
Droplet Microarray Based on Superhydrophobic-Superhydrophilic Patterns for Single Cell Analysis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
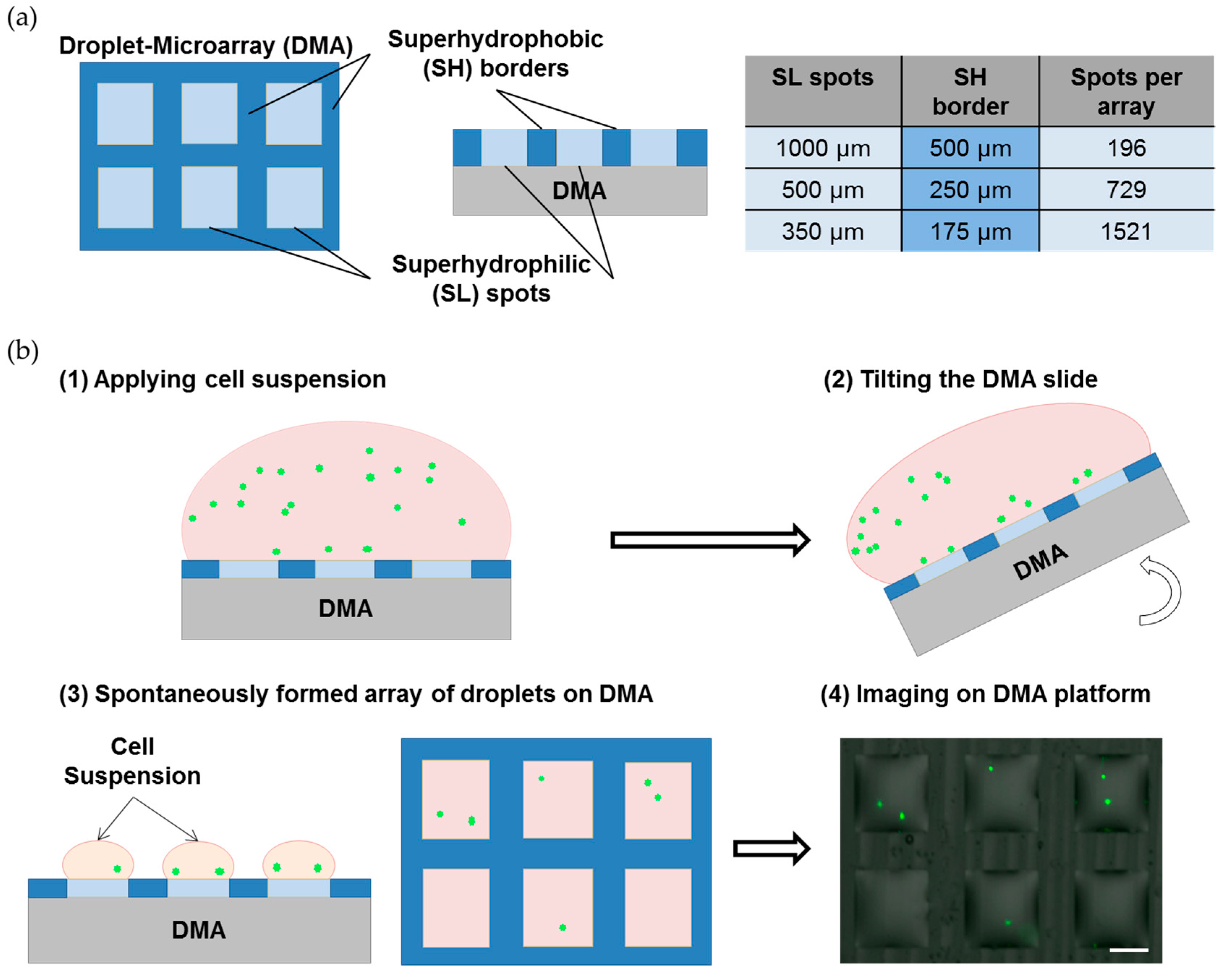
2.1. Preparation of the Droplet Microarray (DMA) Slides
2.2. Cell Culture on the Droplet Microarray
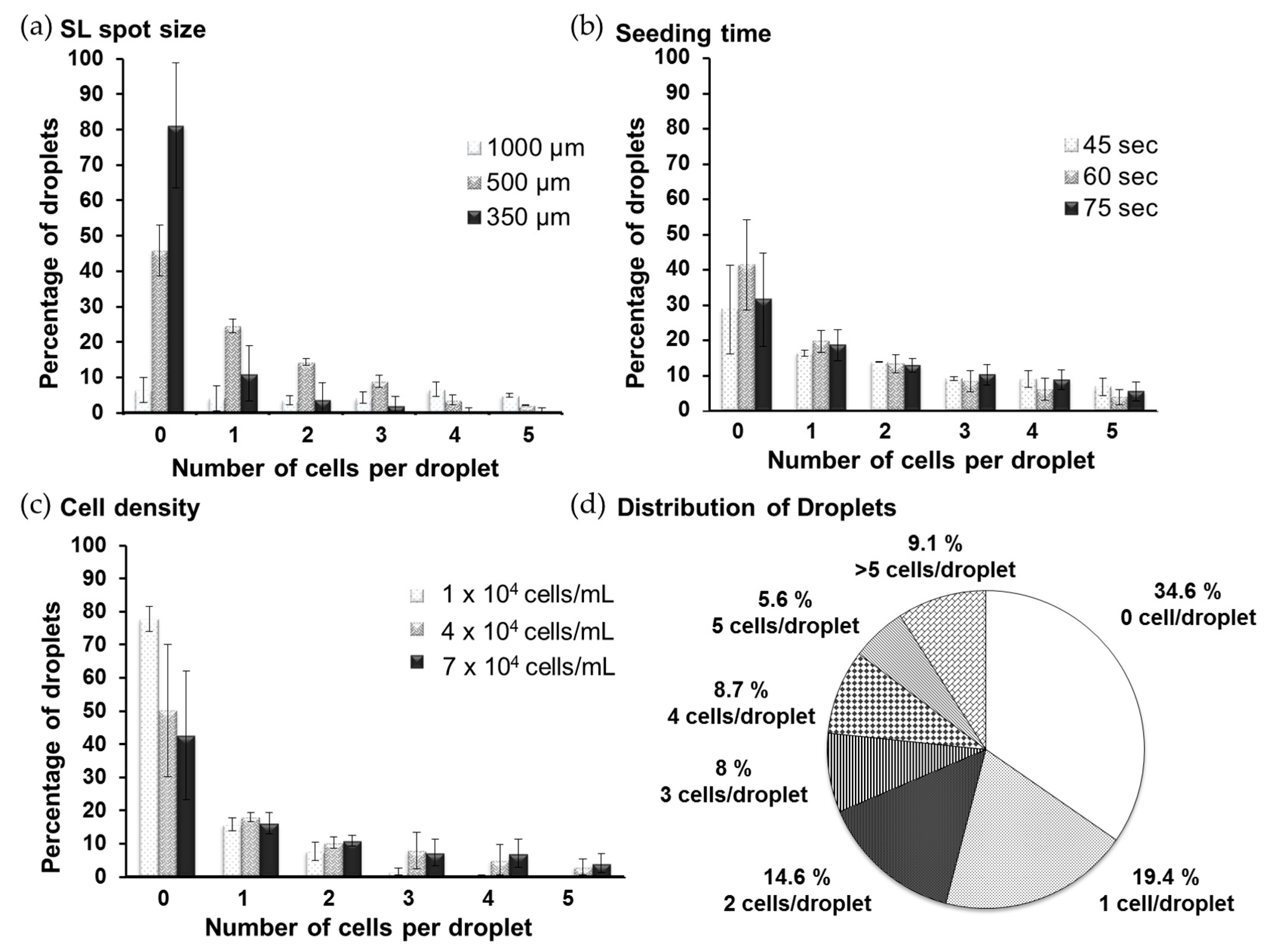
2.3. Analysis of Cell Distribution
2.4. Estimation of Cell Viability and Proliferation Rate
2.5 Statistical Analysis
3. Results
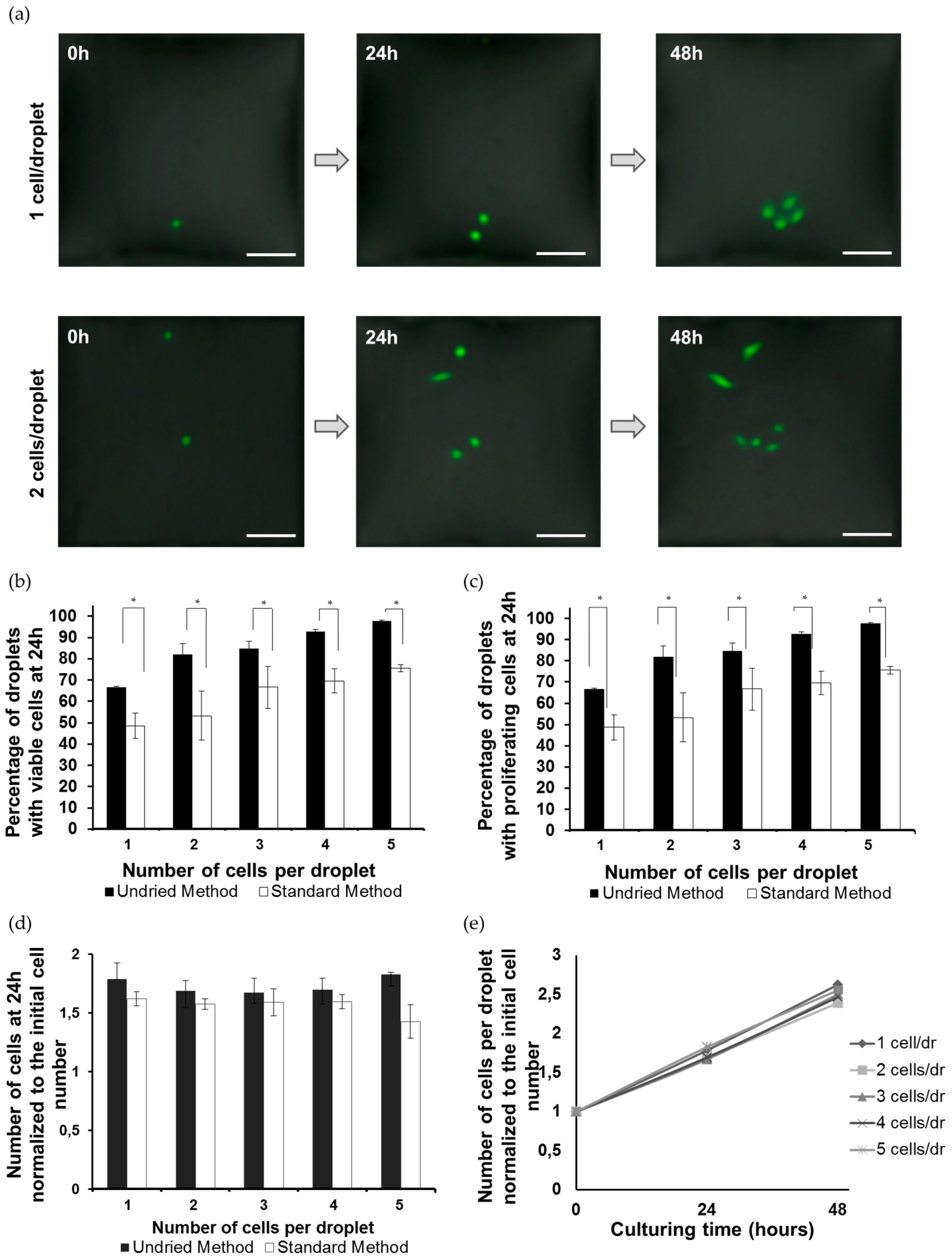
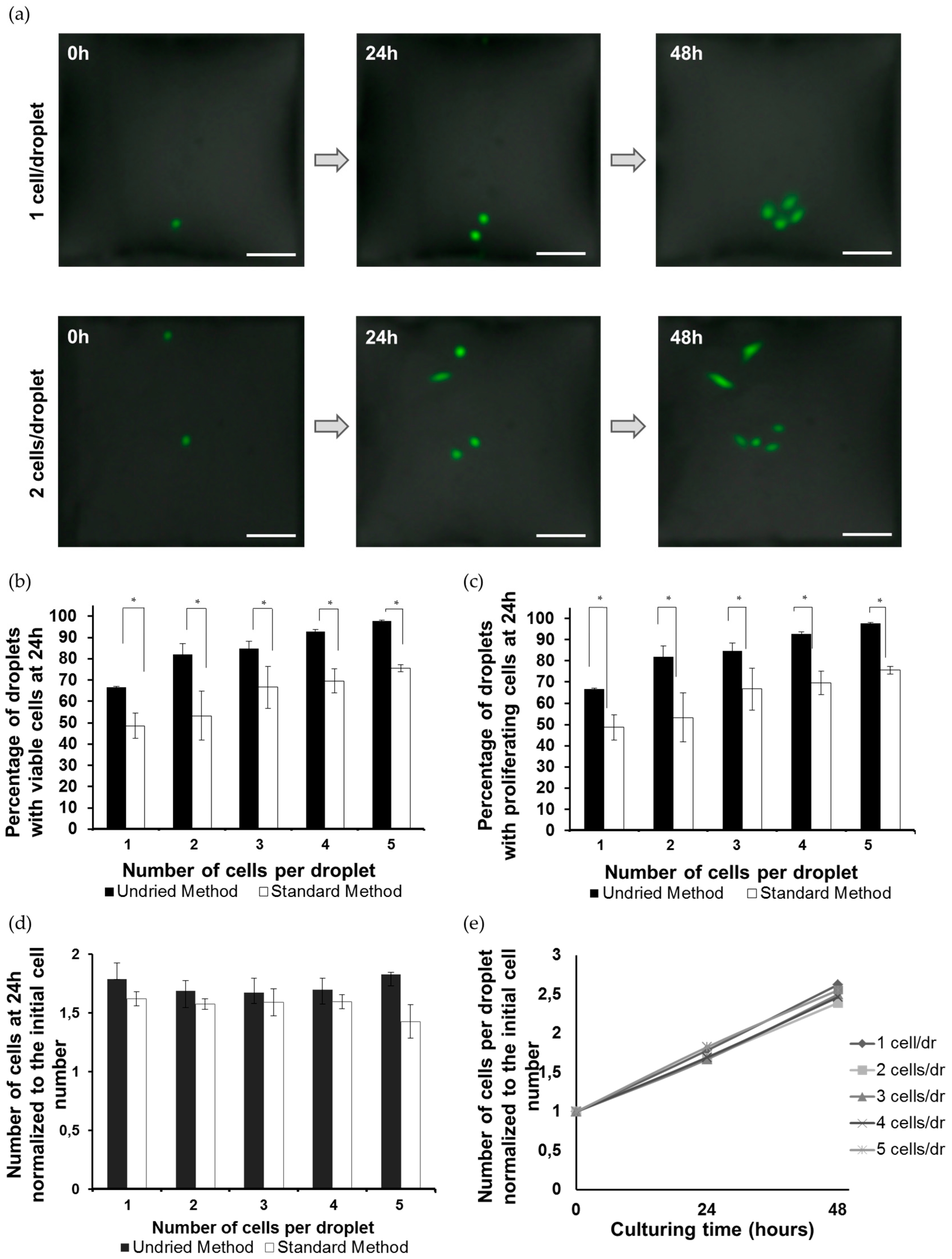
3.1. Formation of the Single-Cell Droplet Microarray (SC-DMA)
3.2. Viability and Growth Rate of Single Cells on the DMA Platform
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, D.; Bodovitz, S. Single cell analysis: The new frontier in ‘omics’. Trends Biotechnol. 2010, 28, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. Cancer genomics: one cell at a time. Genome Biol. 2014, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kalisky, T.; Quake, S.R. Single-cell genomics. Nat. Methods 2011, 8, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Hughey, J.J.; Gutschow, M.V.; Bajar, B.T.; Covert, M.W. Single-cell variation leads to population invariance in nf-kappab signaling dynamics. Mol. Biol. Cell 2015, 26, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, A.; Lai, S.; Guo, G.; Yuan, G.C. Single-cell analysis in cancer genomics. Trends Genet. 2015, 31, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Non-genetic heterogeneity of cells in development: More than just noise. Development 2009, 136, 3853–3862. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Theories of carcinogenesis: An emerging perspective. Semin. Cancer Biol. 2008, 18, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Mukherjee, U.K.; Harding, T.; Jang, J.S.; Stessman, H.; Li, Y.; Abyzov, A.; Jen, J.; Kumar, S.; Rajkumar, V.; et al. Single-cell analysis of targeted transcriptome predicts drug sensitivity of single cells within human myeloma tumors. Leukemia 2016, 30, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.R.; Ribas, A.; Mischel, P.S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 2016, 15, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Berg, D.A.; Zhu, Y.; Shin, J.Y.; Song, J.; Bonaguidi, M.A.; Enikolopov, G.; Nauen, D.W.; Christian, K.M.; Ming, G.-L.; et al. Single-cell RNA-seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell 2015, 17, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.G.; Coletta, M.; et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 2004, 95, 911. [Google Scholar] [CrossRef] [PubMed]
- Young, G.T.; Gutteridge, A.; Fox, H.D.E.; Wilbrey, A.L.; Cao, L.; Cho, L.T.; Brown, A.R.; Benn, C.L.; Kammonen, L.R.; Friedman, J.H.; et al. Characterizing human stem cell-derived sensory neurons at the single-cell level reveals their ion channel expression and utility in pain research. Mol. Ther. 2014, 22, 1530–1543. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, E.; Tsuji, T. Growing bioengineered teeth from single cells: Potential for dental regenerative medicine. Expert Opin. Biol. Ther. 2008, 8, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular stress responses: Cell survival and cell death. Int. J. Cell Biol. 2010, 2010, 23. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R.; Lesperance, J.; Kim, M.; Terskikh, A.V. Efficient propagation of single cells Accutase-dissociated human embryonic stem cells. Mol. Reprod. Dev. 2008, 75, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Pan, X. Single Cell Analysis: From technology to Biology and Medicine. Single Cell Biol. 2014, 3, 106. [Google Scholar] [PubMed]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for single-cell isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yan, S.; Yuan, D.; Alici, G.; Nguyen, N.-T.; Ebrahimi Warkiani, M.; Li, W. Fundamentals and applications of inertial microfluidics: A review. Lab Chip 2016, 16, 10–34. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Marshall, D. Microfluidics for single cell analysis. Curr. Opin. Biotechnol. 2012, 23, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Khalili, A.A.; Ahmad, M.R. A review of cell adhesion studies for biomedical and biological applications. Int. J. Mol. Sci. 2015, 16, 18149–18184. [Google Scholar] [CrossRef] [PubMed]
- Alakomi, H.L.; Matto, J.; Virkajarvi, I.; Saarela, M. Application of a microplate scale fluorochrome staining assay for the assessment of viability of probiotic preparations. J. Microbiol. Methods 2005, 62, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Wan, Z.; Carr, A.; Rizvi, A.H.; Vieira, G.; Pe’er, D.; Sims, P.A. Scalable microfluidics for single-cell RNA printing and sequencing. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Li, L.; Ueda, E.; Li, J.; Heißler, S.; Welle, A.; Trapp, O.; Levkin, P.A. Surface patterning via thiol-yne click chemistry: An extremely fast and versatile approach to superhydrophilic-superhydrophobic micropatterns. Adv. Mater. Interfaces 2014, 1, 1400269. [Google Scholar] [CrossRef]
- Geyer, F.L.; Ueda, E.; Liebel, U.; Grau, N.; Levkin, P.A. Superhydrophobic-superhydrophilic micropatterning: Towards genome-on-a-chip cell microarrays. Angew Chem. Int. Ed. Engl. 2011, 50, 8424–8427. [Google Scholar] [CrossRef] [PubMed]
- Efremov, A.N.; Stanganello, E.; Welle, A.; Scholpp, S.; Levkin, P.A. Micropatterned superhydrophobic structures for the simultaneous culture of multiple cell types and the study of cell-cell communication. Biomaterials 2013, 34, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Ueda, E.; Geyer, F.L.; Nedashkivska, V.; Levkin, P.A. Dropletmicroarray: Facile formation of arrays of microdroplets and hydrogel micropads for cell screening applications. Lab Chip 2012, 12, 5218–5224. [Google Scholar] [CrossRef] [PubMed]
- Ueda, E.; Levkin, P.A. Emerging applications of superhydrophilic-superhydrophobic micropatterns. Adv. Mater. 2013, 25, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.A.; Schillo, S.M.; Demir, K.; Ueda, E.; Nesterov-Mueller, A.; Levkin, P.A. Droplet-array (DA) sandwich chip: A versatile platform for high-throughput cell screening based on superhydrophobic-superhydrophilic micropatterning. Adv. Mater. 2015, 27, 5217–5222. [Google Scholar] [CrossRef] [PubMed]
- Popova, A.A.; Demir, K.; Hartanto, T.G.; Schmitt, E.; Levkin, P.A. Droplet-microarray on superhydrophobic–superhydrophilic patterns for high-throughput live cell screenings. RSC Adv. 2016, 6, 38263–38276. [Google Scholar] [CrossRef]
- Alcor, D.; Calleja, V.; Larijani, B. Revealing signaling in single cells by single- and two-photon fluorescence lifetime imaging microscopy. Methods Mol. Biol. 2009, 462, 307–343. [Google Scholar] [PubMed]
- Staszewski, R. Cloning by limiting dilution: An improved estimate that an interesting culture is monoclonal. Yale J. Biol. Med. 1984, 57, 865–868. [Google Scholar] [PubMed]
- Collins, D.J.; Neild, A.; deMello, A.; Liu, A.Q.; Ai, Y. The Poisson distribution and beyond: Methods for microfluidic droplet production and single cell encapsulation. Lab Chip 2015, 15, 3439–3459. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.J.; Novak, B. Control of cell growth, division and death: Information processing in living cells. Interface Focus 2014, 4, 20130070. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Avarbock, M.R.; Brinster, R.L. Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol. Reprod. 2004, 71, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xiong, H.-R. Culture conditions and types of growth media for mammalian cells. In Biomedical Tissue Culture; Ceccherini, L., Matteoli, B., Eds.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Shen, F.M.; Zhu, L.; Ye, H.; Yang, Y.J.; Pang, D.W.; Zhang, Z.L. A high throughput micro-chamber array device for single cell clonal cultivation and tumor heterogeneity analysis. Sci. Rep. 2015, 5, 11937. [Google Scholar] [CrossRef] [PubMed]
- Heldt, F.S.; Kupke, S.Y.; Dorl, S.; Reichl, U.; Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza a virus infection. Nat. Commun. 2015, 6, 8938. [Google Scholar] [CrossRef] [PubMed]
- Michler-Stuke, A.; Wolff, J.R.; Bottenstein, J.E. Factors influencing astrocyte growth and development in defined media. Int. J. Dev. Neurosci. 1984, 2, 575–584. [Google Scholar] [CrossRef]
- Di Carlo, D.; Wu, L.Y.; Lee, L.P. Dynamic single cell culture array. Lab Chip 2006, 6, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Bowra, S.; Vincze, E. The development and evaluation of single cell suspension from wheat and barley as a model system; a first step towards functional genomics application. BMC Plant Biol. 2010, 10, 239. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jogia, G.E.; Tronser, T.; Popova, A.A.; Levkin, P.A. Droplet Microarray Based on Superhydrophobic-Superhydrophilic Patterns for Single Cell Analysis. Microarrays 2016, 5, 28. https://doi.org/10.3390/microarrays5040028
Jogia GE, Tronser T, Popova AA, Levkin PA. Droplet Microarray Based on Superhydrophobic-Superhydrophilic Patterns for Single Cell Analysis. Microarrays. 2016; 5(4):28. https://doi.org/10.3390/microarrays5040028
Chicago/Turabian StyleJogia, Gabriella E., Tina Tronser, Anna A. Popova, and Pavel A. Levkin. 2016. "Droplet Microarray Based on Superhydrophobic-Superhydrophilic Patterns for Single Cell Analysis" Microarrays 5, no. 4: 28. https://doi.org/10.3390/microarrays5040028
APA StyleJogia, G. E., Tronser, T., Popova, A. A., & Levkin, P. A. (2016). Droplet Microarray Based on Superhydrophobic-Superhydrophilic Patterns for Single Cell Analysis. Microarrays, 5(4), 28. https://doi.org/10.3390/microarrays5040028