004, 64, 175–184. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Anand, A.A.; Visser, L.J.; de Groot, R.J.; Walter, P.; van Kuppeveld, F.J.M. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2097–2102. [Google Scholar] [CrossRef] [PubMed]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Zou, W.; Yue, P.; Khuri, F.R.; Sun, S.Y. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008, 68, 7484–7492. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef]
- Wu, M.; Xu, L.G.; Zhai, Z.; Shu, H.B. SINK is a p65-interacting negative regulator of NF-kappaB-dependent transcription. J. Biol. Chem. 2003, 278, 27072–27079. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR Analysis | ||||
| Gene Symbol | Forward Primer | Backward Primer | Accession Number | Product Size (bp) |
| ATF4 | 5′-GTTCTCCAGCGACAAGGCTA-3′ | 5′-TCTCCAACATCCAATCTGTCC-3′ | NM_001675.4 | 119 |
| BTG1 | 5′-AGCGGATTGGACTGAGCAG-3′ | 5′-GGTGCTGTTTTGAGTGCTAC-3′ | NM_001731.2 | 161 |
| CHOP | 5′-GCACCTCCCAGAGCCCTCACTCTCC-3′ | 5′-GTCTACTCCAAGCCTTCCCCCTGCG-3′ | NG_027674.1 | 422 |
| GAPDH | 5′-GAAGGTGAAGGTCGGAGTC-3′ | 5′-GAAGATGGTGATGGGATTTC-3′ | NM_002046.4 | 226 |
| GRP78 | 5′-TGCTTGATGTATGTCCCCTTA-3′ | 5′-CCTTGTCTTCAGCTGTCACT-3′ | NM_005347.5 | 303 |
| PTGS2 | 5′-ATATGTTCTCCTGCCTACTGGA-3′ | 5′-GCCCTTCACGTTATTGCAGAT-3′ | NM_000963.3 | 108 |
| SLC7A11 | 5′-TGCTGGGCTGATTTATCTTCG-3′ | 5′-GAAAGGGCAACCATGAAGAGG-3′ | NM_014331.4 | 114 |
| TRIB3 | 5′-TGTCTTCGCTGACCGTGAGA-3′ | 5′-ACGCGTGCTTGTCCCACAGG-3′ | NM_021158.5 | 101 |
| Construction of Recombinant Plasmids | ||||
| Plasmid Name | Forward Primer | Backward Primer | ||
| hpBTG1-luc | 5′-CTCGAGACCCTGTCTTAGGCCTAATCG-3′ | 5′-AGATCTTCCAGCTCCGCAGCATTCGAA-3′ | ||
| mpBTG1-luc | 5′-CTCGAGGTGGTGTGTATTGCATCTGATGACC-3′ | 5′-AGATCTCACATCGCTCGGACCTCCCCAGCC-3′ | ||
| * mpBTG1- ΔA4RE-luc | 5′-GCCTCGAGGTGGTGTGTGATGACCAACTAAACTC-3′ 5′-GGATGAGAGGGAGGTGCTTGTAAACAAATAAACCCCC-3′ | 5′-GAGTTTAGTTGGTCATCACACACCACCTCGAGGC-3′ 5′-GGGGGTTTATTTGTTTACAAGCACCTCCCTCTCATCC-3′ | ||
| Species | Chromosomal Location | Matrix ID | Score | Relative Score | * Binding Site (bp) | ** Orientation | DNA Sequence |
|---|---|---|---|---|---|---|---|
| Homo sapiens | chr12:92,145,847–92,147,046 | MA0833.1 | 13.566 | 0.9104 | −197 to −185 | – | 5′-AGCTGACGTAATC-3′ |
| MA0833.2 | 10.3809 | 0.8701 | −198 to −185 | – | 5′-AGCTGACGTAATCC-3′ | ||
| MA0833.1 | 11.1549 | 0.8767 | −115 to −103 | – | 5′-TTGTGACGCTTGC-3′ | ||
| Mus musculus | chr10:96,615,806–96,617,005 | MA0833.2 | 10.6486 | 0.8737 | −1013 to −1000 | – | 5′-ATCAGATGCAATAC-3′ |
| MA0833.1 | 9.3852 | 0.8519 | −1012 to −1000 | – | 5′-ATCAGATGCAATA-3′ | ||
| MA0833.1 | 13.566 | 0.9104 | −197 to −185 | – | 5′-AGCTGACGTAATC-3′ | ||
| MA0833.2 | 11.1591 | 0.8806 | −198 to −185 | – | 5′-AGCTGACGTAATCT-3′ | ||
| Rattus norvegicus | chr7:37,811,631–37,812,830 | MA0833.1 | 13.566 | 0.9104 | −516 to −504 | – | 5′-AGCTGACGTAATC-3′ |
| MA0833.2 | 11.1591 | 0.8806 | −517 to −504 | – | 5′-AGCTGACGTAATCT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, I.-J.; Kim, D.; Kim, E.-O.; Jegal, K.-H.; Kim, J.-K.; Park, S.-M.; Zhao, R.; Ki, S.-H.; Kim, S.-C.; Ku, S.-K. Cystine and Methionine Deficiency Promotes Ferroptosis by Inducing B-Cell Translocation Gene 1. Antioxidants 2021, 10, 1543. https://doi.org/10.3390/antiox10101543
Cho I-J, Kim D, Kim E-O, Jegal K-H, Kim J-K, Park S-M, Zhao R, Ki S-H, Kim S-C, Ku S-K. Cystine and Methionine Deficiency Promotes Ferroptosis by Inducing B-Cell Translocation Gene 1. Antioxidants. 2021; 10(10):1543. https://doi.org/10.3390/antiox10101543
Chicago/Turabian StyleCho, Il-Je, Doyeon Kim, Eun-Ok Kim, Kyung-Hwan Jegal, Jae-Kwang Kim, Sang-Mi Park, Rongjie Zhao, Sung-Hwan Ki, Sang-Chan Kim, and Sae-Kwang Ku. 2021. "Cystine and Methionine Deficiency Promotes Ferroptosis by Inducing B-Cell Translocation Gene 1" Antioxidants 10, no. 10: 1543. https://doi.org/10.3390/antiox10101543
APA StyleCho, I.-J., Kim, D., Kim, E.-O., Jegal, K.-H., Kim, J.-K., Park, S.-M., Zhao, R., Ki, S.-H., Kim, S.-C., & Ku, S.-K. (2021). Cystine and Methionine Deficiency Promotes Ferroptosis by Inducing B-Cell Translocation Gene 1. Antioxidants, 10(10), 1543. https://doi.org/10.3390/antiox10101543