
Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery

Abstract
:1. Introduction
2. Integrative Transcriptomic Data Analysis
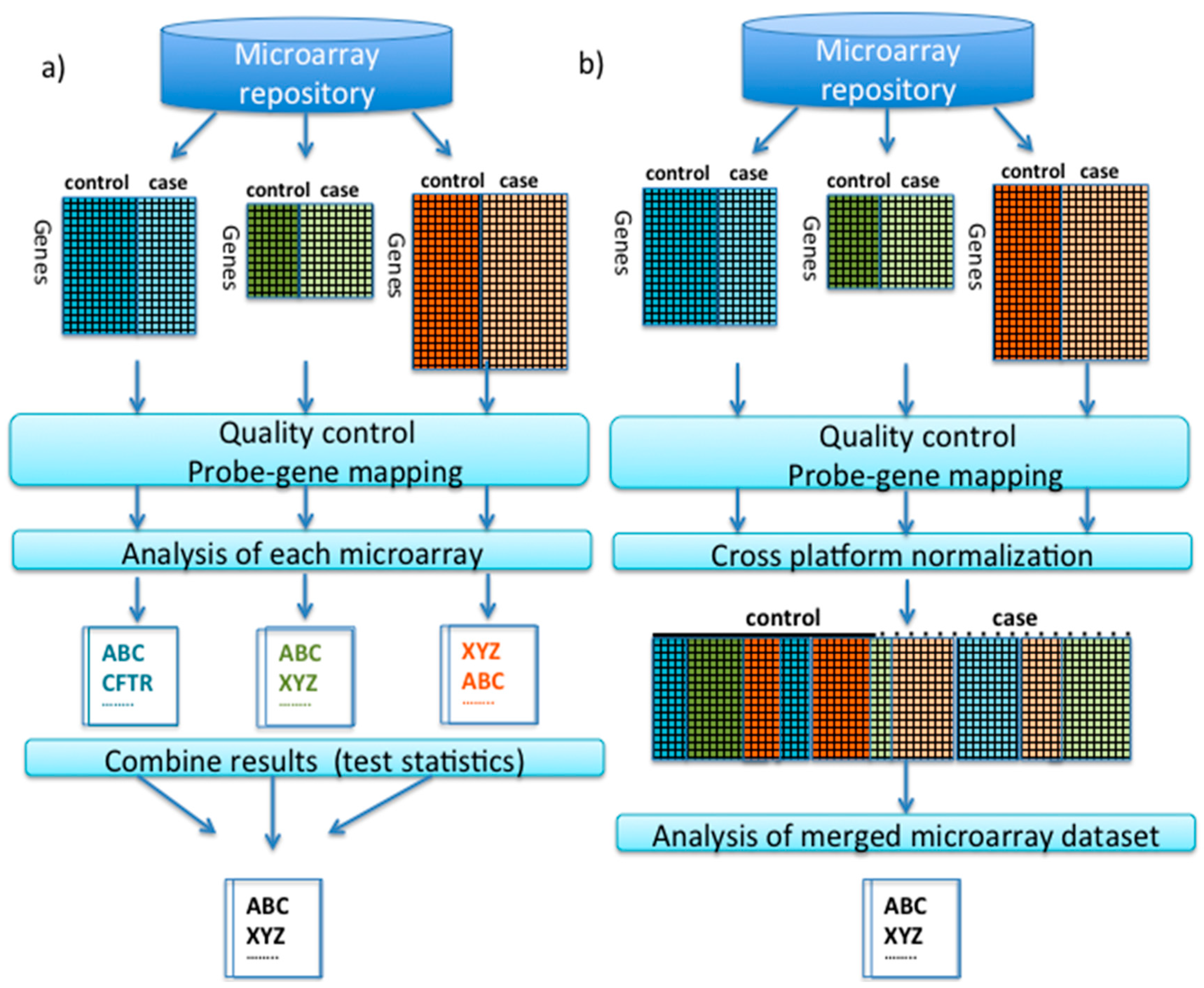
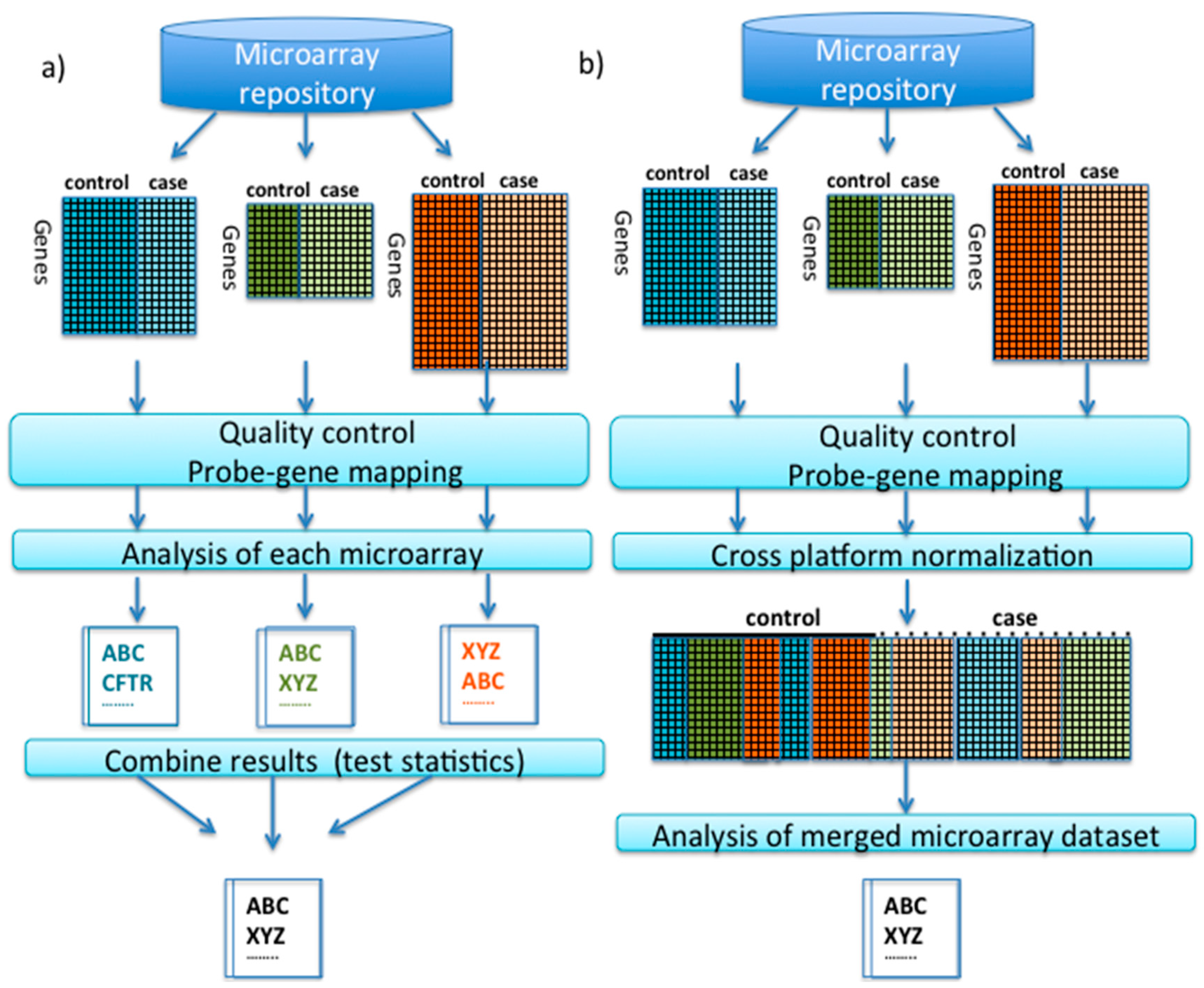
2.1. Pre-Processing and Quality Control Prior to Integrative Analysis
2.2. Meta-Analysis
2.2.1. Comparison of Meta-Analysis Methods
2.2.2. Association of Meta-Analysis Method to Outcome Variable
2.3. Cross-Platform Normalization
2.3.1. Comparison of Cross-Normalization Methods
2.3.2. Software and Websites Implementing Microarray Meta-Analysis and Cross-Platform Merging/Normalization

{kind=link}
| Microarray Meta-Analysis (Command Line Packages) | ||
|---|---|---|
| Software Name | Language | Features |
| metaDE (metaOmics) | R | Implements 12 major meta-analysis methods [37] |
| MAMA | R | Implements combined effect size, combined p-values, combined ranks |
| metaMA | R | Implements combined moderated effect size, combined p-values |
| metaGEM | R | Implements combined effect size, combined p-values, vote counting [24] |
| metahdep | R | Effect size estimates particularly when hierarchical dependence is present |
| GeneMeta | R | Implements combined effect size [47] |
| OrderedList | R | Combine ranks with or without expression data |
| RankProd | R | Implements Product of Ranks method |
| RankAggreg | R | Aggregation of ordered lists based on the ranks using several different algorithms |
| Automated web applications for microarray meta-analysis/normalization | ||
| Software Name | Features and URL | |
| INMEX | Meta-analysis. Support for 45 microarray platforms for human, mouse rat. Combines p-values, effect sizes, rank order, others http://www.inmex.ca/INMEX/ | |
| Network Analyst | Meta-analysis. Combines p-values, effect sizes, rank order. Significantly altered genes are then presented within the context of protein-protein interaction networks. http://www.networkanalyst.ca/NetworkAnalyst/faces/home.xhtml | |
| A-MADMAN | Affymetrix platform normalization using quantile distribution transformation http://compgen.bio.unipd.it/bioinfo/amadman/ | |
| MAAMD | Affymetrix meta-analysis http://www.biokepler.org/use_cases/maamd-workflow-standardize-meta-analyses-affymetrix-microarray-data | |
| Microarray cross-platform merging/normalization (command line packages) | ||
| Software Name | Language | Features |
| mergeMaid | R | Implements Probability of Expression transformation (POE) [62] |
| metaArray | R | Implements POE [62] |
| CONOR | R | Implements XPN, Empirical Bayes (EB), Quantile normalization (QN), Quantile discretization (QD), others [2] |
| VirtualArray | R | Implements EB, QN, QD, others [71] |
| inSilico Merging | R | Implements XPN, EB, DWD, others [23] |
| Automated Microarray Data Analysis v2.13 | R | Implements. Allows analysis of Illumina, Affymetrix and Agilent. |
| XPN | R | Implements Cross Platform Normalization [26] |
| DWD | JAVA, R MATLAB | Implements Distance Weighted Discrimination method [65] |
| Combat | R | Implements Empirical Bayes methods [64] |
| PLIDA | MATLAB | Normalizes an arbitrary number of platforms [60] |
| metAnalyzeAll | R | Elastic net classifier [42] |
2.4. Comparison of Meta-Analysis vs. Cross-Platform Normalization
3. Promising Transcriptomic Biomarkers Identified Using Meta-Analysis Approaches
4. Confounding Adjustment
5. Conclusions
Conflicts of Interest
References
- Pepe, M.S.; Feng, Z. Improving biomarker identification with better designs and reporting. Clin. Chem. 2011, 57, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Rudy, J.; Valafar, F. Empirical comparison of cross-platform normalization methods for gene expression data. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Rung, J.; Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013, 14, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression Omnibus. Secondary. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 5 May 2015).
- Miller, M.B.; Tang, Y.W. Basic concepts of microarrays and potential applications in clinical microbiology. Clin. Microbiol. Rev. 2009, 22, 611–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Calin, G.A.; Volinia, S.; Croce, C.M. MicroRNA expression profiling using microarrays. Nat. Protoc. 2008, 3, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Ptacek, J.; Snyder, M. Protein microarray technology. Mech. Ageing Dev. 2007, 128, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Xu, Y.; Feng, Z.; Yang, X.J.; Wang, X.G.; Gao, X. Multiple-platform data integration method with application to combined analysis of microarray and proteomic data. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Wang, J.; Zhao, C.; Fostel, J.; Tong, W.; Bushel, P.R.; Deng, Y.; Pusztai, L.; Symmans, W.F.; Shi, T. Maximizing biomarker discovery by minimizing gene signatures. BMC Genom. 2011, 12. [Google Scholar] [CrossRef]
- McCollum, E.D.; Preidis, G.A.; Maliwichi, M.; Fostel, J.; Tong, W.; Bushel, P.R.; Deng, Y.; Pusztai, L.; Symmans, W.F.; Shi, T. Clinical versus rapid molecular HIV diagnosis in hospitalized African infants: A randomized controlled trial simulating point-of-care infant testing. J. Acquir. Immune. Defic. Syndr. 2014, 66, e23–e30. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhang, Y.; Lin, S.; Wang, T.H.; Yang, S. Advances in microfluidic PCR for point-of-care infectious disease diagnostics. Biotechnol. Adv. 2011, 29, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Director’s Challenge Consortium for the Molecular Classification of Lung A; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, R.; Flinchum, R.; Brown, N.; Ramsey, J.; Riccitelli, S.; Heuck, C.; Barlogie, B.; Shaughnessy, J.D., Jr. Translating a gene expression signature for multiple myeloma prognosis into a robust high-throughput assay for clinical use. BMC Med. Genom. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Gesthalter, Y.B.; Vick, J.; Steiling, K.; Spira, A. Translating the transcriptome into tools for the early detection and prevention of lung cancer. Thorax 2015, 70, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Chinnaiyan, A.M.; Ghosh, D. Pathway analysis reveals functional convergence of gene expression profiles in breast cancer. BMC Med. Genom. 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Campbell, G.; Jones, W.D.; Campagne, F.; Wen, Z.; Walker, S.J.; Su, Z.; Chu, T.M.; Goodsaid, F.M.; Pusztai, L.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 2010, 28, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Simon, R. Genomic biomarkers in predictive medicine: An interim analysis. EMBO Mol. Med. 2011, 3, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P. Cancer biomarkers: can we turn recent failures into success? J. Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G. Improving the biomarker pipeline to develop and evaluate cancer screening tests. J. Natl. Cancer Inst. 2009, 101, 1116–1169. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.; Wishart, D. Applications of machine learning in cancer prediction and prognosis. Cancer Inf. 2006, 2, 59–77. [Google Scholar]
- Michiels, S.; Koscielny, S.; Hill, C. Prediction of cancer outcome with microarrays: A multiple random validation strategy. Lancet 2005, 365, 488–492. [Google Scholar] [CrossRef]
- Hamid, J.S.; Hu, P.; Roslin, N.M.; Ling, V.; Greenwood, C.T.; Beyene, J. Data integration in genetics and genomics: Methods and challenges. Hum. Genom. Proteom. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Taminau, J.; Lazar, C.; Meganck, S.; Nowé, A. Comparison of merging and meta-analysis as alternative approaches for integrative gene expression analysis. ISRN Bioinform. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Greenwood, C.M.; Beyene, J. Integrative analysis of multiple gene expression profiles with quality-adjusted effect size models. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A.; Tjelmeland, H.; Fan, C.; Perou, C.M.; Nobel, A.B. Merging two gene-expression studies via cross-platform normalization. Bioinformatics 2008, 24, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Tseng, G.C.; Ghosh, D.; Feingold, E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012, 40, 3785–3799. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Wang, X.; Haitsma, J.J.; Furmli, S.; Masoom, H.; Liu, M.; Imai, Y.; Slutsky, A.S.; Beyene, J.; Greenwood, C.M.; et al. Microarray meta-analysis identifies acute lung injury biomarkers in donor lungs that predict development of primary graft failure in recipients. PLoS ONE 2012, 7, e45506. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Morgun, A.; Shulzhenko, N. Microarrays for cancer diagnosis and classification. Adv. Exp. Med. Biol. 2007, 593, 74–85. [Google Scholar] [PubMed]
- Xia, J.; Gill, E.E.; Hancock, R.E. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, R.R.; Sabine, V.S.; Simen, A.A.; Dixon, J.M.; Bartlett, J.M.; Sims, A.H. Relative impact of key sources of systematic noise in Affymetrix and Illumina gene-expression microarray experiments. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.K.; Kitchen, R.R.; Larionov, A.A.; Renshaw, L.; Dixon, J.M.; Sims, A.H. Direct integration of intensity-level data from Affymetrix and Illumina microarrays improves statistical power for robust reanalysis. BMC Med. Genom. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Lin, H.M.; Sibille, E.; Tseng, G.C. Meta-analysis methods for combining multiple expression profiles: Comparisons, statistical characterization and an application guideline. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Miller, C.J. Simpleaffy: A BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics 2005, 21, 3683–3685. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B. affyPLM: Model Based QC Assessment of Affymetrix GeneChips. Available online: http://www.cse.unsw.edu.au/~mike/myrlibrary.old/affyPLM/doc/QualityAssess.pdf (accessed on 16 April 2015).
- Kang, D.D.; Sibille, E.; Kaminski, N.; Tseng, G.C. MetaQC: Objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kang, D.D.; Shen, K.; Song, C.; Lu, S.; Chang, L.C.; Liao, S.G.; Huo, Z.; Tang, S.; Ding, Y.; et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics 2012, 28, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Morais, N.L.; Dunning, M.J.; Samarajiwa, S.A.; Darot, J.F.; Ritchie, M.E.; Lynch, A.G.; Tavaré, S. A re-annotation pipeline for Illumina BeadArrays: Improving the interpretation of gene expression data. Nucleic Acids Res. 2010, 38. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Cannistra, S.A.; Fountzilas, H.; Culhane, A.; Pillay, K.; Rueda, B.; Cramer, D.; Seiden, M.; Birrer, M.; Coukos, G.; Zhang, L.; et al. Integrated analysis of multiple microarray datasets identifies a reproducible survival predictor in ovarian cancer. PLoS ONE 2011, 6, e18202. [Google Scholar] [CrossRef] [PubMed]
- Hughey, J.J.; Butte, A.J. Robust meta-analysis of gene expression using the elastic net. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y.; Song, C.; Culhane, A.; Pillay, K.; Rueda, B.; Cramer, D.; Seiden, M.; Birrer, M.; Coukos, G.; et al. Detecting disease-associated genes with confounding variable adjustment and the impact on genomic meta-analysis: With application to major depressive disorder. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Sabine, V.S.; Sims, A.H.; Macaskill, E.J.; Renshaw, L.; Thomas, J.S.; Dixon, J.M.; Bartlett, J.M. Gene expression profiling of response to mTOR inhibitor everolimus in pre-operatively treated post-menopausal women with oestrogen receptor-positive breast cancer. Breast Cancer Res. Treat. 2010, 122, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Beyene, J.; Greenwood, C.M. Tests for differential gene expression using weights in oligonucleotide microarray experiments. BMC Genom. 2006, 7. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl. Acad. Sci. USA 2004, 101, 9309–9314. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Yu, U.; Kim, S.; Yoo, O.J. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 2003, 19, i84–i90. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Barrette, T.; Rubin, M.A.; Ghosh, D.; Chinnaiyan, A.M.; et al. Meta-analysis of microarrays: Interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002, 62, 4427–4433. [Google Scholar] [PubMed]
- Song, C.; Tseng, G.C. Hypothesis setting and order statistic for robust genomic meta-analysis. Ann. Appl. Stat. 2014, 8, 777–800. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Huang, J. Regularized gene selection in cancer microarray meta-analysis. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, J.; Song, C.; Tseng, G.C. Biomarker detection in the integration of multiple multi-class genomic studies. Bioinformatics 2010, 26, 333–340. [Google Scholar]
- Campain, A.; Yang, Y.H. Comparison study of microarray meta-analysis methods. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Breitling, R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics 2008, 24, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Herold, T.; He, C.; Valk, P.J.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.D.; Maharry, K.S.; Sun, M.; et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: An international collaborative study. J. Clin. Oncol. 2013, 31, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.H.; Smethurst, G.J.; Hey, Y.; Okoniewski, M.J.; Pepper, S.D.; Howell, A.; Miller, C.J.; Clarke, R.B. The removal of multiplicative, systematic bias allows integration of breast cancer gene expression datasets—Improving meta-analysis and prediction of prognosis. BMC Med. Genom. 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tan, A.C.; Winslow, R.L.; Geman, D. Merging microarray data from separate breast cancer studies provides a robust prognostic test. BMC Bioinformatics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Hu, J.; Kalakrishnan, M.; Huang, H.; Zhou, X.J. Integrative disease classification based on cross-platform microarray data. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genom. 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Deshwar, A.G.; Morris, Q. PLIDA: Cross-platform gene expression normalization using perturbed topic models. Bioinformatics 2014, 30, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Deng, Y.; Chen, H.S.; Tao, L.; Sha, Q.; Chen, J.; Tsai, C.J.; Zhang, S. Joint analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes. BMC Bioinform. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Ghosh, D.; Chinnaiyan, A.M. Prognostic meta-signature of breast cancer developed by two-stage mixture modeling of microarray data. BMC Genom. 2004, 5. [Google Scholar] [CrossRef]
- Parmagiani, G.; Garret-Mayer, E.S.; Anbazhagan, R.; Gabrielson, E. A cross-study comparison of gene expression studies for the molecular classificaiton of lung cancer. Clin. Cancer Res. 2004, 10, 2922–2927. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lu, X.; Liu, Y.; Haaland, P.; Marron, J.S. R/DWD: Distance-weighted discrimination for classification, visualization and batch adjustment. Bioinformatics 2012, 28, 1182–1183. [Google Scholar] [CrossRef] [PubMed]
- WebArray, D.B. Secondary. Available online: http://www.webarraydb.org/webarray/index.html. (accessed on 12 May 2015).
- Benito, M.; Parker, J.; Du, Q.; Wu, J.; Xiang, D.; Perou, C.M.; Marron, J.S. Adjustment of systematic microarray data biases. Bioinformatics 2003, 20, 105–114. [Google Scholar] [CrossRef]
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing batch effects in analysis of expression microarray data: An evaluation of six batch adjustment methods. PLoS ONE 2011, 6, e17238. [Google Scholar] [CrossRef] [PubMed]
- R, Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012. [Google Scholar]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; de Schaetzen, V.; Weiss Solís, D.Y.; Bersini, H.; et al. Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Heider, A.; Alt, R. virtualArray: A R/bioconductor package to merge raw data from different microarray platforms. BMC Bioinform. 2013, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Warnat, P.; Eils, R.; Brors, B. Cross-platform analysis of cancer microarray data improves gene expression based classification of phenotypes. BMC Bioinform. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Fielden, M.R.; Nie, A.; McMillian, M.; Yi, Y.; Morrison, C.; Yang, P.; Sun, Z.; Szoke, J.; Gerald, W.L.; Watson, M.; et al. Interlaboratory evaluation of genomic signatures for predicting carcinogenicity in the rat. Toxicol. Sci. 2008, 103, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lemon, W.; Liu, P.Y.; Yi, Y.; Morrison, C.; Yang, P.; Sun, Z.; Szoke, J.; Gerald, W.L.; Watson, M.; et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006, 3, e467. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.E.; Shidham, A.; Wong, H.R.; Khatri, P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Potashkin, J.A. Network-based metaanalysis identifies HNF4A and PTBP1 as longitudinally dynamic biomarkers for Parkinson’s disease. Proc. Natl. Acad. Sci USA 2015, 112, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Yu, A.; Kim, S.; Kang, J.; Hong, S.M. Robust likelihood-based survival modeling with microarray data. J. Stat. Softw. 2009, 29, 1–16. [Google Scholar]
- Chikina, M.D.; Sealfon, S.C. Increasing consistency of disease biomarker prediction across datasets. PLoS ONE 2014, 9, e91272. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.J.; Wu, M.-J. The Synthesis of Regression Slopes in Meta-Analysis. Stat. Sci. 2007, 22, 414–429. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walsh, C.J.; Hu, P.; Batt, J.; Santos, C.C.D. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays 2015, 4, 389-406. https://doi.org/10.3390/microarrays4030389
Walsh CJ, Hu P, Batt J, Santos CCD. Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays. 2015; 4(3):389-406. https://doi.org/10.3390/microarrays4030389
Chicago/Turabian StyleWalsh, Christopher J., Pingzhao Hu, Jane Batt, and Claudia C. Dos Santos. 2015. "Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery" Microarrays 4, no. 3: 389-406. https://doi.org/10.3390/microarrays4030389
APA StyleWalsh, C. J., Hu, P., Batt, J., & Santos, C. C. D. (2015). Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery. Microarrays, 4(3), 389-406. https://doi.org/10.3390/microarrays4030389