
SNP Array in Hematopoietic Neoplasms: A Review

Department of Hematopathology and Laboratory Medicine, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL 33612, USA
*
Author to whom correspondence should be addressed.
Microarrays 2016, 5(1), 1; https://doi.org/10.3390/microarrays5010001
Submission received: 31 July 2015
/
Revised: 13 November 2015
/
Accepted: 14 December 2015
/
Published: 22 December 2015
(This article belongs to the Special Issue SNP Array)
Abstract
:Cytogenetic analysis is essential for the diagnosis and prognosis of hematopoietic neoplasms in current clinical practice. Many hematopoietic malignancies are characterized by structural chromosomal abnormalities such as specific translocations, inversions, deletions and/or numerical abnormalities that can be identified by karyotype analysis or fluorescence in situ hybridization (FISH) studies. Single nucleotide polymorphism (SNP) arrays offer high-resolution identification of copy number variants (CNVs) and acquired copy-neutral loss of heterozygosity (LOH)/uniparental disomy (UPD) that are usually not identifiable by conventional cytogenetic analysis and FISH studies. As a result, SNP arrays have been increasingly applied to hematopoietic neoplasms to search for clinically-significant genetic abnormalities. A large numbers of CNVs and UPDs have been identified in a variety of hematopoietic neoplasms. CNVs detected by SNP array in some hematopoietic neoplasms are of prognostic significance. A few specific genes in the affected regions have been implicated in the pathogenesis and may be the targets for specific therapeutic agents in the future. In this review, we summarize the current findings of application of SNP arrays in a variety of hematopoietic malignancies with an emphasis on the clinically significant genetic variants.
1. Introduction
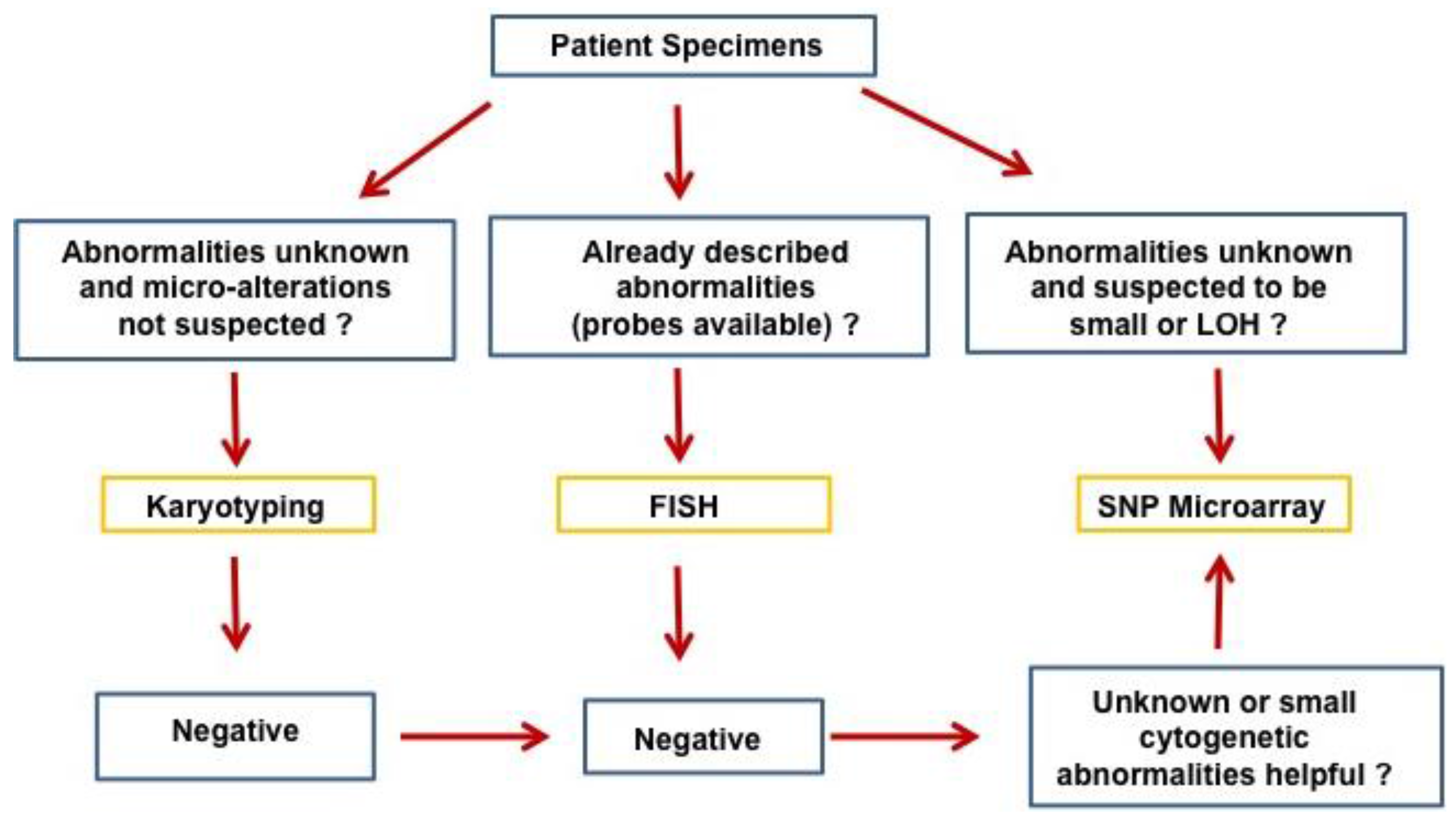
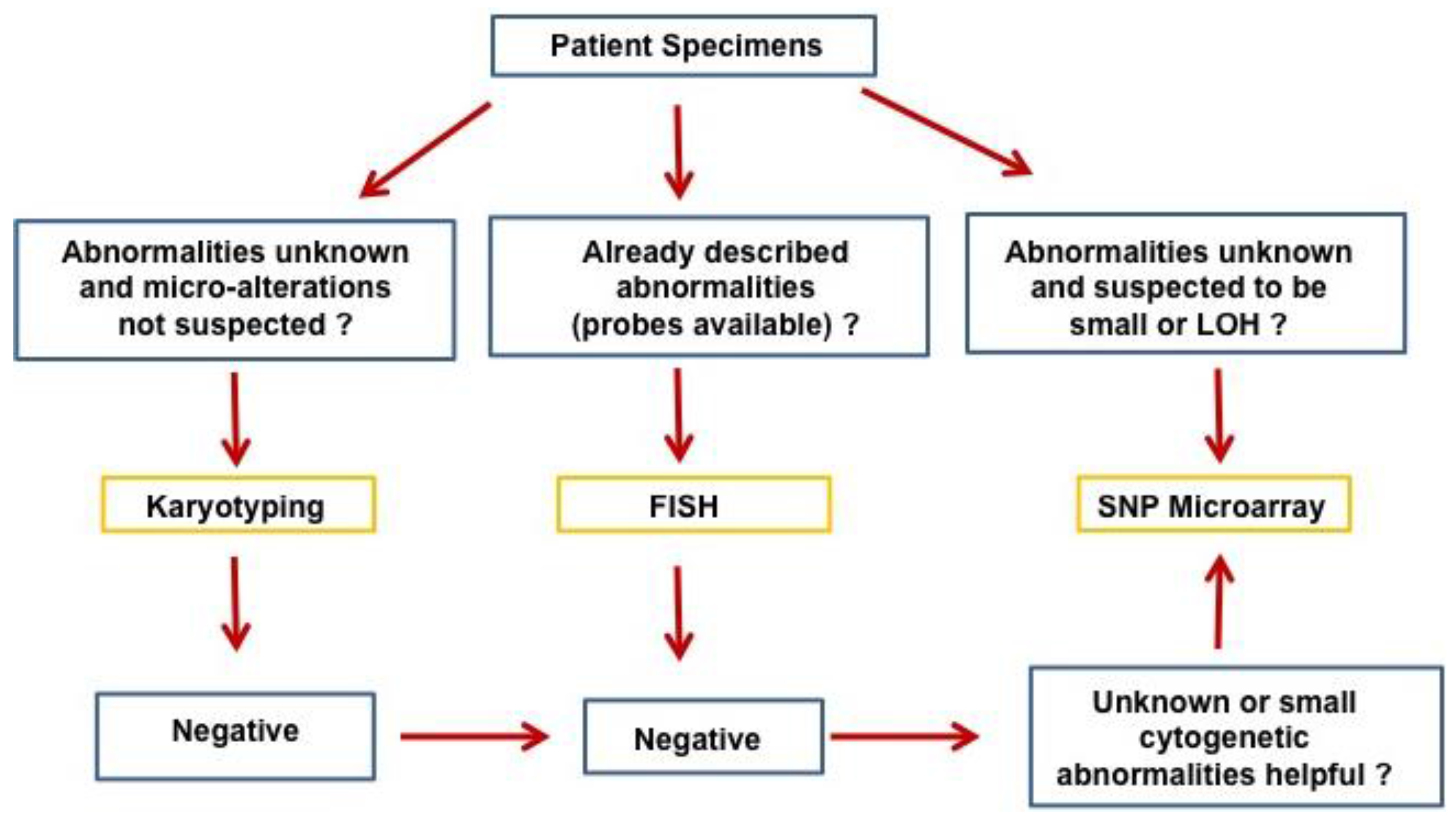
The progressive accumulation of genetic changes plays an essential role in the tumorigenesis and evolution of human cancers. The genetic changes commonly seen in human cancers include chromosomal translocations, amplifications, allelic loss, loss of heterozygosity, deletions, mutations, and epigenetic changes/DNA methylation affecting oncogenes and tumor suppressor genes [1]. The resolution of genetic alterations identified in clinical specimens has been pushed to the single nucleotide level over the decades with advancements in genetic technologies. Conventional cytogenetic analysis with G-banding karyotyping, a routine clinical analysis in cytogenetic labs, allows differentiation of approximately 400–500 bands per haploid genome [2]. At this level of resolution, chromosomal change over 10 Mb can be detected. Fluorescence in situ hybridization (FISH) offers high sensitivity and specificity of detecting genetic abnormalities such as translocations, aneuploidy, deletions, inversions, or amplifications by using DNA probes targeted to known DNA sequences [3]. FISH can identify genetic changes at a resolution up to a few kilobases (kb), but is not suited for identification of unknown genetic changes or global chromosomal abnormalities. Array-based comparative genomic hybridization (aCGH) developed in the early 1990s offers efficient high-throughput analysis of the entire genome for identification of copy number variations/aberrations (CNVs/CNAs) that are usually not detectable by conventional karyotyping or targeted FISH studies, and has an improved resolution down to 100 kb [4,5,6,7]. Single nucleotide polymorphism (SNP) arrays, manufactured by Affymetrix and Illumina, were initially designed for high-throughput SNP genotyping, but were quickly applied to cancer genomics [8,9,10,11]. In contrast to aCGH, SNP arrays are able to detect both CNVs/CNAs and loss of heterozygosity (LOH) or copy-neutral LOH/uniparental disomy (UPD), which are frequently involved in the development of cancers. With the advance in technology and marked improvements in resolution, the new SNP array offers over 90% coverage of known copy number variants by using more than 946,000 probes and an average inter-marker distance of 680 base pairs. This high level of resolution of cytogenetic changes has only recently been surpassed by next generation sequencing (NGS) technology developed in the last decade [12,13]. Ever since the invention of SNP arrays, they have been extensively applied to various hematologic malignancies. While currently there are no clinical guidelines on the use of SNP array in hematopoietic malignancies, SNP array will certainly be useful in difficult cases, especially in myelodysplastic syndrome (MDS) diagnosis, when other methodologies fail to identify cytogenetic abnormalities. A proposed flow chart for the application of SNP array in hematopoietic malignancies is presented in Figure 1. In this review, we summarize the important findings of chromosomal changes in hematopoietic malignancies identified by SNP array analysis.
Figure 1.
Proposed application of SNP array in hematopoietic malignancies.
2. Acute Lymphoblastic Leukemia/Lymphoma
Acute lymphoblastic leukemia/lymphoma is the most frequent pediatric malignancy, affecting 20–40 patients per million children in developed countries [14], and accounts for 20% of all acute leukemias in adults. B-lymphoblastic leukemia/lymphoma (B-ALL) is the most common type of acute lymphoblastic leukemia, and comprises genetically distinct subtypes including B-ALL with Philadelphia chromosome t(9;22)(q34;q11.2) (BCR-ABL1), t(v;11q23) (MLL rearranged), t(12;21)(p13;q22) (TEL-AML1), t(5;14)(q31;q32) (IL3-IGH), t(1;19)(q23;p13.3) (E2A-PBX1), hyperdiploidy, hypodiploidy, and about 25% cases without defined cytogenetic abnormalities [15]. Pediatric B-ALL has a favorable prognosis with approximately 80% rate of cure, while adult B-ALL has an inferior prognosis with about 40% rate of cure [16,17,18,19,20,21,22,23,24]. B-ALL with t(9;22)(q34;q11.2) (BCR-ABL1) is associated with the worst prognosis in both children and adults. Studies have shown that the currently identified chromosomal translocations are early initiating genetic events but are not sufficient to induce ALL [25].
To identify additional genetic lesions important for leukaemogenesis, a SNP array is well-suited for global genomic mapping of B-ALL. Irving et al. [26] first applied The Affymetrix 10K SNP array with resolution of 100 to 200 kb was used in 10 cases of pediatric B-ALL and demonstrated the usefulness of this technique in studying B-ALL. Of the 10 cases, LOH was detected in eight cases with the most frequent abnormality (50%) in chromosome 9p harboring the CDKN2A/B (INK4) gene locus. The loss of INK4 gene locus was only observed at relapse in three of the four cases, suggesting its association with treatment failure. Subsequently, Mullighan et al. [27] performed the first large-scale study of 242 cases of paediatric ALL, including 192 B-ALL and 50 T-ALL, by using Affymetrix SNP arrays that examine over 350,000 loci with an average resolution of less than 5 kb. Matched remission samples allowed the identification of somatic CNAs and LOH in leukemic blasts. The SNP arrays showed a low number of somatic copy number alterations (mean of 6.46) per case in ALL, with deletions outnumbering amplifications almost 2:1. The frequency of CNAs varied significantly between different cytogenetically defined ALL subtypes, with deletions more frequent than gains of DNA. Chromosomal deletions occurred more frequently in B-ALL with ETV6–RUNX1 and hypodiploidy with average of six deletions per case, up to 21 deletions, and only one deletion in MLL rearranged B-ALL. Gains of DNA occurred most frequently in hyperdiploid B-ALL (average of 10 gains), and uncommon in other types of ALL. The study identified 54 recurring regions of deletion that were mostly focal with the minimal deletion less than 1 Mb, and 24 deletions harboring only one single gene. The most important finding was that genes regulating normal B-cell development were deleted or mutated in approximately 40% cases of B-ALL. Copy number changes of PAX5, which is essential for B-cell differentiation, occurred in about 30% cases of B-ALL, making PAX5 the most frequently altered gene in B-ALL. These changes resulted in either reduced level or hypomorphic alleles of PAX5. Sequencing studies also identified a variety of somatic mutations in PAX5 resulting in either lost or altered DNA-binding or transcriptional functions. Other important genes deleted in ALL included EBF1, TCF3, LEF1, IKZF1 (IKAROS), and IKZF3 (AIOLOS). These findings suggest that ALL is not a neoplasm characterized by chromosomal instability, and genetic alterations in genes controlling B-cell development (PAX5, EBF1 and IKZF1) are common and play important roles in B-ALL leukaemogenesis. In a separate study, Kawamata et al. [28] studied 399 pediatric ALL samples with matched remission marrow using Affymetrix 50K SNP arrays, and identified three most common genetic alterations: deletion of ETV6, deletion of CDKN2A/p16INK4A, and hyperdiploidy. This study also confirmed the deletions of PAX5 (9p13), EBF (5q33), IKAROS (7p12.2), AIOLOS (17q12), LEF1 (4q25), RAG1 (11p12), and RAG2 (11p12) in pediatric ALL, albeit with a lower frequency except PAX5. Uniparental disomy (UPD) was frequently identified, especially in chromosome 9. In addition, hyperdiploid ALL without gains of chromosomes 17 and 18 was found to have poor prognosis. The common deletions of CDKN2A at 9p21 (29%) and ETV6 (TEL) at 12p13 (3/24, 12%) were also confirmed by Bungaro et al. [29] in a separate study.
T-ALL comprises about 25% cases of adult ALL and approximately 15% cases of childhood ALL and is most commonly present in adolescents as mediastinal lymphoma. The most frequent recurrent cytogenetic abnormalities involve translocation of T-cell receptor gene locus (14q11.2, 7q35, and 7p14-15) with a variety of partner genes such as HOX11, MYC, TAL1, RBTN1/2, and LYL1 [30,31] and, thus, T-ALL is genetically more heterogeneous. In their SNP array analysis of 50 cases of T-ALL, Mullighan et al. [32] identified multiple new genomic changes in T-ALL, including deletions of TAL1, RB1, and PTEN, and duplications of protooncogene MYB. Most recently, Karrman et al. [33] investigated 47 cases of T-ALL with an Illumina HumanOmni1-Quad BeadChip containing >1 million markers and a median marker interval of 1.5 kb. Copy number changes and UPD were identified in the majority of cases (92%), with a median of three changes/per case. This study identified recurring region of deletion harboring genes CDKN2A, CDKN2B, LEF1, PTEN, RBI, and STIL. In terms of uniparental disomy (UPD), the T-ALL lacked whole chromosome UPD, but showed segmental UPDs (sUPDs) in 42% of cases, with a high proportion of sUPD 9p (30% of the cases) associated with homozygous CDKN2A deletion. Therefore, disruption of the p53 and RB1 pathways through deletion of INK4/ARF on CDKN2A gene locus appears to be important in the pathogenesis of T-ALL.
The genomic changes that may explain the difference in survival between pediatric and adult ALL were addressed by Okamoto et al. [34] with Affymetrix 50K or 250K SNP arrays. The authors studied 75 cases of adult ALL and 399 cases of pediatric ALL. This study showed 572 genomic alterations with a mean of 7.6 genomic changes per case in adult ALL. The genomic changes in adult ALL were comparable to those identified in pediatric ALL, including deletions of 3p14.2 (FHIT), 5q33.3 (EBF), 6q, 9p21.3 (CDKN2A/B), 9p13.2 (PAX5), 13q14.2 (RB1), and 17q11.2 (NF1). The recurrent genomic alterations had similar rate of occurrence in pediatric and adult ALL. As the adult ALL cases were all non-hyperdiploid, the pediatric ALL cases were divided into hyperdiploid (HD) and non-hyperdiploid groups. There was no significant difference between adult and non-HD pediatric ALL in terms of deletions of 3p14.2 (FHIT), 9p21.3 (CDKN2A/B), 9p13.2 (PAX5), 13q14.2 (RB1), and 17q11.2 (NF1), and adult ALL showed more frequent deletion of 17p (TP53) and duplication of 17q than non-HD pediatric ALL (11% vs. 2%, and 9% vs. 1%, respectively). Overall, there were no unequivocal CNAs identified by SNP array that can account for the differences of prognosis between adult and pediatric ALL. The characteristic deletions of CDKN2A, PAX5, IKZF1, ETV6, RB1, and EBF1 genes were also identified by Safavi et al. [35] in adult ALL using SNP array covering 5 million markers and a resolution of 10 kb. A number of novel recurrent cryptic genetic changes involving BCAT1, SERP2, RAB30, SRPR, ST3GAL4, ASS1, RASSF3, FUBP3, BCL11A, GAB1, LINGO2, TOX, and CXCR4 genes, and partial and whole-chromosome UPDs were also discovered in adult ALL. Their significance in pediatric and adult ALL remains to be determined.
SNP arrays were designed for genome-wide association (GWA) study, and it was naturally applied in ALL for germline SNPs that may have an association with ALL. Trevino et al. [36] studied 317 cases of pediatric ALL along with 17,958 control cases with an Affymetrix 500K Mapping array, and found two SNPs at chromosome 10q21 (rs10821936 and rs10994982) located in intron 3 of the ARID5B gene to be associated pediatric ALL. In addition, both SNPs discriminated hyperdiploid B-ALL from other major ALL subtypes. A genome-wide association study by Papaemmanuil et al. [37] on two case-control series with 907 ALL cases and 2398 controls also identified an association between a SNP at 10q21.2 in the ARID5B gene (rs7089424) and pediatric ALL. The 10q21.2 (ARID5B) risk association was selective for hyperdiploid B-ALL. In addition, they also found two additional risk loci for ALL at 7p12.2 (IKZF1, rs4132601), and 14q11.2 (CEBPE, rs2239633). The association between genetic variations at 7p12.2 (IKZF1), 10q21.2 (ARIDB5), and 14q11.2 (CEBPE) with pediatric ALL was replicated by Prasad et al. [38] in genotyping 1384 cases of pediatric B-ALL and 1877 controls. These findings indicate that common germline variants contribute to the risk of development of pediatric ALL. As both ARID5B and IKZF1 play important roles in B-lymphocyte growth and differentiation, the possibility of SNPs in these genes predispose the patients to the development of B-ALL is high.
3. Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is genetically heterogeneous, ranging from cases with recurrent cytogenetic abnormalities to approximately 40% cases with normal karyotype, some of which have prognostically significant somatic mutations. AMLs with t(15;17), inv(16) and t(8;21) respond well to chemotherapy and have good prognosis, while AMLs with Philadelphia chromosome t(9;22), complex karyotype, −5/−5q, −7/−7q, and 11q23 translocations have poor prognosis. In AMLs with normal karyotype, FLT3 internal tandem duplications (ITDs) are associated with poor prognosis [39], while mutations of NPM1 and CEBPA are associated with favorable prognosis [40]. In 2005, Raghavan et al. [41] first applied a 10K SNP array in 64 cases of AML and identified partial uniparental disomy (pUPD) in approximately 20% cases. A similar study with 10K SNP array demonstrated homozygous mutations involving genes FLT3, CEBPA, and RUNX1 in approximately 50% patients with UPD involving corresponding chromosomal regions [42]. Subsequently, Gupta et al. [43] expanded the study with 10K SNP array on 454 cases of AML from young adults, and found nonrandom acquired UPD (aUPD) in 17% cases, preferentially affecting chromosomes 13q, 11p, and 11q, similar to the findings by Raghavan et al. [43], and additional recurrent aUPDs at 2p, 17p, 2q, 17q, 1p, and Xq. AMLs with FLT3-ITD had aUPD 13q involving the FLT3 gene, while AMLs with FLT3-ITD-/FLT3-TKD+ mutation did not have aUPD 13q.
These early studies suffered from the low resolution of the SNP arrays available at that time. With advances in SNP array technology, Bullinger et al. [44] were able to use 50 and 500K Affymetrix SNP arrays on 157 cases of cytogenetically normal AML. The cohort showed 12% aUPDs with chromosomal regions 6p, 11p, and 13q most commonly affected and all aUPDs were >29 Mb. The aUPDs were associated with mutations in NPM1 or CEBPA, which suggested that aUPDs may affect genes critical for hematopoiesis. In terms of aCNAs, as expected, the 500K SNP array was much more sensitive than 50K SNP array, which missed approximately 60%–70% of CNAs detected by the 500K SNP array. aCNAs were identified in 49% cases, with genomic losses (1.21/case) more frequent than gains (0.25/case). The recurrent genomic deletions included bands 3p14.1-p13, 6q27, 8q23.3, 10q11.21, 11q25, 12p13.2, and 15q21.3, that harbor genes FOXP1 and RYBP (3p14.1-p13), RPS6KA2 (6q27), TRPS1 (8q23.3), HNRPF (10q11.21), ETV6 (12p13.2) and RFXDC2 (15q21.3). In a separate study, Walter et al. [45] applied a 1.85 million SNP array from Affymetrix in 86 adult patients with de novo AML, and identified a total of 201 aCNAs in 44% of the cases with a mean of 2.34 CNAs per genome. Acute erythroid leukemia and acute megakaryocytic leukemia had more CNAs (10–29 CNAs per genome) than other morphologic variants of AML. CNAs were detected in 24% AMLs with normal cytogenetics and in 40% AMLs with abnormal karyotype. 12 chromosomal regions (eight deletions and four amplifications) containing at least one gene implicated in AML or MDS (deletions of 3p14.1: FHIT, 5q31.1: CTNNA1, 12p12.3: ETV6, 16q22.1: CBFB, 17p13.1: TP53, 17q11.2: NF1, and amplifications of 8q23.2: MYC, 11q23.3: MLL, and 21q22.2: ETS2) were identified in multiple AML cases. CNAs in chromosomal regions 17q11.2 and 21q22.2 (CNAs spanning NF1 and ETS2) were found in at least five cases, most of which had complex karyotypes, and associated with worse overall survival. aUPD was infrequent occurring in eight of 86 genomes, with most in AML with normal cytogenetics. 50% of the AML studied had no CNA or UPD at this resolution. The identified CNAs did not predict overall or event-free survival independent of cytogenetics. Radtke et al [46] showed similarly very low burden of genomic alterations in pediatric de novo AML, with a mean of only 2.38 somatic copy-number alterations per case. These studies indicate that AMLs are not genomically unstable and the genes implicated in these CNAs likely play important role in the leukemogenesis of AML.
While Walter’s study did not identify any prognostically significant CNAs or UPDs, three groups clearly showed prognostic significance of SNP array lesions in AML, likely due to utilization of more stringent SNP lesion detection algorithms. Parkin et al. [47] examined 114 previously untreated prospectively enrolled AML patients with Affymetrix SNP 6.0 arrays, and showed that ≥2 genomic lesions detected by SNP 6.0 array almost doubled the risk of death after controlling for age- and karyotype-based risk by multivariate analyses. P53 mutations, or P53 mutations coupled with 17p-LOH conferred an independent negative prognosis. Tiu et al. [48] performed 250 K and 6.0 SNP arrays on 140 cases of primary (p) and secondary (s) AML, and demonstrated that patients with genomic lesions including acquired somatic UPD identified by SNP array had worse overall survival (OS) and event-free survival (EFS) in pAML with normal cytogenetics and in pAML/sAML with abnormal cytogenetics. The SNP array lesions, AML type, and metaphase cytogenetics had independent predictive value for OS by multivariate analyses. In a study of 133 cases of AML with normal cytogenetics by Genome-Wide Human SNP 6.0 Array, Yi et al. [49] found at least one abnormal SNP lesion in 32.3% cases. Detection of abnormal SNP lesions by SNP-A karyotyping conferred an unfavorable prognosis for overall survival by multivariate analyses. All these studies confirmed the clinical relevance as well as prognostic significance of SNP lesions in AML, especially AML with normal cytogenetics, which would allow a better prognostic stratification of patients with AML for appropriate treatment.
4. Myelodysplastic Syndrome
Myelodysplastic syndrome (MDS) is a group of clonal hematopoietic neoplasm characterized by ineffective hematopoiesis, cytopenia, morphologic dysplasia, and potential progression to acute myeloid leukemia [50]. Most patients with MDS die of bone marrow failure rather than transformation to acute myeloid leukemia. MDS is diagnosed based on the World Health Organization (WHO) classification, but the prognosis for survival is stratified based on the international prognostic scoring system (IPSS) and revised IPSS (IPSS-R) that incorporate cytogenetic abnormalities, percentage of bone marrow myeloblasts, and number of cytopenias [51,52]. As part of the IPSS-R system, cytogenetic abnormalities are stratified from very good with single del(11q) and −Y to very poor with complex karyotype (>3 abnormalities). Approximately half of patients with MDS have normal karyotype that is associated with good prognosis in the IPSS-R system, but MDS patients with normal karyotype are still heterogeneous genetically. SNP arrays were therefore performed on MDS to identify additional occult genetic abnormalities, especially in patients with normal karyotype.
Gondek et al. [53,54] was the first group to study MDS with SNP array. They applied an Affymetrix 50K SNP Assay to 66 and 72 patients with MDS, and found chromosomal defects in 82% of MDS patients, including 68% patients with normal karyotype, 81% patients with abnormal karyotypes, with chromosomes 8, 7, 5, and 11 most frequently involved. Segmental uniparental disomy (sUPD) was found in 33% of patients with MDS, and usually in regions frequently affected by deletions detected by metaphase cytogenetic analysis, including 7q and 11q. In a similar study, Mohamedali et al. [55] studied 119 patients with low-risk MDS with 50 K, 250 K and 500 K SNP arrays and identified deletions in 10%, amplifications in 8%, and UPD in 46% of cases. Nowak et al. [56] showed that CNAs and LOH can be identified in the CD34 positive blasts. These early studies suffered from absence of paired normal tissue for each case, which made distinction of inherited CNAs and LOH from somatic acquired ones difficult. Heinrichs et al. [57] performed a prospective study of matched pairs of bone marrow and buccal cell (normal) DNA from 51 patients with MDS by 250K SNP array, and identified somatically acquired genomic abnormalities in 41% patients, including 15% in MDS with normal karyotypes. UPDs affecting chromosome 7q was associated with rapidly progressive clinical course despite a low-risk IPSS score [57]. Similarly, Tiu et al. [58] analyzed 250 cases of MDS by 250K and Affymetrix SNP Array 6.0 with paired bone marrow and CD3+ lymphocytes to distinguish germline lesions. In this study, they showed that combined metaphase cytogenetics and SNP array had a higher diagnostic yield of chromosomal defects (74% vs. 44%), compared with conventional karyotyping. The genetic abnormalities detected by SNP arrays were deletions and aUPD involving chromosomes 1, 5, 7, 11, 17, and 21. While Mohamedali’s study failed to show independent prognostic significance of the genetic lesions identified by SNP array on multivariate analysis, Tiu showed that the presence of new genetic lesions detected by SNP array was predictive of poor prognosis in MDS by univariate and multivariate analyses [58]. These studies proved the utility of SNP array in detecting submicroscopic genetic lesions in MDS as a complement to metaphase cytogenetics, and the lesions identified by SNP arrays can further help prognostic stratification of MDS patients.
MDS can be difficult to diagnose clinically due to many mimickers of the disease and the lack of significant morphologic dysplasia in a small subset of cases. For example, severe aplastic anemia (AA) may be difficult to distinguish from hypoplastic MDS morphologically and cytogenetically. Afable et al. [59] demonstrated the utility of SNP analysis in AA to complement metaphase cytogenetics for the detection of clonal chromosomal lesions. Combined metaphase cytogenetics and SNP array identified chromosomal lesions in 19% of AA and 54% of hypoplastic MDS. Persistent detection of chromosomal lesions by SNP array would be highly suspicious for hypoplastic MDS and less response to immunotherapy (ATG/cyclosporine) for AA. Therefore, in diagnostically-challenging and equivocal cases of MDS, SNP array can be used to establish the presence of clonal hematopoiesis in patients with normal karyotype and allow appropriate management of the patients.
With the advent of next generation sequencing, SNP arrays are not likely to be used for the identification of genes involved in disease. In the last decade, SNP arrays have played an important role in the identification of individual genes important for the pathogenesis of MDS. TET2 gene was identified by SNP array genomic profiling and genomic sequencing in 102 patients with MDS, and acquired deletions, missense and nonsense mutations in the TET2 gene were found in 26% cases of MDS [60]. Recurrent aUPD and microdeletion of chromosome 7q led to identification of EZH2 gene and mutations in MDS [61,62]. The TET2 gene mutations have been found to be associated with better response to hypomethylating agents [63], while EZH2 mutations are poor prognostic marker for MDS. By combining SNP-array and gene expression profiling, Merkerova et al. [64] identified BMP2 and TRIB3 genes located in 20p UPD as potential candidate genes for the pathogenesis of MDS.
5. Chronic Myelogenous Leukemia
Chronic myelogenous leukemia (CML) is characterized by the presence of Philadelphia chromosome t(9;22)(q34;q11) (BCR-ABL1), for which targeted therapy with tyrosine kinase inhibitors (TKI) have revolutionized the treatment of CML. In an effort to define genetic lesions that cooperate with BCR-ABL1 to transform to Philadelphia chromosome positive acute leukemia, Mullighan et al. [65] studied 304 cases of ALL, including 23 CML cases with a 250K Affymetrix SNP array, and found only 0.47 copy number alterations per case in chronic phase CML (range 0–8), which suggested that chronic phase CML is genomically stable and BCR–ABL1 is sufficient to induce CML. However, a separate study by Khorashad et al. [66] on 10 chronic phase CML patients with high resolution 2.1 million oligonucleotide array comparative genomic hybridization (CGH) showed an average of 53 CNAs per patient (range: 4–166) with majority being amplifications. The difference in CNAs detected by the two techniques is most likely due to the marked difference in resolutions of the arrays. In Mullighan’s study, IKZF1 was deleted in CML lymphoid blast phase, but not in CML chronic phase [65]. The IKZF1 encodes IKAROS which is an essential transcription factor for normal lymphoid development. Deletion of IKZF1 results in monoallelic, expression of dominant-negative form, or loss of expression of IKAROS. Deletion of PAX5 and CDKN2A/B, together with loss of IKFZ1 in lymphoid but not myeloid blast phase of CML indicates that these genes play important role in the transformation of CML to lymphoid blast phase of CML [32]. A subset of the CML patients is resistant to TKI therapy, mainly due to BCR-ABL1 mutations or amplification. In a study to identify genetic alterations in 45 TKI-resistant CML by 250K SNP array, Nowak et al. [67] found recurrent submicroscopic alterations, including aUPD in chromosomes 1, 8, 9, 17, 19, and 22. Recurrent deletions of IGLC1 locus on chromosome 22 were identified in three patients with previous blast crisis, suggesting dedifferentiation into immature progenitors as a possible mechanism of TKI resistance.
6. Polycythemia Vera, Essential Thrombocythemia and Primary Myelofibrosis
Polycythemia (Rubra) vera (PV) is a clonal hematopoietic neoplasm characterized by increased red cell mass and JAK2 V617F mutation in 95% cases, and potential to progress to myelofibrosis or transformation to acute myeloid leukemia. Essential thrombocythemia (ET) is a chronic myeloproliferative neoplasm affecting the megakaryocytic lineage and characterized by abnormally large megakaryocytes in the marrow and persistent thrombocytosis. Primary myelofibrosis (PMF) is characterized by megakaryocytic and myeloid proliferation and progressive fibrosis in the marrow. JAK2 V617F mutation is identified in approximately 40%–50% cases of ET and approximately 50% of PMF. Of note, JAK2 V617F mutation can be detected in healthy individuals by high sensitivity methods. CNAs were rare in PV and ET and approximately one third cases of PMF showed small genomic losses (<5 Mb) with 250K SNP array [68]. In terms of copy-neutral aberrations (UPD), recurrent changes were only observed on chromosome 9p. Rice et al. [69] confirmed that chromosome 9 abnormalities including 9p LOH, trisomy 9, amplifications of 9p13.3–23.3, 9q33.1–34.13, and 9q34.13 were most frequent in myeloproliferative neoplasms when analyzing 87 myeloproliferative neoplasmas (MPN) cases with an Affymetrix 250K SNP array. Other less frequent recurrent genetic alterations included gains of 1p36.31–36.33, 17q21.2–q21.31, and 17q25.1–25.3, and deletions affecting 18p11.31–11.32. In the study of genetic profiles of myeloproliferative neoplasms by 50K SNP array, Kawamata et al. [70] found rare genomic abnormalities in ET, and deletion of chromosomal regions harboring RB (13q14) or NF1 (17q11) in 25% PMF cases. aUPD involving JAK2 was found in five PV cases with homozygous JAK2 V617F [70]. A subpopulation with 9p aUPD was detected in 30% of PV and approximately 50% PMF cases, and UPD at 1p was identified in one case of PV. The relationship between JAK2 V617F mutation and 9p aUPD in PV was further addressed by Wang et al. [71]. They investigated 31 PV patients with SNP array and whole genome sequencing and validated the findings in 59 additional PV patients [71]. They defined four PV subgroups based on the quantitative relationship between JAK2 V617F and 9p aUPD: 42% of patients with heterozygous JAK2 V617F and no detectable 9p aUPD (subgroup I); 45% of patients with homozygous JAK2 V617F and an allelic fraction directly proportional to the level of 9p aUPD (subgroup II); 10% with 9p aUPD at approximately twice the level of heterozygous JAK2 V617F allelic burden (subgroup III) and 3% with trisomy 9p and two copies of JAK2 V617F allele (subgroup IV), which likely suggest different pathways leading to PV phenotype. These genomic profiling studies indicated that ET and PV are genomically stable and JAK2 on chromosome 9p is critical for the pathogenesis of MPN. A genome-wide associate study seems to support this, with the identification of a SNP in the JAK2 locus (rs10974944), which predisposed to the development of JAK2 V617F-positive myeloproliferative neoplasm [72].
A subset of the patients with MPNs eventually progresses to acute myeloid leukemia. Identification of acquired genetic alterations facilitating this transformation would be valuable for patient stratification. In a study comparing genome profiles of 88 cases of MPN and 71 cases of MPN-blasts phase with 50 and 250 K SNP arrays, leukemic transformation of MPN was accompanied by up to three-fold more genomic alterations per case than chronic phase [73]. The genomic regions commonly affected during leukemic transformation harbored established genes such as ETV6, TP53, and RUNX1, and also new candidate genes on 7q, 16q, 19p, and 21q. Trisomy 8 or amplification of 8q24 (MYC) was identified exclusively in JAK2 V617F(−) MPN-blast phase. A poor prognosis after leukemic transformation was associated with copy number-neutral loss of heterozygosity (CNN-LOH) on either 7q or 9p including homozygous JAK2 V617F. With higher resolution SNP 6.0 arrays, Rumi et al. [74] showed a close relationship between UPD and/or gain of chromosome 9p with progression from PV to post-PV myelofibosis, and genetic aberrations of chromosome 5, 7, or 17p associated with progression to AML and overall survival.
7. Myelodysplastic/Myeloproliferative Neoplasms
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) are characterized by the presence of features of both MDS and MPN. Chronic myelomonocytic leukemia (CMML) is the most common form of MDS/MPN and characterized by persistent monocytosis, dysplasia, and transformation to acute myeloid leukemia. SNP Array 6.0 identified genomic alterations in 60% of patients with CMML with cryptic CN-LOH in 71% and microdeletions in 45% cases [75]. CN-LOH was frequent on 7q harboring EZH2, 11q harboring CBL, and 4q harboring TET2. The presence of multiple chromosomal defects detected by SNP array was associated with a worse overall survival by univariate analysis [75]. In a separate study, UPD occurred in 48% of CMML by 250K SNP array analysis, with the most frequently affected chromosomal region in 11q harboring proto-oncogene c-CBL [76]. All patients with UPD 17q and UPD 4q were found to have CMML or M5 primary AML. These studies indicated that CMML is genetically heterogeneous with different pathways to a common disease phenotype, and CBL mutations may activate the RAS pathway and aberrant pSTAT5 activation in CMML. A recent study suggested that abnormal SNP array lesions were associated with an inferior complete and partial remission rate, and worse overall survival when compared with patients without SNP lesions after decitabine therapy [77]. This finding, if confirmed, would certainly help better prognostic stratification and treatment of CMML patients.
Juvenile myelomonocytic leukemia (JMML) is a rare clonal hematopoietic disorder of childhood characterized by monocytosis and loss of function of neurofibromatosis 1 (NF1) or somatic mutations of genes in RAS/MAPK pathway. Flotho et al. [78] first applied SNP array on 16 cases of JMML with normal karyotype and identified large regions of UPD on chromosome 17 spanning approximately 55 Mb, which contained the locus of the NF1 tumor suppressor gene on 17q11.2, in four of five patients with JMML and NF1, but not in other cases without NF1. Inactivating NF1 lesion on both alleles was found by mutational analysis in each case. This study indicates that 17q UPD with homozygous loss of normal NF1 plays a critical role for the pathogenesis of JMML in NF1 patients.
8. Classical Hodgkin Lymphoma
Classical Hodgkin lymphoma (cHL) is characterized by presence of small numbers of neoplastic Reed-Sternberg/Hodgkin cells admixed with mixed inflammatory cells. Metaphase cytogenetic study is typically unsuccessful due to low numbers of neoplastic cells. For the same reason, SNP array is not expected to yield useful information on patient samples. Instead, SNP array 6.0 was performed on cHL cell lines and showed a UPD of chromosome 14q, which was associated with biallelic deletion of TRAF3 in one cell line, and a gain of copy number for MAP3K14 in three other cell lines [79]. With primary cHL tissues, interphase cytogenetic analyses confirmed monoallelic deletion of TRAF3 in 3/20 cases and gains of MAP3K14 in 5/16 cases. Both TRAF3 and MAP3K14 are regulators of the NF-κB pathway, which is constitutively activated in cHL. The study suggested that genetic alterations of the components of the NF-κB pathways contributed to the pathogenesis of cHL, at least in a subset of cases.
A genome-wide association study identified five SNPs on chromosome 6p21.32 associated with nodular sclerosis cHL (NSHL), which is a common subtype of cHL [80]. Two of the SNPs, rs6903608 and rs2858870, were significantly associated with NSHL. The extended haplotype containing these five SNPs was the strongest overall predictor of risk for NSHL [80]. The haplotype with all five risk alleles for the SNPs (Hap3: AGGCT) was associated with a 70% increased risk of NSHL; while the haplotype with all five protective alleles (Hap6: GAATC) was associated with a 60% decreased risk. The DRB1*07:01 allele, which was carried by all individuals with haplotype 6 (GAATC), was associated with a 50% decreased risk of NSHL, suggesting HLA-DRB1 polymorphisms likely implicated in NSHL susceptibility.
9. Mature B-Cell Lymphoproliferative Disorder
Mature B-cell neoplasms account for over 90% lymphomas, with the most common types being diffuse large B-cell lymphoma and follicular lymphoma. Diffuse large B-cell lymphoma (DLBCL) is composed of large neoplastic B-cells and genetically heterogeneous. Based on gene expression profiles, DLBCL can be further classified into germinal center B-cell like (GCB) type with good prognosis and activated B-cell like (ABC) type with poor prognosis. Scholtysik et al. [81] applied 250K SNP array on 148 cases of DLBCL and found recurrent genomic gains in 24 regions and recurrent genomic losses in 38 regions, with a median of 19 imbalances per case in GCB-DLBCL and 25 per case in ABC-DLBCL. A number of genetic alterations showed different frequencies in GCB and ABC-DLBCL, such as gains of HDAC7A on chromosome 12 predominantly in GCB-DLBCL (38% of cases) and losses of BACH2 and CASP8AP2 on chromosome 6 predominantly in ABC-DLBCL (35%), suggesting different pathways to lymphomagenesis in the two subtypes of DLBCL. Two new potential tumor suppressor genes CASP3 and IL5RA were identified in the analysis and showed no somatic mutations, suggesting a haploinsufficiency effect of the genes. Another study showed high frequency of LOH over chromosomal region 11p11.2 harboring the gene encoding the protein tyrosine phosphatase receptor type J (PTPRJ), which regulates a number of survival pathways [82]. The combination of SNP array and whole exome/whole transcriptome sequencing was especially productive and identified ARID1B, ROBO2, and MRS1 as potential tumor suppressor genes and KLHL6, IL31, and LRP1 as oncogenes in DLBCL [83]. The impact of genomic alterations on clinical course was studied by 250 SNP array in 124 patients treated with R-CHOP [84]. 20 recurrent genetic lesions were shown to have an impact on the clinical course, in which loss of 8p23.1 had the strongest statistical significance. In this study, five clusters of DLBCL showed distinct genetic profiles, clinical features, and outcomes.
Follicular lymphoma (FL) is a mature B-cell lymphoma composed of malignant germinal center B-cells with some cases eventually transforming to diffuse large B-cell lymphoma. Early study with 10K SNP array on 26 cases of FL revealed recurrent aUDP on 6p, 9p, 12q, and 17p [85]. Homozygosity of 9p and 17p were found predominantly in transformed FL with homozygosity of pre-existing mutation of either CDKN2A or TP53 identified in a subset of cases. Interestingly, 10 cases showed chromosomal regions of homozygosity in FL that were absent in the subsequent transformed FL, suggesting the transformed FL derived from a common malignant precursor but not from step-wise clonal evolution of the preceding FL, at least in some cases [85]. The prognostic significance of aUPD was investigated in 185 cases of FL by 10K SNP array [86]. This study found genetic abnormalities in 65% cases, and more than three abnormalities were associated with inferior overall survival. Recurrent aUPD were detected on 6p, 16p, 12q, 1p36, 10q, and 6q, which were confirmed by other studies [87,88]. O’Shea et al. [86] showed that aUPD on 1p36 was correlated with shorter overall survival on multivariate analysis, and aUPD on 16p predicted transformation and poorer progression free survival. Somatic mutations of TNFRSF14 gene was identified by exon sequencing of the minimum region of deletion of ∼97 kb within 1p36.32, and TNFRSF14 mutations and 1p36 deletions were associated with inferior overall survival and disease specific survival after adjustment for the International Prognostic Index [89]. Individual genes including CDKN2A, CDKN2B, FHIT, KIT, PEX14, and PTPRD, which were associated with canonical pathways, were implicated by SNP array analysis in FL [87]. FL is characterized by t(14;18)(q32;q21) with IGH/BCL2 fusion, but a small subset of cases of FL is negative for t(14;18). The genetic profiles of t(14;18) positive and negative FL were compared by SNP array, and showed that gains/amplifications of the BCL2 gene locus at 18q were only present in the t(14;18)-positive FL [90].
Chronic lymphocytic leukemia is the most common leukemia in adults, and characterized by proliferation of small mature lymphoid cells in the peripheral blood, bone marrow, spleen and lymph nodes. Risk stratification based on FISH findings are routinely performed clinically. Early studies with 10 K and 50 K Affymetrix SNP arrays showed chromosomal imbalances in 65.6% and 82% cases, respectively, including UPD in 20% cases [91,92]. The cytogenetic changes commonly identified by FISH studies including trisomy 12, deletions of TP53 (17p13), ATM (11q22), and 13q14 were readily identified by SNP arrays. SNP arrays found a total of 45 CNAs in 45% cases excluding the four common cytogenetic changes identifiable by FISH [92]. High resolution Affymetrix SNP array 6.0 study on 353 samples of chronic lymphocytic leukemia (CLL) showed similar findings [93], with CN-LOH in 6% of CLL cases, most frequently on 13q, 17p, and 11q. Minimally-deleted regions were identified on 13q14 to the DLEU1 and DLEU2 genes, 11q22.3 to ATM, 2p16.1-2p15 to a 1.9-Mb fragment containing nine genes, and 8q24.21 to a 486 kb region proximal to the MYC locus. Breakpoint cluster regions flanking 13q deletions were found. A 3.5-Mb gain at 2p16 harboring REL and BCL11A oncogenes and deletion at 6q21 that involved the AIM1 gene were identified in a subset of cases [91,92], suggesting involvement of these genes in the pathogenesis of CLL. UPD was detected in 7% cases, with 50% involving whole chromosome 13 resulting in homozygous deletion of micro-RNA-15a (miR-15a)/miR-16-1 [92], which was confirmed by another study [94]. The genomic complexity identified by SNP array was an independent risk factor for aggressive CLL and short survival on multivariate analysis [94,95,96,97]. Large genomic aberrations identified by SNP array but not covered by the standard FISH panel was found to be an independent prognosticator of a shorter time to first treatment in CLL, by multivariate analysis [98]. Clonal evolution was shown to developed in 33% patients with unmutated IGHV, and in 16% treated patients with mutated IGHV, and included known recurrent aberrations such as del(13q) [94]. Analysis of clonal diversity by SNP array for genomic alterations and mosaic distribution of clones was also shown to be predictive of disease progression [99]. Del13q14 is the most frequent cytogenetic abnormality in CLL and associated with good prognosis. Ouillette et al. [100] showed that del13q14 was heterogeneous and composed of multiple subtypes, with deletion of RB or the miR15a/miR16 loci as anatomic landmarks. Large (type II) 13q14 deletions spanning the RB gene were associated with elevated genomic complexity, accelerated clinical course and short survival [101]. Similarly a separate study was able to classify CLL with del13q14 into two separate clusters characterized by short/biallelic deletion with loss of the miR-15a/16-1 versus wide/monoallelic 13q14 deletions [102]. Therefore, despite the established good prognosis of del13q14 by FISH, SNP arrays are clinically useful to identify a subset of cases with del13q14 that has poor prognosis. SNP arrays were shown to be able to supplement FISH studies with unusual signal patterns [103].
Mantle cell lymphoma is an aggressive B-cell lymphoma characterized by IGH/CCND1 translocation resulting in overexpression of cyclin D1. A large numbers of genomic abnormalities have been identified through metaphase cytogenetics and comparative genomic hybridization in addition to the characteristic t(11;14)(q13;q32). With 250K SNP array, Kawamata et al. [104] confirmed the presence of known genetic alterations, including deletion of INK4A/ARF, duplication/amplification of MYC, deletion of ATM, and deletion of TP53 in 33 samples of MCL. A duplication/amplification at 13q involving oncogenic microRNA, miR17-92, other genomic abnormalities, including duplication/amplification of cyclin D1, del(1p), del(6q), dup(3q) and dup(18q), and a number of aUPD sites, including whole chromosome 9 aUPD and 9p aUPD were identified by the SNP array. To identify the target genes in the genomic lesions, Beà et al. [105] combined SNP array and gene expression profiling and detected high number of partial UPDs with UPD 17p one of the most common associated with TP53 gene inactivation. 4 known tumor suppressor genes (CDKN2C, BCL2L11, CDKN2A, and RB1) and six new genes (FAF1, MAP2, SP100, MOBKL2B, ZNF280A, and PRAME) were identified in homozygous deletions. The most recurrent amplifications were at 11q13.3–q13.5, 13q31.3, and 18q21.33, targeting CCND1, C13orf25, and BCL2, which may be important for the lymphomogenesis of MCL.
Marginal zone lymphoma is a diverse group of B-cell lymphoma derived from post-germinal center B-cells. SNP array studies in this group of lymphoma are limited. Flossbach et al. [106] examined a series of marginal zone B-cell lymphomas of the gastrointestinal tract including ones with large cell transformation by SNP array. They found increase of genomic complexity with lymphoma progression to large cell lymphoma. Gains of protooncogenes such as REL, BCL11A, ETS1, PTPN1, PTEN, and KRAS were identified exclusively in the large cell variants. Copy numbers of ADAM3A, SCAPER and SIRPB1 were also associated with progression from small to large cell lymphoma. The gene for tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20), a negative regulator of NF-κB was found to be deleted at chromosomal region 6q23 in a small subset of marginal zone lymphomas by SNP array analysis [107]. In ocular adnexal MALT lymphoma, one study showed CNAs in approximately 70% cases and UPD detected on 6q (14%) and 3q (10%) [108]. The UPD on 6q likely involves the A20 gene. In this study, chromosomal gains were most commonly trisomy 3 (31%), trisomy 18 (17%), and 6p and 21q (14%), and the most frequent copy neutral (CN) loss regions were 6q and 9p (7%). Importantly, CNAs were not detected in reactive lymphoid hyperplasia, suggesting that SNP may be useful in difficult cases for diagnostic purpose.
Burkitt lymphoma (BL) is an aggressive B-cell lymphoma mainly in pediatric patients and characterized cytogenetically by t(8;14)(q24;q32) involving the MYC gene. Illumina 1M SNP array was applied to 20 cases of BL to identify additional genomic lesions in addition to t(8;14)(q24;q32) [109]. Genomic imbalances were found in 95% cases by SNP array, including recurrent losses of 6q14.1–q22.33, 9p21.3, and 13q14.2–q14.3, gains of 1q23.3–q31.3, chromosome 7, 13q31.3, and partial UPD for 6p12.2-pter, 9p23-pter, and 17p11.2-pter. These genetic alterations resulted in deletion of CDKN2A and TP53 genes, and gains/losses of other genes including MIR17HG and E2F2K that are involved in the MYC pathway, suggesting dysregulation of the MYC pathway by 8q24/MYC translocation or secondary genomic alterations are essential for development of Burkitt lymphoma.
Hairy cell leukemia (HCL) is an indolent B-cell neoplasm with “hairy cells” in the peripheral blood and bone marrow aspirates, and characteristic BRAF V600E mutation. With current therapy, patients with HCL have an excellent prognosis and near normal life span. HCL was found to have a remarkably stable genome by a 250K SNP array analysis [110]. With high resolution SNP Array 6.0, Rinaldi et al. [111] confirmed this finding. In this study, the 19 cases of HCL showed an extremely low numbers of CNAs with only two heterozygous losses detected (11%). These studies indicate very limited genetic damages in HCL, which may partly explain the excellent treatment response in current therapy.
Multiple myeloma (MM) is a neoplasm of terminally differentiation B-cells (plasma cells) characterized by multisystem damage and presence of M-Spike in the peripheral blood. In a study of 30 samples of patients with newly diagnosed MM by a 50K SNP array, genomic alterations at 1p, 1q, 6q, 8p, 13, and 16q were most frequent [112]. Multiple regions of UPD were identified for the first time and were found to be interspersed throughout the genome, with a median of three UPD per sample (range, 0–19). These findings were largely confirmed by Agnelli et al. [113] in analyzing 41 cases of MM and four cases of plasma cell leukemia. Using unsupervised clustering methods five main groups of genetic imbalances were identified and showed strict correlation with transcriptional expression: cluster I with hyperdiploidy, particularly trisomy 11; cluster II with no or limited alterations; cluster III with 1q gain and chromosome 13 deletion; cluster IV with deletions of 1p, 13, 14, plus deletions of 8p and 22; and cluster V with near-tetraploidy. In an effort to address the prognostic significance of genetic lesions detected by high resolution SNP array, Avet-Loiseau et al. [114] showed deletions and amplifications in 98% of patients with MM. Amplifications in 1q and deletions in 1p, 12p, 14q, 16q, and 22q were frequently associated with adverse prognosis, and recurrent amplifications of chromosomes 5, 9, 11, 15, and 19 was associated with a favorable prognosis. Amp(1q23.3), amp(5q31.3), and del(12p13.31) retained independent prognostic value in multivariate analysis. Del(12p13.31) alone, or amp(5q31.3) and del(12p13.31), and high Sβ2M predicted a very poor prognosis. The prognostic significance of 1q amplification was confirmed by two other groups even after removing cases with the most adverse cytogenetic factors such as translocations involving FGFR3/MMSET, MAF, and MAFB, and del17p [115,116]. Walker et al. [115] identified UPD on 1q (8%), 16q (9%), and X (20%), that was associated with regions of gain and loss. Kamada et al. [116] showed accumulation of deletions and UPD at 22q12.1 associated with poor prognosis in hyperdiploid MM. With a 500K SNP array, Jenner et al. [117] identified LOH at 16q involving CYLD, a negative regulator of the NF-κB pathway, and WWOX, a tumor suppressor involved in apoptosis, that were independently associated with poor prognosis in MM. Multiple myeloma nowadays is typically treated with hypomethylating agent, bortezomib plus melphalan and prednisone. Kim et al. [118] showed that increasing genomic complexity identified by SNP arrays correlated with the outcome of the bortezomib plus melphalan and prednisone therapy. Patients with deletion of 1p and gain of 3q did not achieve very good partial response, while complex karyotype and gain of 3q were associated with progressive disease after therapy. Finally, López-Corral et al. [119] showed progressive increase in the incidence of CNAs from precursor monoclonal gammopathy of undetermined significance (MGUS) to MM. Gains on 1q, 3p, 6p, 9p, 11q, 19p, 19q, and 21q along with deletions of 1p, 16q, and 22q were significantly less frequent in MGUS than in MM. The frequency of UPD was higher in active MM than in the asymptomatic MM. As expected, the increasing genomic complexity from MGUS to MM is consistent with acquisition of additional genomic lesions as essential pathway to the progression from MGUS to MM.
10. Mature T/NK-Cell Lymphoproliferative Disorders
The T/NK-cell lymphoproliferative disorders comprise a diverse group of mature lymphomas or leukemias with variable etiology and clinical course. SNP array studies in this group of neoplasms are limited to a few selected types, largely due to the much lower frequency of occurrence of mature T/NK-cell leukemia/lymphoma overall. Peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS) is the most common T-cell lymphoma and morphologically, phenotypically and cytogenetically heterogeneous. Hartmann et al studied 47 cases of PTCL, NOS with a 250K SNP array, and found genomic alterations in 47% of cases, including recurrent gains of chromosome regions 1q32–43, 2p15–16, 7, 8q24, 11q14–25, 17q11–21 and 21q11–21 and losses of chromosome regions 1p35–36, 5q33, 6p22, 6q16, 6q21–22, 8p21–23, 9p21, 10p11–12, 10q11–22, 10q25–26, 13q14, 15q24, 16q22, 16q24, 17p11, 17p13, and Xp22 [120]. Genomic gains of REL gene locus at 2p15–16 and nuclear expression of the REL protein by immunohistochemistry were identified in approximately 25% cases of PTCL, NOS, suggesting pathogenetic relevance of REL in a subset of PTCL, NOS cases.
Angioimmunoblastic T-cell lymphoma (AILT) is the second most common mature T-cell lymphoma and characterized by proliferation of malignant follicular T-helper cells associated with Epstein-Barr virus infection. In a study comparing the genomic profiles of 40 cases of AILT and 33 cases of PTCL, NOS by 50 K SNP array, three quarters of the cases had relatively stable genomes, while the remaining one quarter had CNAs of various sizes [121]. The presence of CNAs was associated with poor prognosis. Highly-recurrent chromosomal gains in both AILT and PTCL, NOS were clustered at three distinct regions of 8q, 9p, and 19q, and genomic losses at two distinct regions of 3q and 9p. The most common region of LOH was identified in a 440-kb region at 2q32.3. AILT- or PTCL NOS-specific CNAs or LOH were present at 21 regions. Furthermore, overexpression of CARMA1 at 7p22 and MYCBP2 at frequently amplified 13q22 predicted poor prognosis. A novel isoform of IKZF2 was identified in the LOH region at 2q34, which likely acted as a dominant negative form to participate in the transformation to AILT or PTCL, NOS.
Adult T-cell leukemia/lymphoma is a well-defined malignant T-cell neoplasm caused by human T-cell leukemia virus type 1 (HTLV-1). To understand the genetic events occurring after HTLV-1 infection, spectral karyotyping and SNP array of 61 ATLL cases revealed a 2-Mb deletion region breakpoint in 10p11.2 in 35% cases [122]. Transcription Factor 8 (TCF8) was identified within this region by gene expression studies as a possible tumor suppressor for ATLL. Loss of TCF8 resulted in resistance to transforming growth factor β1 (TGF-β1) mediated growth inhibition in ATLL cells, which likely contributed to the pathogenesis of ATLL. The same group in a subsequent study showed localization of the breakpoints at 10p11.2 within the EPC1 locus by SNP array in two cases [123]. EPC1 is a member of the polycomb group gene family and participate in chromatin formation and gene regulation. EPC1/ASXL2 and truncated EPC1 were identified in the two cases respectively, and both were able to induce cellular proliferation in in vitro studies, implicating EPC1 in the pathogenesis of ATLL in some cases.
T-cell prolymphocytic leukemia (T-PLL) is an aggressive T-cell leukemia characterized by proliferation of prolymphocytes in the peripheral blood, bone marrow, liver, spleen and lymph nodes. The most frequent genetic change is inversion of chromosome 14. Gains in 6p (3/12), 8q (10/12), and of losses in 6q (5/12), 8p (7/12), 10p (4/12), 11q (3/12), and 18p (3/12) were identified by a 50K SNP array when analyzing 11 T-PLL with inv(14)(q11q32) or t(14;14)(q11;q32) and one T-PLL case without inv(14)/t(14;14) [124]. Recurrent UPD in 3q, and non-recurrent partial UPD on chromosomes 3, 6, 9, 11, and 13 were identified. In a subsequent study of 18 cases of T-PLL by 250K SNP array, Nowak et al. [125] identified abundant copy number alterations, and confirmed the characteristic genetic lesions described before. Recurrent microdeletions targeting microRNA 34b/c, ETS1, and FLI1 were implicated in losses in chromosome 11, and PLEKHA2, NBS1, NOV and MYST3 genes were found to be involved in the breakpoints of chromosome 8. New recurrent lesions were identified on chromosomes 5p, 12p, 13q, 17, and 22 including aUPD on chromosome 17q, with genes DNAH5, ETV6, miR-15a, and miR-16-1, p53, BIRC5, and SOCS3 implicated in the regions. Future studies of the implicated genes identified by SNP array are likely going to further our understanding of the pathogenesis and provide potential targets for therapy.
Sézary syndrome (SS) is rare but aggressive neoplasm characterized by erythroderma, generalized adenopathy, and infiltrate of cerebriform Sézary cells in the peripheral blood, skin and lymph nodes. A low resolution 10K SNP array of eight patients with SS identified frequent SNP copy number changes and LOH involving 1, 2p, 3, 4q, 5q, 6, 7p, 8, 9, 10, 11, 12q, 13, 14, 16q, 17, and 20 [126]. SNP copy number loss was most frequent at FAT gene at 4q35 (75%), followed by VEGFC at 4q34.1q34.3 (50%), NFIB at chromosome 12 (38%), and TRIM16 at 17p11.2 (38%). SNP LOH gene clusters at chromosome regions of 9q31q34, 10p11q26, and 13q11q12 were only present in SS but not in normal controls, suggesting their involvement in SS pathology.
11. Conclusions
In summary, SNP array studies have contributed significantly in our understanding of the genomics of various hematopoietic malignancies. A summary of the common genomic abnormalities identified by SNP arrays is presented in Table 1. Several oncogenes have been mapped through application of SNP array and were shown to be important in clinical applications. As the cost of next generation sequencing (NGS) continues to drop, NGS will be increasingly applied in clinical labs. As a result, SNP array will gradually phase out. However, as a mature technology with fully developed data analysis and relatively low cost, SNP array will continue to play a role in the clinical lab, especially in situations where diagnostic and prognostic significance of SNP lesions were well established.

{kind=link}
| Disease | CNVs/CNAs and/or Associated Genes | LOH/UPD and/or Associated Genes | Prognostic Association | Ref. |
|---|---|---|---|---|
| B-ALL | Deletion of PAX5, EBF1, TCF3, LEF1, IKZF1 (IKAROS), IKZF3 (AIOLOS), ETV6, and CDKN2A/p16INK4A | 9p (CDKN2A/B) | - | [26,27,28,29,35] |
| T-ALL | Deletion of TAL1, RB1, PTEN, CDKN2A, CDKN2B, LEF1, and STIL; Gains of MYB | 9p (CDKN2A) | - | [32] |
| AML | Deletion of 3p14.1–p13 (FOXP1, RYBP, FHIT), 6q27 (RPS6KA2), 8q23.3 (TRPS1), 10q11.21 (HNRPF), 11q25, 12p13.2 (ETV6), 15q21.3 (RFXDC2), 5q31.1 (CTNNA1), 16q22.1 (CBFB), 17p13.1 (TP53), 17q11.2 (NF1) | 13q (FLT3), 11p (WT1, PU1) and 11q (MLL), 19q (CEBPA), 6p and 21q (RUNX1) | Worse prognosis: ≥2 genomic lesions detected by SNP array | [42,43,44,45,47,48,49] |
| Amplifications of 8q23.2 (MYC), 11q23.3 (MLL), and 21q22.2 (ETS2) | ||||
| MDS | Deletion and aUPD of chromosomes 1, 5q, 7, 11, 17, and 21 | Worse prognosis: UPDs of 7q; New genetic lesions detected by SNP array; EZH2 mutations | [57,58,60,61,62,63,64] | |
| Deletion of EZH2 and TET2 | ||||
| UPD 20p (BMP2 and TRIB3) | ||||
| CML | Frequent amplifications in chronic phase | 1, 8, 9, 17, 19, and 22 in TKI-resistant CML | - | [66,67] |
| Deletion of IKFZ1 in lymphoid blast phase | ||||
| MPN | Rare in ET and PV | 9p (JAK2) | Worse prognosis: CNN-LOH on 7q or 9p (JAK2 V617F); Genetic aberrations of chromosome 5, 7, or 17p | [68,69,70,73,74] |
| Deletion of 13q14 (RB) or 17q11 (NF1) in PMF | ||||
| CMML | Frequent microdeletions | 7q (EZH2), 11q (CBL), and 4q (TET2) | Worse prognosis: Multiple chromosomal defects detected by SNP array | [75,76] |
| cHL | Gain of MAP3K14 | 14q (TRAF3) | - | [79] |
| DLBCL | Frequent gains and deletions; gains HDAC7A on chromosome 12 predominantly in GCB-DLBCL, losses of BACH2 and CASP8AP2 on chromosome 6 predominantly in ABC-DLBCL; Potential tumor suppressor genes: CASP3, IL5RA ARID1B, ROBO2 and MRS1; Potential oncogenes: KLHL6, IL31 and LRP1 | 11p11.2 (PTPRJ), | Worse prognosis: Loss of 8p23.1 | [81,82,83,84] |
| FL | CDKN2A, CDKN2B, FHIT, KIT, PEX14, and PTPRD | 1p36 (TNFRSF14), 6p, 6q, 9p (CDKN2A), 10q, 12q, 16p, and 17p (TP53) | Worse prognosis: >3 SNP abnormalities; aUPD and deletion of 1p36, aUPD of 16p | [85,86] |
| CLL | Deletion of 17p13 (TP53), 11q22 (ATM) and 13q14 (DLEU1 and DLEU2), 2p16.1–2p15, 8q24.21, 6q21 (AIM1) | 13q, 13 (miR-15a/miR-16-1), 17p, and 11q | Worse prognosis: Genomic complexity; large genomic aberrations; large (type II) 13q14 deletions | [91,92,93,101] |
| Gain of 12, 2p16 (REL, BCL11A) | ||||
| MCL | Deletion of INK4A/ARF, ATM, TP53, 1p, 6q, CDKN2C, BCL2L11, CDKN2A, and RB1, FAF1, MAP2, SP100, MOBKL2B, ZNF280A, and PRAME. | 9p, 9, 17p (TP53) | - | [104,105] |
| Amplification of MYC, 11q13(cyclin D1), 13q (miR17-92, C13 or f25), dup(3q), 18q (BCL2) | ||||
| MZL | Deletion of 6q23 (TNFAIP3, A20), 9p | 6q (A20), 3q | - | [106,107,108] |
| Gain of 3, 18, 6p and 21q | ||||
| Gains of REL, BCL11A, ETS1, PTPN1, PTEN and KRAS in transformation to DLBCL | ||||
| BL | Losses of 6q14.1–q22.33, 9p21.3 (CDKN2A), and 13q14.2–q14.3 | 6p12.2-pter, 9p23-pter, and 17p11.2-pter (TP53). | - | [109] |
| Gains of 1q23.3–q31.3, 7, 13q31.3, | ||||
| MM | Genomic alterations at 1p, 1q, 6q, 8p, 13, and 16q | 1q, 16q (CYLD), and X | Worse prognosis: Amplifications in 1q and deletions in 1p, 12p, 14q, 16q, and 22q | [111,112,115,116,117] |
| PTCL, NOS | Losses of 1p35-36, 3q, 5q33, 6p22, 6q16, 6q21–22, 8p21–23, 9p21, 10p11–12, 10q11-22, 10q25–26, 13q14, 15q24, 16q22, 16q24, 17p11, 17p13 and Xp22 | 2q32.3 | - | [120,121] |
| Gains of 1q32–43, 2p15–16 (REL), 7, 8q24, 11q14–25, 17q11–21 and 21q11–21, 9p and 19q | ||||
| AILT | Loss of 3q and 9p | 2q32.3 | Worse prognosis: The presence of CNAs; overexpression of CARMA1 at 7p22 and MYCBP2 at 13q22 | [121] |
| Gains of 8q, 9p and 19q | ||||
| ATLL | Deletion of 10p11.2 ( TCF8) | - | - | [122] |
| T-PLL | Loss in 6q, 8p, 10p, 11q (microRNA 34b/c, ETS1 and FLI1), and 18p and Gains of 6p, 8q | 3q, 17q | - | [124,125] |
| Aberrations in 5p, 12p, 13q, 17 and 22 (DNAH5, ETV6, miR-15a and miR-16-1, p53, BIRC5, and SOCS3) | ||||
| SS | Loss at 4q35 (FAT), 4q34 (VEGFC), 12 (NFIB), and 17p11.2 (TRIM16) | 9q31q34, 10p11q26, and 13q11q12 | - | [126] |
CNV, copy number variations; CAN, copy number aberrations; LOH, loss of heterozygosity; UPD, uniparental disomy; Ref, reference; B-ALL, B lymphoblastic leukemia; T-ALL, T lymphoblastic leukemia; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome; CML, chronic myelogenous leukemia; MPN, myeloproliferative neoplasm; PV, polycythemia (Rubra) vera; ET, essential thrombocythemia; PMF, primary myelofibrosis; CMML, chronic myelomonocytic leukemia; cHL, classical Hodgkin lymphoma; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MZL, marginal zone lymphoma; BL, Burkitt lymphoma; MM, multiple myeloma; PTCL, NOS, peripheral T-cell lymphoma, NOS; AILT, angioimmunoblastic T-cell lymphoma; ATLL, adult T-cell leukemia/lymphoma; T-PLL, T-cell prolymphocytic leukemia; SS, Sezary syndrome.
Author Contributions
All authors performed literature searches and wrote this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hahn, W.C.; Weinberg, R.A. Rules for making human tumor cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Bayani, J.S.J. Traditional banding of chromosomes for cytogenetic analysis. Curr. Protoc. Cell Biol. 2004, S23. [Google Scholar] [CrossRef]
- Levsky, J.M.; Singer, R.H. Fluorescence in situ hybridization: Past, present and future. J. Cell Sci. 2003, 116, 2833–2838. [Google Scholar] [CrossRef] [PubMed]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Du Manoir, S.; Speicher, M.R.; Joos, S.; Schrock, E.; Popp, S.; Dohner, H.; Kovacs, G.; Robert-Nicoud, M.; Lichter, P.; Cremer, T. Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum. Genet. 1993, 90, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Albertson, D.G. Comparative genomic hybridization. Annu Rev. Genomics Hum. Genet. 2005, 6, 331–354. [Google Scholar] [CrossRef] [PubMed]
- De Ravel, T.J.; Devriendt, K.; Fryns, J.P.; Vermeesch, J.R. What’s new in karyotyping? The move towards array comparative genomic hybridisation (CGH). Eur. J. Pediatr. 2007, 166, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.C.; Matsuzaki, H.; Dong, S.; Liu, W.M.; Huang, J.; Liu, G.; Su, X.; Cao, M.; Chen, W.; Zhang, J.; et al. Large-scale genotyping of complex DNA. Nat. Biotechnol. 2003, 21, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Tanenbaum, D.M.; Daly, M.J.; Winchester, E.; Lui, W.O.; Villapakkam, A.; Stanton, S.E.; Larsson, C.; Hudson, T.J.; Johnson, B.E.; et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat. Biotechnol. 2000, 18, 1001–1005. [Google Scholar] [PubMed]
- Wong, K.K.; Tsang, Y.T.; Shen, J.; Cheng, R.S.; Chang, Y.M.; Man, T.K.; Lau, C.C. Allelic imbalance analysis by high-density single-nucleotide polymorphic allele (SNP) array with whole genome amplified DNA. Nucleic Acids Res. 2004, 32. [Google Scholar] [CrossRef] [PubMed]
- La Framboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Parkin, D.M. Geographic and ethnic variations in the incidence of childhood cancer. Br. Med. Bull. 1996, 52, 682–703. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. Who Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; pp. 167–178. [Google Scholar]
- Schrappe, M.; Reiter, A.; Ludwig, W.D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM study group. Blood 2000, 95, 3310–3322. [Google Scholar] [PubMed]
- Gaynon, P.S.; Trigg, M.E.; Heerema, N.A.; Sensel, M.G.; Sather, H.N.; Hammond, G.D.; Bleyer, W.A. Children’s cancer group trials in childhood acute lymphoblastic leukemia: 1983–1995. Leukemia 2000, 14, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Harms, D.O.; Janka-Schaub, G.E. Co-operative study group for childhood acute lymphoblastic leukemia (COALL): Long-term follow-up of trials 82, 85, 89 and 92. Leukemia 2000, 14, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.B.; Gelber, R.D.; Dalton, V.K.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Hurwitz, C.A.; Moghrabi, A.; Samson, Y.; Schorin, M.A.; et al. Improved outcome for children with acute lymphoblastic leukemia: Results of dana-farber consortium protocol 91–01. Blood 2001, 97, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, G.; Schmiegelow, K.; Forestier, E.; Clausen, N.; Glomstein, A.; Jonmundsson, G.; Mellander, L.; Makipernaa, A.; Nygaard, R.; Saarinen-Pihkala, U.M. Improving outcome through two decades in childhood all in the nordic countries: The impact of high-dose methotrexate in the reduction of CNS irradiation. Nordic society of pediatric haematology and oncology (NOPHO). Leukemia 2000, 14, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Sandlund, J.T.; Pei, D.; Rivera, G.K.; Howard, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Razzouk, B.I.; Hudson, M.M.; Cheng, C.; et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 2003, 290, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Gokbuget, N.; Hoelzer, D. Recent approaches in acute lymphoblastic leukemia in adults. Rev. Clin. Exp. Hematol. 2002, 6, 114–141. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Smith, T.L.; Cortes, J.; Giles, F.J.; Beran, M.; Pierce, S.; Huh, Y.; Andreeff, M.; Koller, C.; et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J. Clin. Oncol. 2000, 18, 547–561. [Google Scholar] [PubMed]
- Linker, C.; Damon, L.; Ries, C.; Navarro, W. Intensified and shortened cyclical chemotherapy for adult acute lymphoblastic leukemia. J. Clin. Oncol. 2002, 20, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.F.; Wiemels, J. Origins of chromosome translocations in childhood leukaemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Irving, J.A.; Bloodworth, L.; Bown, N.P.; Case, M.C.; Hogarth, L.A.; Hall, A.G. Loss of heterozygosity in childhood acute lymphoblastic leukemia detected by genome-wide microarray single nucleotide polymorphism analysis. Cancer Res. 2005, 65, 3053–3058. [Google Scholar] [PubMed]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, N.; Ogawa, S.; Zimmermann, M.; Kato, M.; Sanada, M.; Hemminki, K.; Yamatomo, G.; Nannya, Y.; Koehler, R.; Flohr, T.; et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood 2008, 111, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Bungaro, S.; Dell’Orto, M.C.; Zangrando, A.; Basso, D.; Gorletta, T.; Lo Nigro, L.; Leszl, A.; Young, B.D.; Basso, G.; Bicciato, S.; et al. Integration of genomic and gene expression data of childhood all without known aberrations identifies subgroups with specific genetic hallmarks. Genes Chromosomes Cancer 2009, 48, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Foroni, L. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Rev. Clin. Exp. Hematol. 2002, 6, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Graux, C.; Cools, J.; Michaux, L.; Vandenberghe, P.; Hagemeijer, A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: From thymocyte to lymphoblast. Leukemia 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Williams, R.T.; Downing, J.R.; Sherr, C.J. Failure of CDKN2A/b (INK4A/b-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008, 22, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Karrman, K.; Castor, A.; Behrendtz, M.; Forestier, E.; Olsson, L.; Ehinger, M.; Biloglav, A.; Fioretos, T.; Paulsson, K.; Johansson, B. Deep sequencing and SNP array analyses of pediatric T-cell acute lymphoblastic leukemia reveal NOTCH1 mutations in minor subclones and a high incidence of uniparental isodisomies affecting CDKN2A. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Ogawa, S.; Nowak, D.; Kawamata, N.; Akagi, T.; Kato, M.; Sanada, M.; Weiss, T.; Haferlach, C.; Dugas, M.; et al. Genomic profiling of adult acute lymphoblastic leukemia by single nucleotide polymorphism oligonucleotide microarray and comparison to pediatric acute lymphoblastic leukemia. Haematologica 2010, 95, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Safavi, S.; Hansson, M.; Karlsson, K.; Biloglav, A.; Johansson, B.; Paulsson, K. Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia. Haematologica 2015, 100, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.R.; Yang, W.; French, D.; Hunger, S.P.; Carroll, W.L.; Devidas, M.; Willman, C.; Neale, G.; Downing, J.; Raimondi, S.C.; et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Hosking, F.J.; Vijayakrishnan, J.; Price, A.; Olver, B.; Sheridan, E.; Kinsey, S.E.; Lightfoot, T.; Roman, E.; Irving, J.A.; et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.B.; Hosking, F.J.; Vijayakrishnan, J.; Papaemmanuil, E.; Koehler, R.; Greaves, M.; Sheridan, E.; Gast, A.; Kinsey, S.E.; Lightfoot, T.; et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood 2010, 115, 1765–1767. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schakel, U.; Platzbecker, U.; Wermke, M.; Bornhauser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Frohling, S.; Schlenk, R.F.; Stolze, I.; Bihlmayr, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Dohner, H.; Dohner, K. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: Prognostic relevance and analysis of cooperating mutations. J. Clin. Oncol. 2004, 22, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Lillington, D.M.; Skoulakis, S.; Debernardi, S.; Chaplin, T.; Foot, N.J.; Lister, T.A.; Young, B.D. Genome-wide single nucleotide polymorphism analysis reveals frequent partial uniparental disomy due to somatic recombination in acute myeloid leukemias. Cancer Res. 2005, 65, 375–378. [Google Scholar] [PubMed]
- Fitzgibbon, J.; Smith, L.L.; Raghavan, M.; Smith, M.L.; Debernardi, S.; Skoulakis, S.; Lillington, D.; Lister, T.A.; Young, B.D. Association between acquired uniparental disomy and homozygous gene mutation in acute myeloid leukemias. Cancer Res. 2005, 65, 9152–9154. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Raghavan, M.; Gale, R.E.; Chelala, C.; Allen, C.; Molloy, G.; Chaplin, T.; Linch, D.C.; Cazier, J.B.; Young, B.D. Novel regions of acquired uniparental disomy discovered in acute myeloid leukemia. Genes Chromosomes Cancer 2008, 47, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Kronke, J.; Schon, C.; Radtke, I.; Urlbauer, K.; Botzenhardt, U.; Gaidzik, V.; Cario, A.; Senger, C.; Schlenk, R.F.; et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia 2009, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Payton, J.E.; Ries, R.E.; Shannon, W.D.; Deshmukh, H.; Zhao, Y.; Baty, J.; Heath, S.; Westervelt, P.; Watson, M.A.; et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc. Natl. Acad. Sci. USA 2009, 106, 12950–12955. [Google Scholar] [CrossRef] [PubMed]
- Radtke, I.; Mullighan, C.G.; Ishii, M.; Su, X.; Cheng, J.; Ma, J.; Ganti, R.; Cai, Z.; Goorha, S.; Pounds, S.B.; et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 12944–12949. [Google Scholar] [CrossRef] [PubMed]
- Parkin, B.; Erba, H.; Ouillette, P.; Roulston, D.; Purkayastha, A.; Karp, J.; Talpaz, M.; Kujawski, L.; Shakhan, S.; Li, C.; et al. Acquired genomic copy number aberrations and survival in adult acute myelogenous leukemia. Blood 2010, 116, 4958–4967. [Google Scholar] [CrossRef] [PubMed]
- Tiu, R.V.; Gondek, L.P.; O’Keefe, C.L.; Huh, J.; Sekeres, M.A.; Elson, P.; McDevitt, M.A.; Wang, X.F.; Levis, M.J.; Karp, J.E.; et al. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J. Clin. Oncol. 2009, 27, 5219–5226. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Huh, J.; Kim, H.J.; Kim, S.H.; Kim, Y.K.; Sohn, S.K.; Moon, J.H.; Kim, K.H.; Won, J.H.; Mun, Y.C.; et al. Adverse prognostic impact of abnormal lesions detected by genome-wide single nucleotide polymorphism array-based karyotyping analysis in acute myeloid leukemia with normal karyotype. J. Clin. Oncol. 2011, 29, 4702–4708. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vardiman, J.W. Myelodysplastic syndromes. N. Engl. J. Med. 2009, 361, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Gondek, L.P.; Tiu, R.; Haddad, A.S.; O’Keefe, C.L.; Sekeres, M.A.; Theil, K.S.; Maciejewski, J.P. Single nucleotide polymorphism arrays complement metaphase cytogenetics in detection of new chromosomal lesions in mds. Leukemia 2007, 21, 2058–2061. [Google Scholar] [CrossRef] [PubMed]
- Gondek, L.P.; Haddad, A.S.; O’Keefe, C.L.; Tiu, R.; Wlodarski, M.W.; Sekeres, M.A.; Theil, K.S.; Maciejewski, J.P. Detection of cryptic chromosomal lesions including acquired segmental uniparental disomy in advanced and low-risk myelodysplastic syndromes. Exp. Hematol. 2007, 35, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, A.; Gaken, J.; Twine, N.A.; Ingram, W.; Westwood, N.; Lea, N.C.; Hayden, J.; Donaldson, N.; Aul, C.; Gattermann, N.; et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood 2007, 110, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Nolte, F.; Mossner, M.; Nowak, V.; Baldus, C.D.; Hopfer, O.; Noll, S.; Thiel, E.; Wagner, F.; Hofmann, W.K. Genome-wide DNA-mapping of CD34+ cells from patients with myelodysplastic syndrome using 500k SNP arrays identifies significant regions of deletion and uniparental disomy. Exp. Hematol. 2009, 37, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Heinrichs, S.; Kulkarni, R.V.; Bueso-Ramos, C.E.; Levine, R.L.; Loh, M.L.; Li, C.; Neuberg, D.; Kornblau, S.M.; Issa, J.P.; Gilliland, D.G.; et al. Accurate detection of uniparental disomy and microdeletions by SNP array analysis in myelodysplastic syndromes with normal cytogenetics. Leukemia 2009, 23, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Tiu, R.V.; Gondek, L.P.; O’Keefe, C.L.; Elson, P.; Huh, J.; Mohamedali, A.; Kulasekararaj, A.; Advani, A.S.; Paquette, R.; List, A.F.; et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 2011, 117, 4552–4560. [Google Scholar] [CrossRef] [PubMed]
- Afable, M.G., II; Wlodarski, M.; Makishima, H.; Shaik, M.; Sekeres, M.A.; Tiu, R.V.; Kalaycio, M.; O’Keefe, C.L.; Maciejewski, J.P. SNP array-based karyotyping: Differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood 2011, 117, 6876–6884. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, S.M.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Perez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Merkerova, M.D.; Bystricka, D.; Belickova, M.; Krejcik, Z.; Zemanova, Z.; Polak, J.; Hajkova, H.; Brezinova, J.; Michalova, K.; Cermak, J. From cryptic chromosomal lesions to pathologically relevant genes: Integration of SNP-array with gene expression profiling in myelodysplastic syndrome with normal karyotype. Genes Chromosomes Cancer 2012, 51, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; le Beau, M.M.; Pui, C.H.; et al. BCR-ABl1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Khorashad, J.S.; de Melo, V.A.; Fiegler, H.; Gerrard, G.; Marin, D.; Apperley, J.F.; Goldman, J.M.; Foroni, L.; Reid, A.G. Multiple sub-microscopic genomic lesions are a universal feature of chronic myeloid leukaemia at diagnosis. Leukemia 2008, 22, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Ogawa, S.; Muschen, M.; Kato, M.; Kawamata, N.; Meixel, A.; Nowak, V.; Kim, H.S.; Kang, S.; Paquette, R.; et al. SNP array analysis of tyrosine kinase inhibitor-resistant chronic myeloid leukemia identifies heterogeneous secondary genomic alterations. Blood 2010, 115, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Stegelmann, F.; Bullinger, L.; Griesshammer, M.; Holzmann, K.; Habdank, M.; Kuhn, S.; Maile, C.; Schauer, S.; Dohner, H.; Dohner, K. High-resolution single-nucleotide polymorphism array-profiling in myeloproliferative neoplasms identifies novel genomic aberrations. Haematologica 2010, 95, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.L.; Lin, X.; Wolniak, K.; Ebert, B.L.; Berkofsky-Fessler, W.; Buzzai, M.; Sun, Y.; Xi, C.; Elkin, P.; Levine, R.; et al. Analysis of genomic aberrations and gene expression profiling identifies novel lesions and pathways in myeloproliferative neoplasms. Blood Cancer J. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, N.; Ogawa, S.; Yamamoto, G.; Lehmann, S.; Levine, R.L.; Pikman, Y.; Nannya, Y.; Sanada, M.; Miller, C.W.; Gilliland, D.G.; et al. Genetic profiling of myeloproliferative disorders by single-nucleotide polymorphism oligonucleotide microarray. Exp. Hematol. 2008, 36, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Swierczek, S.I.; Lanikova, L.; Kim, S.J.; Hickman, K.; Walker, K.; Wang, K.; Drummond, J.; Doddapaneni, H.; Reid, J.G.; et al. The relationship of JAK2(V617F) and acquired upd at chromosome 9p in polycythemia vera. Leukemia 2014, 28, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Thoennissen, N.H.; Krug, U.O.; Lee, D.H.; Kawamata, N.; Iwanski, G.B.; Lasho, T.; Weiss, T.; Nowak, D.; Koren-Michowitz, M.; Kato, M.; et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of philadelphia chromosome-negative myeloproliferative neoplasms. Blood 2010, 115, 2882–2890. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Harutyunyan, A.; Elena, C.; Pietra, D.; Klampfl, T.; Bagienski, K.; Berg, T.; Casetti, I.; Pascutto, C.; Passamonti, F.; et al. Identification of genomic aberrations associated with disease transformation by means of high-resolution SNP array analysis in patients with myeloproliferative neoplasm. Am. J. Hematol. 2011, 86, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Makishima, H.; Tiu, R.V.; Szpurka, H.; Huang, Y.; Traina, F.; Visconte, V.; Sugimoto, Y.; Prince, C.; O’Keefe, C.; et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, AND DNMT3A. Blood 2011, 118, 3932–3941. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, A.J.; Gondek, L.P.; O’Keefe, C.L.; Makishima, H.; Rataul, M.S.; Szpurka, H.; Sekeres, M.A.; Wang, X.F.; McDevitt, M.A.; Maciejewski, J.P. 250k single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008, 68, 10349–10357. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Huh, J.; Kim, H.J.; Kim, S.H.; Kim, K.H.; Do, Y.R.; Mun, Y.C.; Kim, H.; Kim, M.K.; Kim, T.; et al. Genome-wide single-nucleotide polymorphism array-based karyotyping in myelodysplastic syndrome and chronic myelomonocytic leukemia and its impact on treatment outcomes following decitabine treatment. Ann. Hematol. 2013, 92, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Steinemann, D.; Mullighan, C.G.; Neale, G.; Mayer, K.; Kratz, C.P.; Schlegelberger, B.; Downing, J.R.; Niemeyer, C.M. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene 2007, 26, 5816–5821. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Giefing, M.; Massow, A.; Vater, I.; Gesk, S.; Schlesner, M.; Richter, J.; Klapper, W.; Hansmann, M.L.; Siebert, R.; et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical hodgkin lymphoma. Br. J. Haematol. 2012, 157, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Cozen, W.; Li, D.; Best, T.; Van Den Berg, D.J.; Gourraud, P.A.; Cortessis, V.K.; Skol, A.D.; Mack, T.M.; Glaser, S.L.; Weiss, L.M.; et al. A genome-wide meta-analysis of nodular sclerosing hodgkin lymphoma identifies risk loci at 6p21.32. Blood 2012, 119, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Scholtysik, R.; Kreuz, M.; Hummel, M.; Rosolowski, M.; Szczepanowski, M.; Klapper, W.; Loeffler, M.; Trumper, L.; Siebert, R.; Kuppers, R. Characterization of genomic imbalances in diffuse large B-cell lymphoma by detailed SNP-chip analysis. Int. J. Cancer 2015, 136, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Aya-Bonilla, C.; Green, M.R.; Camilleri, E.; Benton, M.; Keane, C.; Marlton, P.; Lea, R.; Gandhi, M.K.; Griffiths, L.R. High-resolution loss of heterozygosity screening implicates PTPRJ as a potential tumor suppressor gene that affects susceptibility to non-hodgkin’s lymphoma. Genes Chromosomes Cancer 2013, 52, 467–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonov, V.; Pasqualucci, L.; Dalla Favera, R.; Rabadan, R. Mutcomfocal: An integrative approach to identifying recurrent and focal genomic alterations in tumor samples. BMC Syst. Biol. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Scandurra, M.; Mian, M.; Greiner, T.C.; Rancoita, P.M.; De Campos, C.P.; Chan, W.C.; Vose, J.M.; Chigrinova, E.; Inghirami, G.; Chiappella, A.; et al. Genomic lesions associated with a different clinical outcome in diffuse large B-cell lymphoma treated with R-CHOP-21. Br. J. Haematol. 2010, 151, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbon, J.; Iqbal, S.; Davies, A.; O’Shea, D.; Carlotti, E.; Chaplin, T.; Matthews, J.; Raghavan, M.; Norton, A.; Lister, T.A.; et al. Genome-wide detection of recurring sites of uniparental disomy in follicular and transformed follicular lymphoma. Leukemia 2007, 21, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; O’Riain, C.; Gupta, M.; Waters, R.; Yang, Y.; Wrench, D.; Gribben, J.; Rosenwald, A.; Ott, G.; Rimsza, L.M.; et al. Regions of acquired uniparental disomy at diagnosis of follicular lymphoma are associated with both overall survival and risk of transformation. Blood 2009, 113, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Delaney, A.; Ben-Neriah, S.; Schein, J.; Lee, T.; Shah, S.P.; Cheung, D.; Johnson, N.A.; Mungall, A.J.; Telenius, A.; et al. High resolution analysis of follicular lymphoma genomes reveals somatic recurrent sites of copy-neutral loss of heterozygosity and copy number alterations that target single genes. Genes Chromosomes Cancer 2010, 49, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Rogic, S.; Ben-Neriah, S.; Boyle, M.; Connors, J.M.; Gascoyne, R.D.; Horsman, D.E. SNP analysis of minimally evolved t(14;18)(q32;q21)-positive follicular lymphomas reveals a common copy-neutral loss of heterozygosity pattern. Cytogenet. Genome Res. 2012, 136, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Johnson, N.A.; Affleck, J.G.; Severson, T.; Steidl, C.; Ben-Neriah, S.; Schein, J.; Morin, R.D.; Moore, R.; Shah, S.P.; et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010, 70, 9166–9174. [Google Scholar] [CrossRef] [PubMed]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, D.; Pantic, M.; Skatulla, I.; Rawluk, J.; Kreutz, C.; Martens, U.M.; Fisch, P.; Timmer, J.; Veelken, H. Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays. Blood 2007, 109, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Ogawa, S.; Raynaud, S.D.; Sanada, M.; Nannya, Y.; Ticchioni, M.; Bastard, C.; Kawamata, N.; Koeffler, H.P. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer 2008, 112, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Buhler, A.; Zenz, T.; Bullinger, L.; Kuhn, M.W.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 120, 4783–4794. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, R.; Mansouri, L.; Isaksson, A.; Goransson, H.; Cahill, N.; Jansson, M.; Rasmussen, M.; Lundin, J.; Norin, S.; Buhl, A.M.; et al. Array-based genomic screening at diagnosis and during follow-up in chronic lymphocytic leukemia. Haematologica 2011, 96, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, L.; Ouillette, P.; Erba, H.; Saddler, C.; Jakubowiak, A.; Kaminski, M.; Shedden, K.; Malek, S.N. Genomic complexity identifies patients with aggressive chronic lymphocytic leukemia. Blood 2008, 112, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Ouillette, P.; Collins, R.; Shakhan, S.; Li, J.; Peres, E.; Kujawski, L.; Talpaz, M.; Kaminski, M.; Li, C.; Shedden, K.; et al. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood 2011, 118, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Schweighofer, C.D.; Coombes, K.R.; Majewski, T.; Barron, L.L.; Lerner, S.; Sargent, R.L.; O’Brien, S.; Ferrajoli, A.; Wierda, W.G.; Czerniak, B.A.; et al. Genomic variation by whole-genome SNP mapping arrays predicts time-to-event outcome in patients with chronic lymphocytic leukemia: A comparison of CLL and hapmap genotypes. J. Mol. Diagn. 2013, 15, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.; Rinaldi, A.; Mensah, A.A.; Rossi, D.; Ladetto, M.; Forconi, F.; Marasca, R.; Uhr, M.; Stussi, G.; Kwee, I.; et al. Large genomic aberrations detected by SNP array are independent prognosticators of a shorter time to first treatment in chronic lymphocytic leukemia patients with normal fish. Ann. Oncol. 2013, 24, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Znoyko, I.; Costa, L.J.; Conlin, L.K.; Daber, R.D.; Self, S.E.; Wolff, D.J. Clonal diversity analysis using SNP microarray: A new prognostic tool for chronic lymphocytic leukemia. Cancer Genet. 2011, 204, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Ouillette, P.; Erba, H.; Kujawski, L.; Kaminski, M.; Shedden, K.; Malek, S.N. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res. 2008, 68, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Ouillette, P.; Collins, R.; Shakhan, S.; Li, J.; Li, C.; Shedden, K.; Malek, S.N. The prognostic significance of various 13q14 deletions in chronic lymphocytic leukemia. Clin. Cancer Res. 2011, 17, 6778–6790. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Fabris, S.; Lionetti, M.; Todoerti, K.; Agnelli, L.; Morabito, F.; Cutrona, G.; Andronache, A.; Matis, S.; Ferrari, F.; et al. Integrative genomics analyses reveal molecularly distinct subgroups of B-cell chronic lymphocytic leukemia patients with 13q14 deletion. Clin. Cancer Res. 2010, 16, 5641–5653. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, A.; Parker, H.; Glide, S.; Mould, S.; Robinson, H.; Tracy, I.; Stankovic, T.; Oscier, D.; Strefford, J. A new minimal deleted region at 11q22.3 reveals the importance of interpretation of diminished fish signals and the choice of probe for ATM deletion screening in chronic lymphocytic leukemia. Leuk. Res. 2012, 36, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, N.; Ogawa, S.; Gueller, S.; Ross, S.H.; Huynh, T.; Chen, J.; Chang, A.; Nabavi-Nouis, S.; Megrabian, N.; Siebert, R.; et al. Identified hidden genomic changes in mantle cell lymphoma using high-resolution single nucleotide polymorphism genomic array. Exp. Hematol. 2009, 37, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Salaverria, I.; Armengol, L.; Pinyol, M.; Fernandez, V.; Hartmann, E.M.; Jares, P.; Amador, V.; Hernandez, L.; Navarro, A.; et al. Uniparental disomies, homozygous deletions, amplifications, and target genes in mantle cell lymphoma revealed by integrative high-resolution whole-genome profiling. Blood 2009, 113, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Flossbach, L.; Holzmann, K.; Mattfeldt, T.; Buck, M.; Lanz, K.; Held, M.; Moller, P.; Barth, T.F. High-resolution genomic profiling reveals clonal evolution and competition in gastrointestinal marginal zone B-cell lymphoma and its large cell variant. Int. J. Cancer 2013, 132, E116–E127. [Google Scholar] [CrossRef] [PubMed]
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Usui, Y.; Ueda, S.; Yamakawa, N.; Sato-Otsubo, A.; Sato, Y.; Ogawa, S.; Goto, H. Genome-wide analysis of ocular adnexal lymphoproliferative disorders using high-resolution single nucleotide polymorphism array. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4156–4165. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; Hjorth, L.; Behrendtz, M.; Ehinger, M.; Biloglav, A.; Johansson, B. Submicroscopic genomic imbalances in burkitt lymphomas/leukemias: Association with age and further evidence that 8q24/MYC translocations are not sufficient for leukemogenesis. Genes Chromosomes Cancer 2013, 52, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Forconi, F.; Poretti, G.; Kwee, I.; Sozzi, E.; Rossi, D.; Rancoita, P.M.; Capello, D.; Rinaldi, A.; Zucca, E.; Raspadori, D.; et al. High density genome-wide DNA profiling reveals a remarkably stable profile in hairy cell leukaemia. Br. J. Haematol. 2008, 141, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Kwee, I.; Young, K.H.; Zucca, E.; Gaidano, G.; Forconi, F.; Bertoni, F. Genome-wide high resolution DNA profiling of hairy cell leukaemia. Br. J. Haematol. 2013, 162, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Jenner, M.W.; Li, C.; Gonzalez, D.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; Morgan, G.J. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood 2006, 108, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Mosca, L.; Fabris, S.; Lionetti, M.; Andronache, A.; Kwee, I.; Todoerti, K.; Verdelli, D.; Battaglia, C.; Bertoni, F.; et al. A SNP microarray and fish-based procedure to detect allelic imbalances in multiple myeloma: An integrated genomics approach reveals a wide gene dosage effect. Genes Chromosomes Cancer 2009, 48, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Li, C.; Magrangeas, F.; Gouraud, W.; Charbonnel, C.; Harousseau, J.L.; Attal, M.; Marit, G.; Mathiot, C.; Facon, T.; et al. Prognostic significance of copy-number alterations in multiple myeloma. J. Clin. Oncol. 2009, 27, 4585–4590. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Kamada, Y.; Sakata-Yanagimoto, M.; Sanada, M.; Sato-Otsubo, A.; Enami, T.; Suzukawa, K.; Kurita, N.; Nishikii, H.; Yokoyama, Y.; Okoshi, Y.; et al. Identification of unbalanced genome copy number abnormalities in patients with multiple myeloma by single-nucleotide polymorphism genotyping microarray analysis. Int. J. Hematol. 2012, 96, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Jenner, M.W.; Leone, P.E.; Walker, B.A.; Ross, F.M.; Johnson, D.C.; Gonzalez, D.; Chiecchio, L.; Dachs Cabanas, E.; Dagrada, G.P.; Nightingale, M.; et al. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood 2007, 110, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, S.H.; Kim, J.; Lee, S.E.; Kim, Y.J.; Min, C.K. Copy number variations could predict the outcome of bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. Genes Chromosomes Cancer 2015, 54, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Corral, L.; Sarasquete, M.E.; Bea, S.; Garcia-Sanz, R.; Mateos, M.V.; Corchete, L.A.; Sayagues, J.M.; Garcia, E.M.; Blade, J.; Oriol, A.; et al. SNP-based mapping arrays reveal high genomic complexity in monoclonal gammopathies, from MGUS to myeloma status. Leukemia 2012, 26, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Gesk, S.; Scholtysik, R.; Kreuz, M.; Bug, S.; Vater, I.; Doring, C.; Cogliatti, S.; Parrens, M.; Merlio, J.P.; et al. High resolution SNP array genomic profiling of peripheral T cell lymphomas, not otherwise specified, identifies a subgroup with chromosomal aberrations affecting the REL locus. Br. J. Haematol. 2010, 148, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.I.; Yamashita, Y.; Nakamura, N.; Choi, Y.L.; Ueno, T.; Watanabe, H.; Kurashina, K.; Soda, M.; Enomoto, M.; Hatanaka, H.; et al. High-resolution analysis of chromosome copy number alterations in angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified, with single nucleotide polymorphism-typing microarrays. Leukemia 2008, 22, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Nakahata, S.; Hatakeyama, K.; Hamasaki, M.; Yamashita, K.; Kohno, T.; Arai, Y.; Taki, T.; Nishida, K.; Okayama, A.; et al. Down-regulation of TCF8 is involved in the leukemogenesis of adult T-cell leukemia/lymphoma. Blood 2008, 112, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, S.; Saito, Y.; Hamasaki, M.; Hidaka, T.; Arai, Y.; Taki, T.; Taniwaki, M.; Morishita, K. Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer 2009, 48, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Durig, J.; Bug, S.; Klein-Hitpass, L.; Boes, T.; Jons, T.; Martin-Subero, J.I.; Harder, L.; Baudis, M.; Duhrsen, U.; Siebert, R. Combined single nucleotide polymorphism-based genomic mapping and global gene expression profiling identifies novel chromosomal imbalances, mechanisms and candidate genes important in the pathogenesis of T-cell prolymphocytic leukemia with INV(14)(q11q32). Leukemia 2007, 21, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; le Toriellec, E.; Stern, M.H.; Kawamata, N.; Akagi, T.; Dyer, M.J.; Hofmann, W.K.; Ogawa, S.; Koeffler, H.P. Molecular allelokaryotyping of T-cell prolymphocytic leukemia cells with high density single nucleotide polymorphism arrays identifies novel common genomic lesions and acquired uniparental disomy. Haematologica 2009, 94, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Chaplin, T.; Young, B.D. Integrated genomic analysis of sezary syndrome. Genet. Res. Int. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Song, J.; Shao, H. SNP Array in Hematopoietic Neoplasms: A Review. Microarrays 2016, 5, 1. https://doi.org/10.3390/microarrays5010001
AMA Style
Song J, Shao H. SNP Array in Hematopoietic Neoplasms: A Review. Microarrays. 2016; 5(1):1. https://doi.org/10.3390/microarrays5010001
Chicago/Turabian StyleSong, Jinming, and Haipeng Shao. 2016. "SNP Array in Hematopoietic Neoplasms: A Review" Microarrays 5, no. 1: 1. https://doi.org/10.3390/microarrays5010001
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.