The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration


{kind=link}
Abstract
1. Role of Metals and Oxidative Stress in the Local Origin of Neurodegenerative Diseases
2. Melanins Synthesis and Properties
3. Function and Involvement of Neuromelanin in Disease
4. Crosstalk between Neuromelanin and Lipofuscin in Lipid Peroxidation
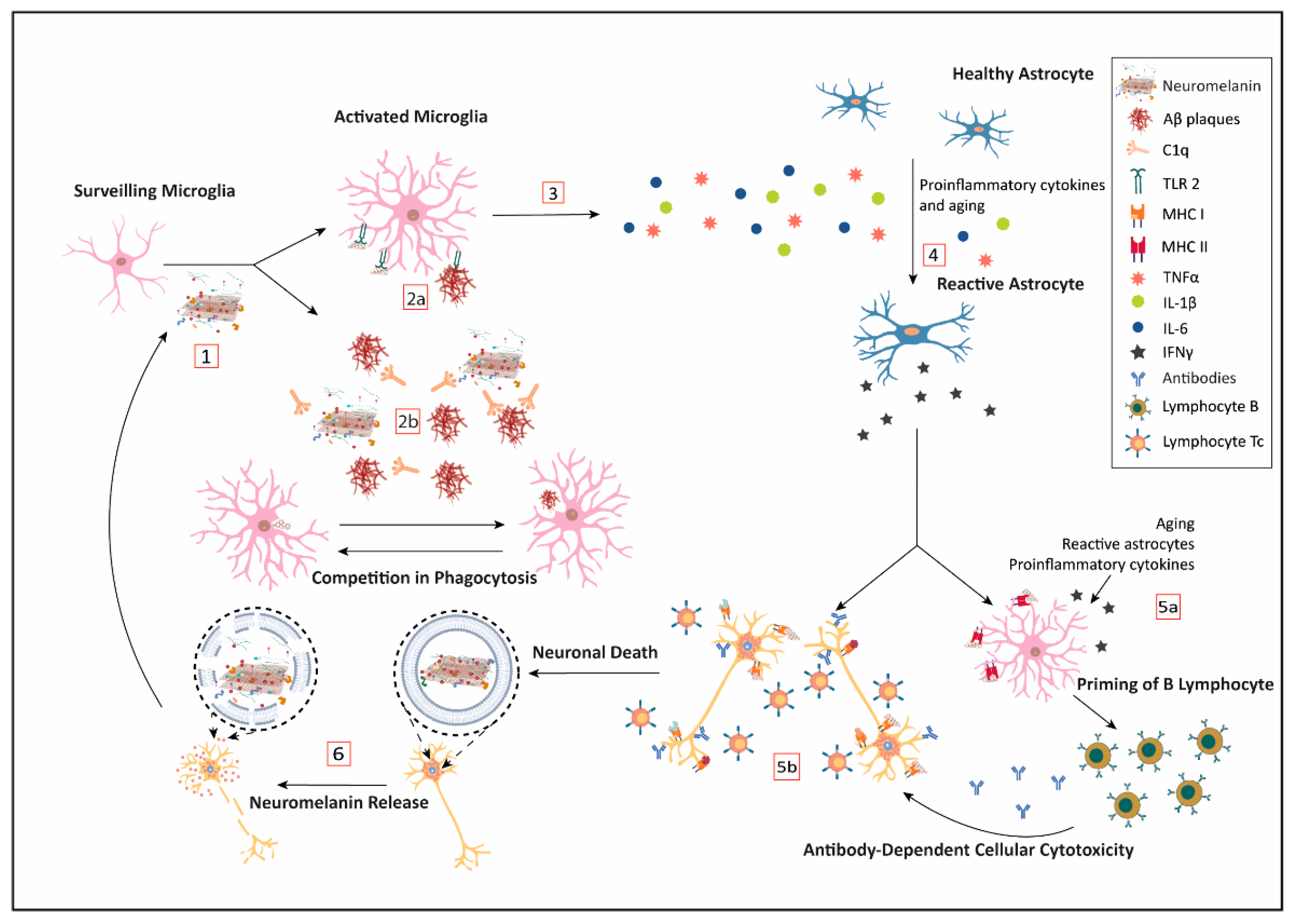
5. Neuromelanin and Immune Response
5.1. Neuromelanin and the Innate Immunity
5.2. Neuromelanin and Acquired Immunity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer´s disease |
| APC | Antigen-presenting cell |
| BAM | Border associated macrophage |
| CNS | Central nervous system |
| DC | Dendritic cell |
| dCLN | Deep cervical lymph node |
| FcγRI | High-affinity IgG receptor |
| IFNγ | Interferon γ |
| IgG | Immunoglobulin G |
| iNOS | Inducible nitric oxygen synthase |
| LC | Locus coeruleus |
| LF | Lipofuscin |
| MHC | Major histocompatibility complex |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NFκB | Nuclear factor κ B |
| NM | Neuromelanin |
| NOD2 | Nucleotide-binding oligomerization domain containing protein 2 |
| PD | Parkinson´s disease |
| PHOX | Phagocyte oxidase |
| PNS | Peripheral nervous system |
| PRR | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| SN | Substantia nigra |
| TLR2 | Toll-like receptor 2 |
| TNFα | Tumor necrosis factor |
References
- Kukull, W.A.; Higdon, R.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Schellenberg, G.D.; van Belle, G.; Jolley, L.; Larson, E.B. Dementia and Alzheimer Disease Incidence: A Prospective Cohort Study. Arch. Neurol. 2002, 59, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 301–307. ISBN 978-0-12-802395-2. [Google Scholar]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative Diseases and Oxidative Stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative Stress and Neurodegeneration: Where Are We Now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative Stress in Neurodegeneration: Cause or Consequence? Nat. Rev. Neurosci. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R. Rise and Fall of Reactive Oxygen Species (ROS): Implications in Aging and Neurodegenerative Disorders. Cell Dev. Biol. 2013, 2. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible to Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Vento, M.; Baquero, M.; Cháfer-Pericás, C. Lipid Peroxidation in Neurodegeneration. Clin. Chim. Acta 2019, 497, 178–188. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free Radicals, Reactive Oxygen Species, Oxidative Stress and Its Classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, J.; Krause, K. Reactive Oxygen Species: From Health to Disease. Swiss Med. Wkly. 2012. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein Oxidation in Aging and Age-Related Diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef]
- Levine, R.L.; Stadtman, E.R. Oxidative Modification of Proteins during Aging. Exp. Gerontol. 2001, 36, 1495–1502. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Kong, Q.; Lin, C.-L.G. Oxidative Damage to RNA: Mechanisms, Consequences, and Diseases. Cell. Mol. Life Sci. 2010, 67, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid Peroxidation Triggers Neurodegeneration: A Redox Proteomics View into the Alzheimer Disease Brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid Peroxidation Products and Their Role in Neurodegenerative Diseases. Ann. Res. Hospit. 2019, 3, 2. [Google Scholar] [CrossRef]
- Muthuraman, A.; Rishitha, N.; Paramakrishnan, N.; Mahendran, B.; Ramesh, M. Role of Lipid Peroxidation Process in Neurodegenerative Disorders. In Lipid Peroxidation Research; Ahmed Mansour, M., Ed.; IntechOpen: London, UK, 2020; ISBN 978-1-83968-547-7. [Google Scholar]
- Reed, T.T. Lipid Peroxidation and Neurodegenerative Disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef]
- Shichiri, M. The Role of Lipid Peroxidation in Neurological Disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Ren, J.-X.; Sun, X.; Yan, X.-L.; Guo, Z.-N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and Cell Death Mechanisms in Parkinson’s Disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Raina, A.K.; Hochman, A.; Zhu, X.; Rottkamp, C.A.; Nunomura, A.; Siedlak, S.L.; Boux, H.; Castellani, R.J.; Perry, G.; Smith, M.A. Abortive Apoptosis in Alzheimer’s Disease. Acta Neuropathologica 2001, 101, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochimica et Biophysica Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Kinowaki, Y.; Kurata, M.; Ishibashi, S.; Ikeda, M.; Tatsuzawa, A.; Yamamoto, M.; Miura, O.; Kitagawa, M.; Yamamoto, K. Glutathione Peroxidase 4 Overexpression Inhibits ROS-Induced Cell Death in Diffuse Large B-Cell Lymphoma. Lab. Investig. 2018, 98, 609–619. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Crichton, R. Inorganic Biochemistry of Iron Metabolism—From Molecular Mechanism to Clinical Consequences, 2nd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2001; 326p. [Google Scholar]
- Dlouhy, A.C.; Outten, C.E. The Iron Metallome in Eukaryotic Organisms. Met. Ions Life Sci. 2013, 12, 241–278. [Google Scholar] [CrossRef]
- Beard, J. Iron Deficiency Alters Brain Development and Functioning. J. Nutr. 2003, 133, 1468S–1472S. [Google Scholar] [CrossRef] [PubMed]
- Todorich, B.; Pasquini, J.M.; Garcia, C.I.; Paez, P.M.; Connor, J.R. Oligodendrocytes and Myelination: The Role of Iron. Glia 2009, 57, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein Carbonylation in Human Diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef]
- Aisen, P.; Enns, C.; Wessling-Resnick, M. Chemistry and Biology of Eukaryotic Iron Metabolism. Int. J. Biochem. Cell Biol. 2001, 33, 940–959. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C.T. Oxidative Stress and Mitochondrial Dysfunction in Neurodegenerative Diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative Stress, Mitochondrial Dysfunction and Neurodegenerative Diseases; a Mechanistic Insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking While the Iron Is Hot: Iron Metabolism and Ferroptosis in Neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- White, A.R.; Kanninen, K.M.; Crouch, P.J. Editorial: Metals and Neurodegeneration: Restoring the Balance. Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Atwood, C.S.; Anderson, V.E.; Siedlak, S.L.; Smith, M.A.; Perry, G.; Carey, P.R. Metal Binding and Oxidation of Amyloid-β within Isolated Senile Plaque Cores: Raman Microscopic Evidence †. Biochemistry 2003, 42, 2768–2773. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Pithadia, A.S.; Lim, M.H. Metal-Associated Amyloid-β Species in Alzheimer’s Disease. Curr. Opin. Chem. Biol. 2012, 16, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Harris, P.L.R.; Sayre, L.M.; Perry, G. Iron Accumulation in Alzheimer Disease Is a Source of Redox-Generated Free Radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Raven, E.P.; Lu, P.H.; Tishler, T.A.; Heydari, P.; Bartzokis, G. Increased Iron Levels and Decreased Tissue Integrity in Hippocampus of Alzheimer’s Disease Detected in Vivo with Magnetic Resonance Imaging. J. Alzheimers Dis. 2013, 37, 127–136. [Google Scholar] [CrossRef]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, Zinc and Copper in the Alzheimer’s Disease Brain: A Quantitative Meta-Analysis. Some Insight on the Influence of Citation Bias on Scientific Opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef]
- Smith, M.A.; Zhu, X.; Tabaton, M.; Liu, G.; McKeel, D.W.; Cohen, M.L.; Wang, X.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased Iron and Free Radical Generation in Preclinical Alzheimer Disease and Mild Cognitive Impairment. J. Alzheimer’s Dis. 2010, 19, 363–372. [Google Scholar] [CrossRef]
- Lucaroni, F.; Ambrosone, C.; Paradiso, F.; Messinese, M.; Di Domenicantonio, R.; Alessandroni, C.; Cerone, G.; Cerutti, F.; Di Gaspare, F.; Morciano, L.; et al. Metals Dyshomeostasis in Alzheimer’s Disease: A Systematic Review. Biomed. Prev. 2017. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content in Postmortem Parkinsonian Brain. Lancet 1987, 330, 1219–1220. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.-P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and Aluminum Increase in the Substantia Nigra of Patients with Parkinson’s Disease: An X-Ray Microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B. Zinc Homeostasis and Neurodegenerative Disorders. Front. Aging Neurosci. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s Disease: Mechanisms and Biochemical Processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Snyder, B.S.; Arosio, P.; Loeffler, D.A.; LeWitt, P. A Quantitative Analysis of Isoferritins in Select Regions of Aged, Parkinsonian, and Alzheimer’s Diseased Brains. J. Neurochem. 1995, 65, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Sofic, E.; Rausch, W.-D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B.H. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Möller, H.E.; Bossoni, L.; Connor, J.R.; Crichton, R.R.; Does, M.D.; Ward, R.J.; Zecca, L.; Zucca, F.A.; Ronen, I. Iron, Myelin, and the Brain: Neuroimaging Meets Neurobiology. Trends Neurosci. 2019, 42, 384–401. [Google Scholar] [CrossRef]
- Biasiotto, G.; Di Lorenzo, D.; Archetti, S.; Zanella, I. Iron and Neurodegeneration: Is Ferritinophagy the Link? Mol. Neurobiol. 2016, 53, 5542–5574. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/Ferroptosis: Iron-related Newcomers in Human Diseases. J. Cell. Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Quiles del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis Is an Autophagic Cell Death Process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Tessari, I.; Mammi, S.; Bubacco, L. Interaction Between α-Synuclein and Metal Ions, Still Looking for a Role in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2009, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Carboni, E.; Lingor, P. Insights on the Interaction of Alpha-Synuclein and Metals in the Pathophysiology of Parkinson’s Disease. Metallomics 2015, 7, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Del Barrio, M.; Borghesani, V.; Hureau, C.; Faller, P. Metal-Binding to Amyloid-β Peptide: Coordination, Aggregation, and Reactive Oxygen Species Production. In Biometals in Neurodegenerative Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 265–281. ISBN 978-0-12-804562-6. [Google Scholar]
- Hane, F.; Leonenko, Z. Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101–116. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Vonsattel, J.-P.; Tanzi, R.E.; Bush, A.I. Zinc-Induced Alzheimer’s Aβ1–40 Aggregation Is Mediated by Conformational Factors. J. Biol. Chem. 1997, 272, 26464–26470. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.R.; Shin, H.-J.; Lee, J.-H.; Chang, C.-S.; Kim, J. Copper(II)-Induced Self-Oligomerization of α-Synuclein. Biochem. J. 1999, 340, 821–828. [Google Scholar] [CrossRef]
- Rana, M.; Kumar Sharma, A. Cu and Zn Interactions with Aβ Peptides: Consequence of Coordination on Aggregation and Formation of Neurotoxic Soluble Aβ Oligomers. Metallomics 2019, 11, 64–84. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-Triggered Structural Transformations, Aggregation, and Fibrillation of Human α-Synuclein a Possible Molecular Link Between Parkinson′s Disease and Heavy Metal Exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Fernández, R.D.; Murgas, C.J.; Heitger, D.R.; Forney, A.K.; Crozier, M.K.; Lucas, H.R. Iron Redox Chemistry Promotes Antiparallel Oligomerization of α-Synuclein. J. Am. Chem. Soc. 2018, 140, 5028–5032. [Google Scholar] [CrossRef]
- Young, T.R.; Pukala, T.L.; Cappai, R.; Wedd, A.G.; Xiao, Z. The Human Amyloid Precursor Protein Binds Copper Ions Dominated by a Picomolar-Affinity Site in the Helix-Rich E2 Domain. Biochemistry 2018, 57, 4165–4176. [Google Scholar] [CrossRef]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-β: A Chameleon Walking in Two Worlds: A Review of the Trophic and Toxic Properties of Amyloid-β. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A Beta Peptide of Alzheimer’s Disease Directly Produces Hydrogen Peroxide through Metal Ion Reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Chevion, M. The Role of Iron in Beta Amyloid Toxicity. Biochem. Biophys. Res. Commun. 1995, 216, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18. [Google Scholar] [CrossRef]
- Solano, F. Melanins: Skin Pigments and Much More—Types, Structural Models, Biological Functions, and Formation Routes. N. J. Sci. 2014, 2014, 1–28. [Google Scholar] [CrossRef]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the Human Substantia Nigra: An Update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Apte, S.P. The Polymerization of Melanin: A Poorly Understood Phenomenon with Egregious Biological Implications. Melanoma Res. 2006, 16, 3–10. [Google Scholar] [CrossRef]
- Ito, S. Reexamination of the Structure of Eumelanin. Biochim. Biophys. Acta 1986, 883, 155–161. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Quantitative Analysis of Eumelanin and Pheomelanin in Humans, Mice, and Other Animals: A Comparative Review. Pigment Cell Res. 2003, 16, 523–531. [Google Scholar] [CrossRef]
- Prota, G. Recent Advances in the Chemistry of Melanogenesis in Mammals. J. Invest. Dermatol. 1980, 75, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Prota, G. Progress in the Chemistry of Melanins and Related Metabolites. Med. Res. Rev. 1988, 8, 525–556. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Soavi, F.; Santato, C. An Electrochemical Study on the Effect of Metal Chelation and Reactive Oxygen Species on a Synthetic Neuromelanin Model. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Panzella, L.; Gentile, G.; D’Errico, G.; Della Vecchia, N.F.; Errico, M.E.; Napolitano, A.; Carfagna, C.; d’Ischia, M. Atypical Structural and π-Electron Features of a Melanin Polymer That Lead to Superior Free-Radical-Scavenging Properties. Angewandte Chemie International Edition 2013, 52, 12684–12687. [Google Scholar] [CrossRef]
- Tran, M.L.; Powell, B.J.; Meredith, P. Chemical and Structural Disorder in Eumelanins: A Possible Explanation for Broadband Absorbance. Biophys. J. 2006, 90, 743–752. [Google Scholar] [CrossRef]
- Prota, G.; Thomson, R.H. Melanin Pigmentation in Mammals. Endeavour 1976, 35, 32–38. [Google Scholar] [CrossRef]
- Engelen, M.; Vanna, R.; Bellei, C.; Zucca, F.A.; Wakamatsu, K.; Monzani, E.; Ito, S.; Casella, L.; Zecca, L. Neuromelanins of Human Brain Have Soluble and Insoluble Components with Dolichols Attached to the Melanic Structure. PLoS ONE 2012, 7, e48490. [Google Scholar] [CrossRef]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New Melanic Pigments in the Human Brain That Accumulate in Aging and Block Environmental Toxic Metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef]
- Carstam, R.; Brinck, C.; Hindemith-Augustsson, A.; Rorsman, H.; Rosengren, E. The Neuromelanin of the Human Substantia Nigra. Biochimica et Biophysica Acta (BBA) Mol. Basis Dis. 1991, 1097, 152–160. [Google Scholar] [CrossRef]
- Ito, S. Encapsulation of a Reactive Core in Neuromelanin. Proc. Natl. Acad. Sci. USA 2006, 103, 14647–14648. [Google Scholar] [CrossRef]
- Bush, W.D.; Garguilo, J.; Zucca, F.A.; Albertini, A.; Zecca, L.; Edwards, G.S.; Nemanich, R.J.; Simon, J.D. The Surface Oxidation Potential of Human Neuromelanin Reveals a Spherical Architecture with a Pheomelanin Core and a Eumelanin Surface. Proc. Natl. Acad. Sci. USA 2006, 103, 14785–14789. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, K.; Nagatsu, I.; Ito, S.; King, R.A.; Nishimura, A.; Nagatsu, T. Does Tyrosinase Exist in Neuromelanin-Pigmented Neurons in the Human Substantia Nigra? Neurosci. Lett. 1998, 253, 198–200. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin Organelles Are Specialized Autolysosomes That Accumulate Undegraded Proteins and Lipids in Aging Human Brain and Are Likely Involved in Parkinson’s Disease. NPJ Parkinson’s Dis. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin Biosynthesis is Driven by Excess Cytosolic Catecholamines Not Accumulated by Synaptic Vesicles. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Plum, S.; Steinbach, S.; Attems, J.; Keers, S.; Riederer, P.; Gerlach, M.; May, C.; Marcus, K. Proteomic Characterization of Neuromelanin Granules Isolated from Human Substantia Nigra by Laser-Microdissection. Sci. Rep. 2016, 6, 37139. [Google Scholar] [CrossRef]
- Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.-N.; Wirtz, T.; Pezzoli, G.; Zucca, F.A.; Zecca, L.; Schraermeyer, U. Elemental Mapping of Neuromelanin Organelles of Human Substantia Nigra: Correlative Ultrastructural and Chemical Analysis by Analytical Transmission Electron Microscopy and Nano-Secondary Ion Mass Spectrometry. J. Neurochem. 2016, 138, 339–353. [Google Scholar] [CrossRef]
- Zecca, L.; Zucca, F.A.; Wilms, H.; Sulzer, D. Neuromelanin of the Substantia Nigra: A Neuronal Black Hole with Protective and Toxic Characteristics. Trends Neurosci. 2003, 26, 578–580. [Google Scholar] [CrossRef]
- Liang, C.-L.; Nelson, O.; Yazdani, U.; Pasbakhsh, P.; German, D.C. Inverse Relationship between the Contents of Neuromelanin Pigment and the Vesicular Monoamine Transporter-2: Human Midbrain Dopamine Neurons. J. Comp. Neurol. 2004, 473, 97–106. [Google Scholar] [CrossRef]
- He, A.-Y.; Qiu, L.-J.; Gao, Y.; Zhu, Y.; Xu, Z.-W.; Xu, J.-M.; Zhang, Z.-H. The Role of Oxidative Stress in Neuromelanin Synthesis in PC12 Cells. Neuroscience 2011, 189, 43–50. [Google Scholar] [CrossRef]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K. Neuromelanin in Human Dopamine Neurons: Comparison with Peripheral Melanins and Relevance to Parkinson’s Disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Hardy, J.; Duff, K.E. Selective Vulnerability in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. Selective Neuronal Death in Neurodegenerative Diseases: The Ongoing Mystery. Yale J. Biol. Med. 2019, 92, 695–705. [Google Scholar]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal Loss Is Greater in the Locus Coeruleus than Nucleus Basalis and Substantia Nigra in Alzheimer and Parkinson Diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal Vulnerability, Pathogenesis, and Parkinson’s Disease. Mov. Dis. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.H.; Riederer, P. The Relevance of Iron in the Pathogenesis of Parkinson’s Disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nakao, K.; Tanaka, H.; Kitahori, Y.; Tanaka, Y.; Ojika, M.; Ito, S. The Oxidative Pathway to Dopamine–Protein Conjugates and Their Pro-Oxidant Activities: Implications for the Neurodegeneration of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 2575. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The Absolute Concentration of Nigral Neuromelanin, Assayed by a New Sensitive Method, Increases throughout the Life and Is Dramatically Decreased in Parkinson’s Disease. FEBS Lett. 2002, 510, 216–220. [Google Scholar] [CrossRef]
- Halliday, G.M.; Fedorow, H.; Rickert, C.H.; Gerlach, M.; Riederer, P.; Double, K.L. Evidence for Specific Phases in the Development of Human Neuromelanin. J. Neural Trans. 2006, 113, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A.; Yates, P.O. The Effects of Ageing on the Pigmented Nerve Cells of the Human Locus Caeruleus and Substantia Nigra. Acta Neuropathol. 1979, 47, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Pigmentation in the Nucleus Substantiae Nigrae of Mammals. J. Anat. 1961, 95, 256–261. [Google Scholar]
- Youssef, S.A.; Capucchio, M.T.; Rofina, J.E.; Chambers, J.K.; Uchida, K.; Nakayama, H.; Head, E. Pathology of the Aging Brain in Domestic and Laboratory Animals, and Animal Models of Human Neurodegenerative Diseases. Vet. Pathol. 2016, 53, 327–348. [Google Scholar] [CrossRef]
- Haining, R.L.; Achat-Mendes, C. Neuromelanin, One of the Most Overlooked Molecules in Modern Medicine, Is Not a Spectator. Neural Regen. Res. 2017, 12, 372–375. [Google Scholar] [CrossRef]
- Vila, M. Neuromelanin, Aging, and Neuronal Vulnerability in Parkinson’s Disease. Mov. Disord. 2019, 34, 1440–1451. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain Tyrosinase Overexpression Implicates Age-Dependent Neuromelanin Production in Parkinson’s Disease Pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef]
- Kastner, A.; Hirsch, E.C.; Lejeune, O.; Javoy-Agid, F.; Rascol, O.; Agid, Y. Is the Vulnerability of Neurons in the Substantia Nigra of Patients with Parkinson’s Disease Related to Their Neuromelanin Content? J. Neurochem. 1992, 59, 1080–1089. [Google Scholar] [CrossRef]
- Karlsson, O.; Lindquist, N.G. Melanin and Neuromelanin Binding of Drugs and Chemicals: Toxicological Implications. Arch. Toxicol. 2016, 90, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Tampellini, D.; Gatti, A.; Crippa, R.; Eisner, M.; Sulzer, D.; Ito, S.; Fariello, R.; Gallorini, M. The Neuromelanin of Human Substantia Nigra and Its Interaction with Metals. J. Neural. Transm. 2002, 109, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Shima, T.; Stroppolo, A.; Goj, C.; Battiston, G.A.; Gerbasi, R.; Sarna, T.; Swartz, H.M. Interaction of Neuromelanin and Iron in Substantia Nigra and Other Areas of Human Brain. Neuroscience 1996, 73, 407–415. [Google Scholar] [CrossRef]
- Zecca, L.; Tampellini, D.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Sulzer, D. Substantia Nigra Neuromelanin: Structure, Synthesis, and Molecular Behaviour. Mol. Pathol. 2001, 54, 414–418. [Google Scholar] [PubMed]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Gerlach, M.; Schünemann, V.; Trautwein, A.X.; Zecca, L.; Gallorini, M.; Youdim, M.B.H.; Riederer, P.; Ben-Shachar, D. Iron-Binding Characteristics of Neuromelanin of the Human Substantia Nigra. Biochem. Pharmacol. 2003, 66, 489–494. [Google Scholar] [CrossRef]
- Lapierre-Landry, M.; Carroll, J.; Skala, M.C. Imaging Retinal Melanin: A Review of Current Technologies. J. Biol. Eng. 2018, 12. [Google Scholar] [CrossRef]
- Dieguez, H.H.; Romeo, H.E.; Alaimo, A.; González Fleitas, M.F.; Aranda, M.L.; Rosenstein, R.E.; Dorfman, D. Oxidative Stress Damage Circumscribed to the Central Temporal Retinal Pigment Epithelium in Early Experimental Non-Exudative Age-Related Macular Degeneration. Free Radic. Biol. Med. 2019, 131, 72–80. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-Related Macular Degeneration (AMD): Alzheimer’s Disease in the Eye? J. Alzheimer’s Dis. 2011, 24, 615–631. [Google Scholar] [CrossRef]
- De Pedro-Cuesta, J.; Rábano, A.; Martínez-Martín, P.; Ruiz-Tovar, M.; Alcalde-Cabero, E.; Almazán-Isla, J.; Avellanal, F.; Calero, M. Comparative Incidence of Conformational, Neurodegenerative Disorders. PLoS ONE 2015, 10, e0137342. [Google Scholar] [CrossRef]
- De Pedro-Cuesta, J.; Martínez-Martín, P.; Rábano, A.; Alcalde-Cabero, E.; José García López, F.; Almazán-Isla, J.; Ruiz-Tovar, M.; Medrano, M.-J.; Avellanal, F.; Calero, O.; et al. Drivers: A Biologically Contextualized, Cross-Inferential View of the Epidemiology of Neurodegenerative Disorders. J. Alzheimer’s Dis. 2016, 51, 1003–1022. [Google Scholar] [CrossRef] [PubMed]
- De Pedro-Cuesta, J.; Martínez-Martín, P.; Rábano, A.; Ruiz-Tovar, M.; Alcalde-Cabero, E.; Calero, M. Etiologic Framework for the Study of Neurodegenerative Disorders as Well as Vascular and Metabolic Comorbidities on the Grounds of Shared Epidemiologic and Biologic Features. Front. Aging Neurosci. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Price, E.R.; Fisher, D.E. Sensorineural Deafness and Pigmentation Genes: Melanocytes and the Mitf Transcriptional Network. Neuron 2001, 30, 15–18. [Google Scholar] [CrossRef]
- Gi, M.; Shim, D.B.; Wu, L.; Bok, J.; Song, M.H.; Choi, J.Y. Progressive Hearing Loss in Vitamin A-Deficient Mice Which May Be Protected by the Activation of Cochlear Melanocyte. Sci. Rep. 2018, 8, 16415. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Cuesta, S.; Contreras, J.; Zurita, E.; Cediel, R.; Cantero, M.; Varela-Nieto, I.; Montoliu, L. Melanin Precursors Prevent Premature Age-Related and Noise-Induced Hearing Loss in Albino Mice. Pigment Cell Melanoma Res. 2010, 23, 72–83. [Google Scholar] [CrossRef]
- Wolff, D. Melanin in the Inner Ear. Arch. Otolaryngol. 1931, 14, 195–211. [Google Scholar] [CrossRef]
- LaFerriere, K.A.; Arenberg, I.K.; Hawkins, J.E.; Johnsson, L.-G. Melanocytes of the Vestibular Labyrinth and Their Relationship to the Microvasculature. Ann. Otol. Rhinol. Laryngol. 1974, 83, 685–694. [Google Scholar] [CrossRef]
- Dubey, S.; Roulin, A. Evolutionary and Biomedical Consequences of Internal Melanins. Pigment Cell Melanoma Res. 2014, 27, 327–338. [Google Scholar] [CrossRef]
- Lin, B.M.; Li, W.-Q.; Curhan, S.G.; Stankovic, K.M.; Qureshi, A.A.; Curhan, G.C. Skin Pigmentation and Risk of Hearing Loss in Women. Am. J. Epidemiol. 2017, 186, 1–10. [Google Scholar] [CrossRef]
- Helzner, E.P.; Cauley, J.A.; Pratt, S.R.; Wisniewski, S.R.; Zmuda, J.M.; Talbott, E.O.; de Rekeneire, N.; Harris, T.B.; Rubin, S.M.; Simonsick, E.M.; et al. Race and Sex Differences in Age-Related Hearing Loss: The Health, Aging and Body Composition Study. J. Am. Geriatr. Soc. 2005, 53, 2119–2127. [Google Scholar] [CrossRef]
- Agrawal, Y.; Platz, E.A.; Niparko, J.K. Prevalence of Hearing Loss and Differences by Demographic Characteristics among US Adults: Data from the National Health and Nutrition Examination Survey, 1999–2004. Arch. Intern. Med. 2008, 168, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.R.; Thorpe, R.; Gordon-Salant, S.; Ferrucci, L. Hearing Loss Prevalence and Risk Factors among Older Adults in the United States. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.R.; Maas, P.; Chien, W.; Carey, J.P.; Ferrucci, L.; Thorpe, R. Association of Skin Color, Race/Ethnicity, and Hearing Loss among Adults in the USA. J. Assoc. Res. Otolaryngol. 2012, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of Formation and Increase with Age. APMIS 1998, 106, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal Pigmented Autophagic Vacuoles: Lipofuscin, Neuromelanin, and Ceroid as Macroautophagic Responses during Aging and Disease. J. Neurochem. 2008, 106, 24–36. [Google Scholar] [CrossRef]
- Marsden, C.D. Oxidative Enzymes Responsible for the Conversion of 3,4-dihydroxyphenylalanine to Melanin in the Small Intestine of Rodents. J. Histochem. Cytochem. 1966, 14, 182–186. [Google Scholar] [CrossRef]
- Swan, G.A. Chemical Structure of Melanins. Ann. N. Y. Acad. Sci. 1963, 100, 1005–1019. [Google Scholar] [CrossRef]
- Goldfischer, S. The Localization of Copper in the Pericanalicular Granules (Lysosomes) of Liver in Wilson’s Disease (Hepatolenticular Degeneration). Am. J. Pathol. 1965, 46, 977–983. [Google Scholar]
- Van Woert, M.H.; Prasad, K.N.; Borg, D.C. Spectroscopic Studies of Substantia Nigra Pigment in Human Subjects. J. Neurochem. 1967, 14, 707–716. [Google Scholar] [CrossRef]
- Barden, H. The Histochemical Relationship of Neuromelanin and Lipofuscin. J. Neuropathol. Exp. Neurol. 1969, 28, 419–441. [Google Scholar] [CrossRef] [PubMed]
- Park, B.E.; Netsky, M.Q.; Betsill, W.L. Pathogenesis of Pigment and Spheroid Formation in Hallervorden-Spatz Syndrome and Related Disorders: Peroxidation as a Common Mechanism. Neurology 1975, 25, 1172. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Merighi, A. The Relationship between Lipofuscin and Neuromelanin in Some Sites of the Nervous System of the Horse. Exp. Biol. 1986, 46, 89–99. [Google Scholar] [PubMed]
- Julien, S.; Biesemeier, A.; Kokkinou, D.; Eibl, O.; Schraermeyer, U. Zinc Deficiency Leads to Lipofuscin Accumulation in the Retinal Pigment Epithelium of Pigmented Rats. PLoS ONE 2011, 6, e29245. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The Comparative Biology of Neuromelanin and Lipofuscin in the Human Brain. Cell. Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of Active Nerve Cell Degeneration in the Substantia Nigra of Humans Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of Microglia by Human Neuromelanin Is NF-KappaB Dependent and Involves P38 Mitogen-Activated Protein Kinase: Implications for Parkinson’s Disease. FASEB J. 2003, 17, 500–502. [Google Scholar] [CrossRef]
- Zecca, L.; Wilms, H.; Geick, S.; Claasen, J.-H.; Brandenburg, L.-O.; Holzknecht, C.; Panizza, M.L.; Zucca, F.A.; Deuschl, G.; Sievers, J.; et al. Human Neuromelanin Induces Neuroinflammation and Neurodegeneration in the Rat Substantia Nigra: Implications for Parkinson’s Disease. Acta Neuropathol. 2008, 116, 47–55. [Google Scholar] [CrossRef]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin Activates Microglia and Induces Degeneration of Dopaminergic Neurons: Implications for Progression of Parkinson’s Disease. Neurotox Res. 2011, 19, 63–72. [Google Scholar] [CrossRef]
- Depboylu, C.; Schäfer, M.K.-H.; Arias-Carrión, O.; Oertel, W.H.; Weihe, E.; Höglinger, G.U. Possible Involvement of Complement Factor C1q in the Clearance of Extracellular Neuromelanin From the Substantia Nigra in Parkinson Disease. J. Neuropathol. Exp. Neurol. 2011, 70, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.D.; Yang, A.J.; Margol, L.; Garzon-Rodriguez, W.; Glabe, C.G.; Tenner, A.J. Complement Component C1q Modulates the Phagocytosis of Abeta by Microglia. Exp. Neurol. 2000, 161, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Cho, K. Emerging Roles of Complement Protein C1q in Neurodegeneration. Aging Dis. 2019, 10, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Viceconte, N.; Burguillos, M.A.; Herrera, A.J.; De Pablos, R.M.; Joseph, B.; Venero, J.L. Neuromelanin Activates Proinflammatory Microglia through a Caspase-8-Dependent Mechanism. J. Neuroinflamm. 2015, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Apostolakis, S.; Kypraiou, A.-M. Iron in Neurodegenerative Disorders: Being in the Wrong Place at the Wrong Time? Rev. Neurosci. 2017, 28. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B.H. Iron-Melanin Interaction and Lipid Peroxidation: Implications for Parkinson’s Disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Maruyama, W.; Hashizume, Y.; Yoshida, M.; Osawa, T.; Riederer, P.; Naoi, M. In Parkinsonian Substantia Nigra, Alpha-Synuclein is Modified by Acrolein, a Lipid-Peroxidation Product, and Accumulates in the Dopamine Neurons with Inhibition of Proteasome Activity. J. Neural. Transm. 2007, 114, 1559–1567. [Google Scholar] [CrossRef]
- Atassi, M.Z.; Casali, P. Molecular Mechanisms of Autoimmunity. Autoimmunity 2008, 41, 123–132. [Google Scholar] [CrossRef]
- Orr, C.F.; Rowe, D.B.; Mizuno, Y.; Mori, H.; Halliday, G.M. A Possible Role for Humoral Immunity in the Pathogenesis of Parkinson’s Disease. Brain 2005, 128, 2665–2674. [Google Scholar] [CrossRef]
- Double, K.L.; Rowe, D.B.; Carew-Jones, F.M.; Hayes, M.; Chan, D.K.Y.; Blackie, J.; Corbett, A.; Joffe, R.; Fung, V.S.; Morris, J.; et al. Anti-Melanin Antibodies Are Increased in Sera in Parkinson’s Disease. Exp. Neurol. 2009, 217, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Woodroofe, M.N.; Cuzner, M.L. Microglia Are the Major Cell Type Expressing MHC Class II in Human White Matter. J. Neurol. Sci. 1987, 80, 25–37. [Google Scholar] [CrossRef]
- Waisman, A.; Johann, L. Antigen-Presenting Cell Diversity for T Cell Reactivation in Central Nervous System Autoimmunity. J. Mol. Med. 2018, 96, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Oberländer, U.; Pletinckx, K.; Döhler, A.; Müller, N.; Lutz, M.B.; Arzberger, T.; Riederer, P.; Gerlach, M.; Koutsilieri, E.; Scheller, C. Neuromelanin is an Immune Stimulator for Dendritic Cells in Vitro. BMC Neurosci. 2011, 12, 116. [Google Scholar] [CrossRef]
- Das, R.; Chinnathambi, S. Microglial Priming of Antigen Presentation and Adaptive Stimulation in Alzheimer’s Disease. Cell. Mol. Life Sci. 2019, 76, 3681–3694. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Merlini, M.; Späni, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-Cell Brain Infiltration and Immature Antigen-Presenting Cells in Transgenic Models of Alzheimer’s Disease-like Cerebral Amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef]
- Monteiro, S.; Roque, S.; Marques, F.; Correia-Neves, M.; Cerqueira, J.J. Brain Interference: Revisiting the Role of IFNγ in the Central Nervous System. Prog. Neurobiol. 2017, 156, 149–163. [Google Scholar] [CrossRef]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I Expression Renders Catecholaminergic Neurons Susceptible to T-Cell-Mediated Degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Corrêa, J.D.; Starling, D.; Teixeira, A.L.; Caramelli, P.; Silva, T.A. Chemokines in CSF of Alzheimer’s Disease Patients. Arq. Neuropsiquiatr. 2011, 69, 455–459. [Google Scholar] [CrossRef]
- Galimberti, D.; Schoonenboom, N.; Scarpini, E.; Scheltens, P. Dutch-Italian Alzheimer Research Group Chemokines in Serum and Cerebrospinal Fluid of Alzheimer’s Disease Patients. Ann. Neurol. 2003, 53, 547–548. [Google Scholar] [CrossRef]
- Galimberti, D.; Schoonenboom, N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal Chemokine Synthesis in Mild Cognitive Impairment and Alzheimer Disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Koper, O.; Kamińska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and Their Receptor (CXCR3) in Neuroinflammation and Neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Kalkonde, Y.V.; Morgan, W.W.; Sigala, J.; Maffi, S.K.; Condello, C.; Kuziel, W.; Ahuja, S.S.; Ahuja, S.K. Chemokines in the MPTP Model of Parkinson’s Disease: Absence of CCL2 and Its Receptor CCR2 Does Not Protect against Striatal Neurodegeneration. Brain Res. 2007, 1128, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tousi, N.S.; Buck, D.J.; Zecca, L.; Davis, R.L. Neuromelanin Inhibits CXCL10 Expression in Human Astroglial Cells. Neurosci. Lett. 2010, 486, 47–50. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-García, A.; Kun, A.; Calero, M.; Calero, O. The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration. Antioxidants 2021, 10, 124. https://doi.org/10.3390/antiox10010124
Moreno-García A, Kun A, Calero M, Calero O. The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration. Antioxidants. 2021; 10(1):124. https://doi.org/10.3390/antiox10010124
Chicago/Turabian StyleMoreno-García, Alexandra, Alejandra Kun, Miguel Calero, and Olga Calero. 2021. "The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration" Antioxidants 10, no. 1: 124. https://doi.org/10.3390/antiox10010124
APA StyleMoreno-García, A., Kun, A., Calero, M., & Calero, O. (2021). The Neuromelanin Paradox and Its Dual Role in Oxidative Stress and Neurodegeneration. Antioxidants, 10(1), 124. https://doi.org/10.3390/antiox10010124