
Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells



Abstract
:
1. Introduction
2. Experimental Section
2.1. Chemicals and Microarray
2.2. Cell Culture and Adhesion Assay
2.3. Fluorescence Microscopy and Software
3. Results and Discussion
3.1. Live Cell Imaging and Controls
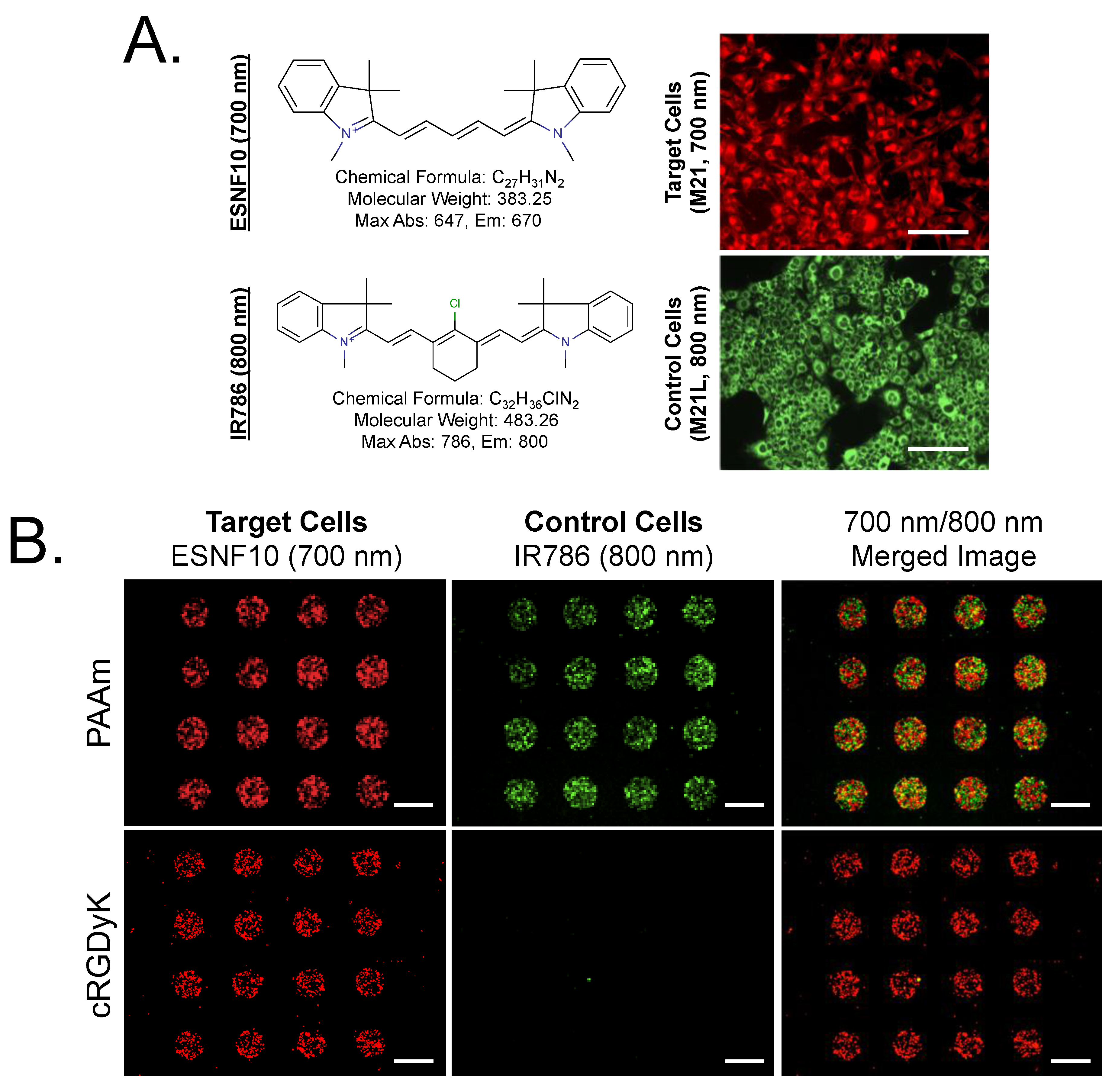
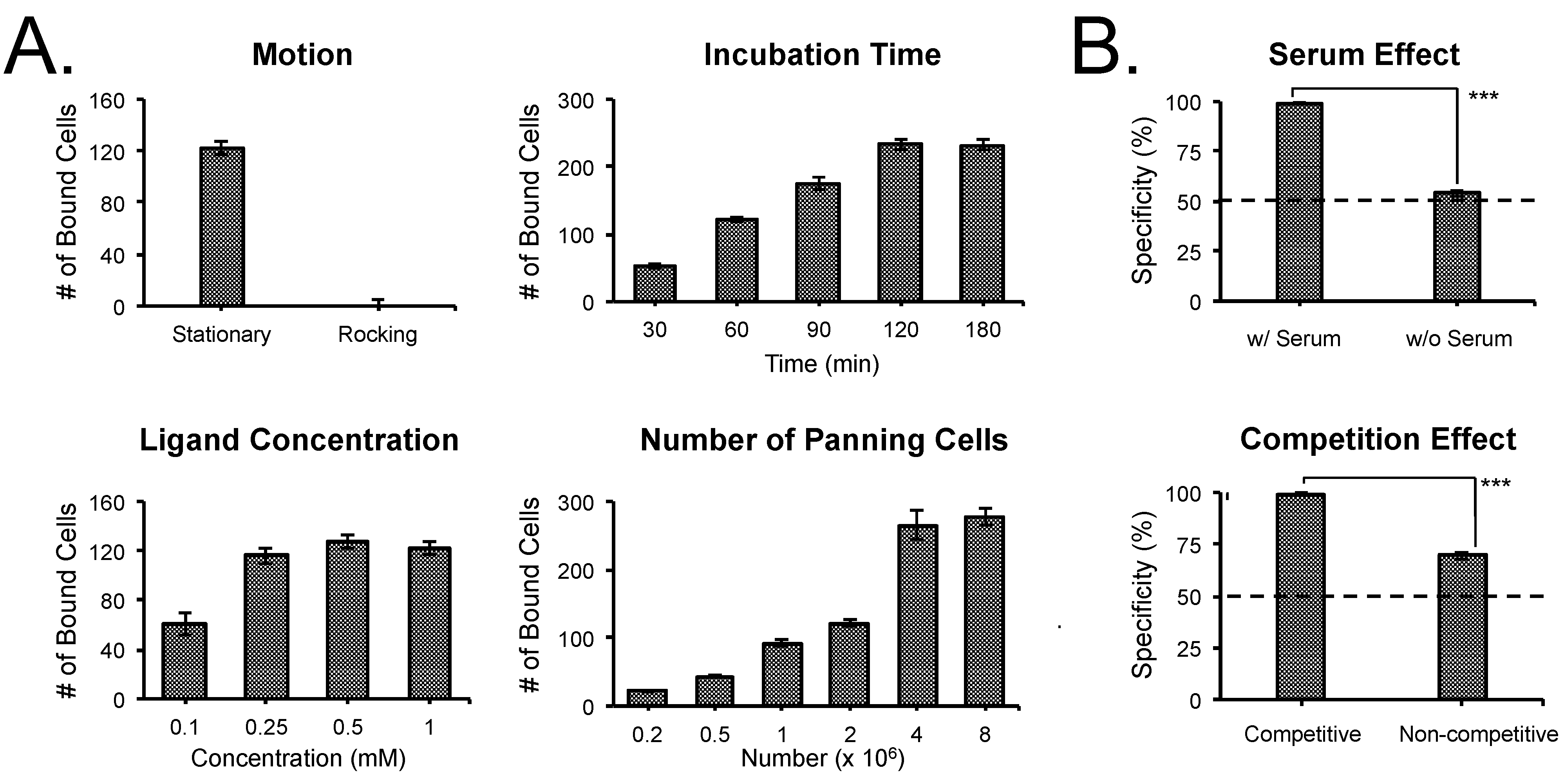
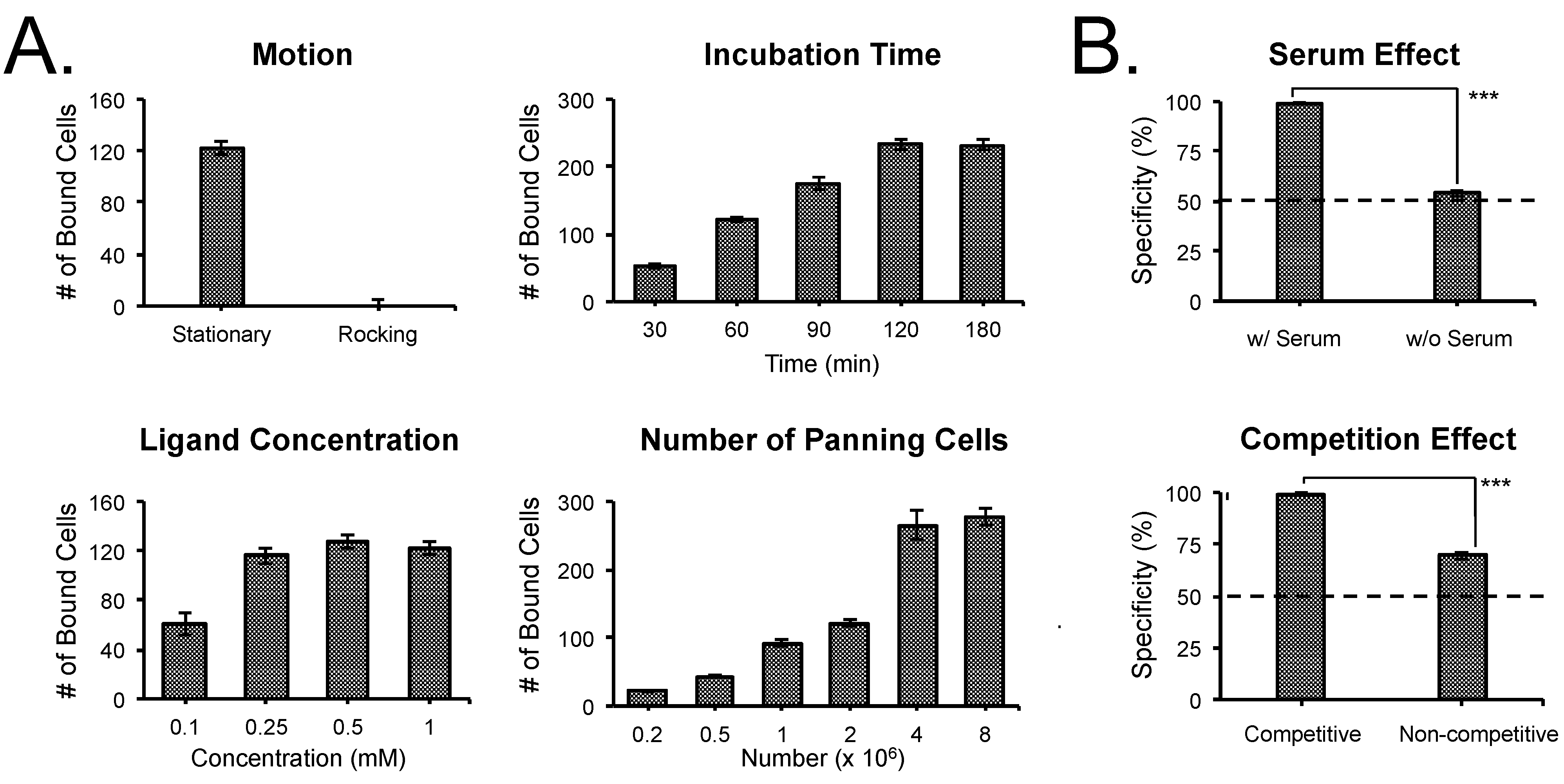
3.2. Optimization of SMM Screening Using Living Cells
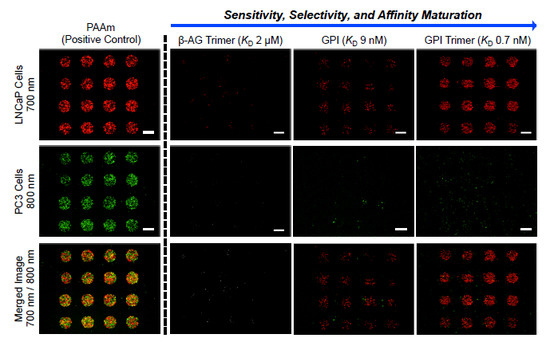
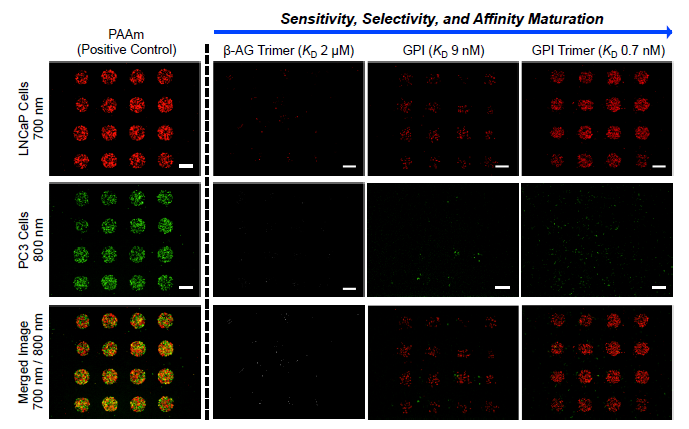
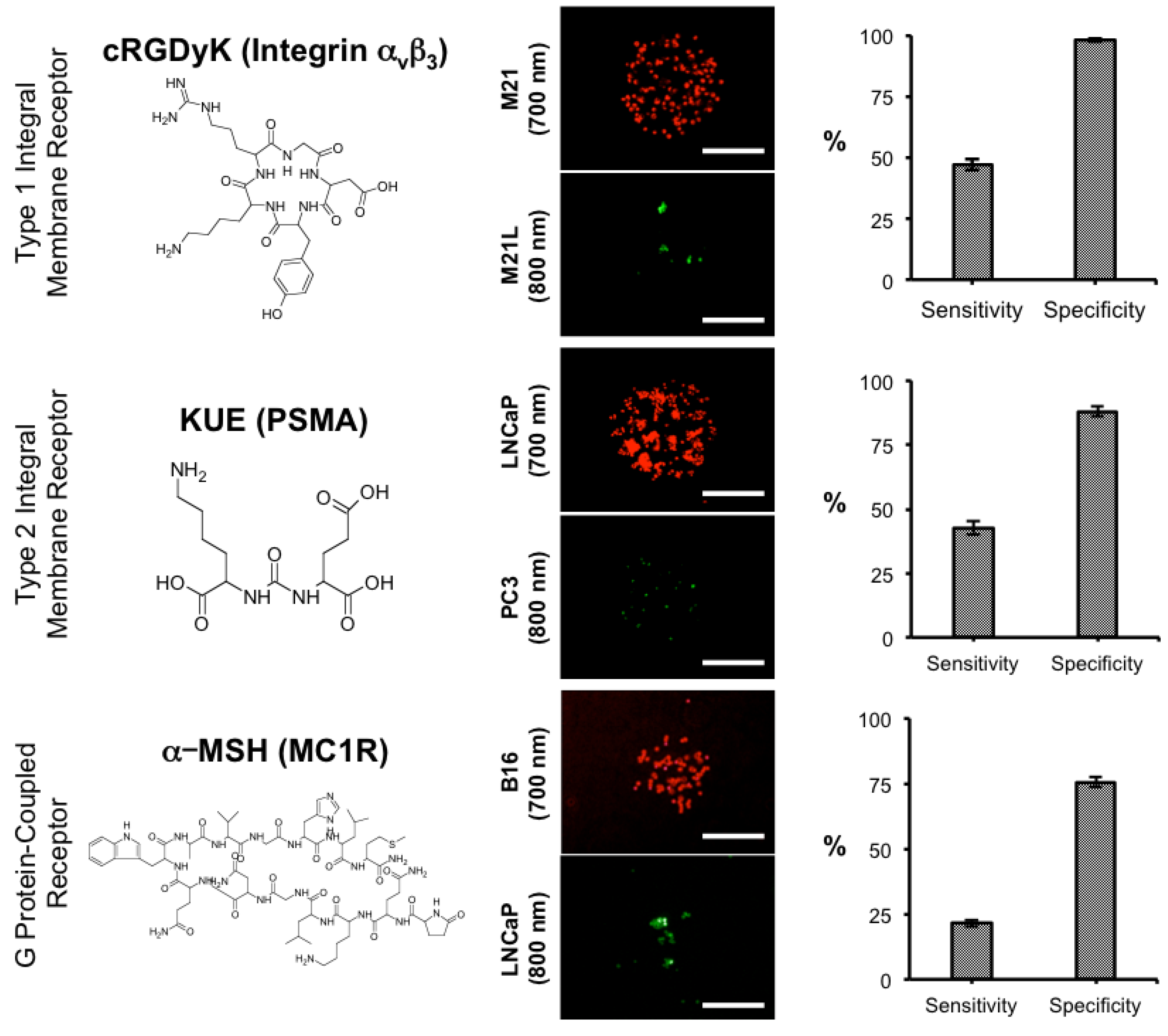
3.3. Screening of Diverse Chemical and Cellular Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M.W.(Da) | Affinity (KD) | Specificity (Receptor) | Tested Cells (Positive/Negative) | Ref. |
|---|---|---|---|---|---|
| β-AG | 248.19 | 2 µM | PSMA | LNCaP/PC3 | [22,23] |
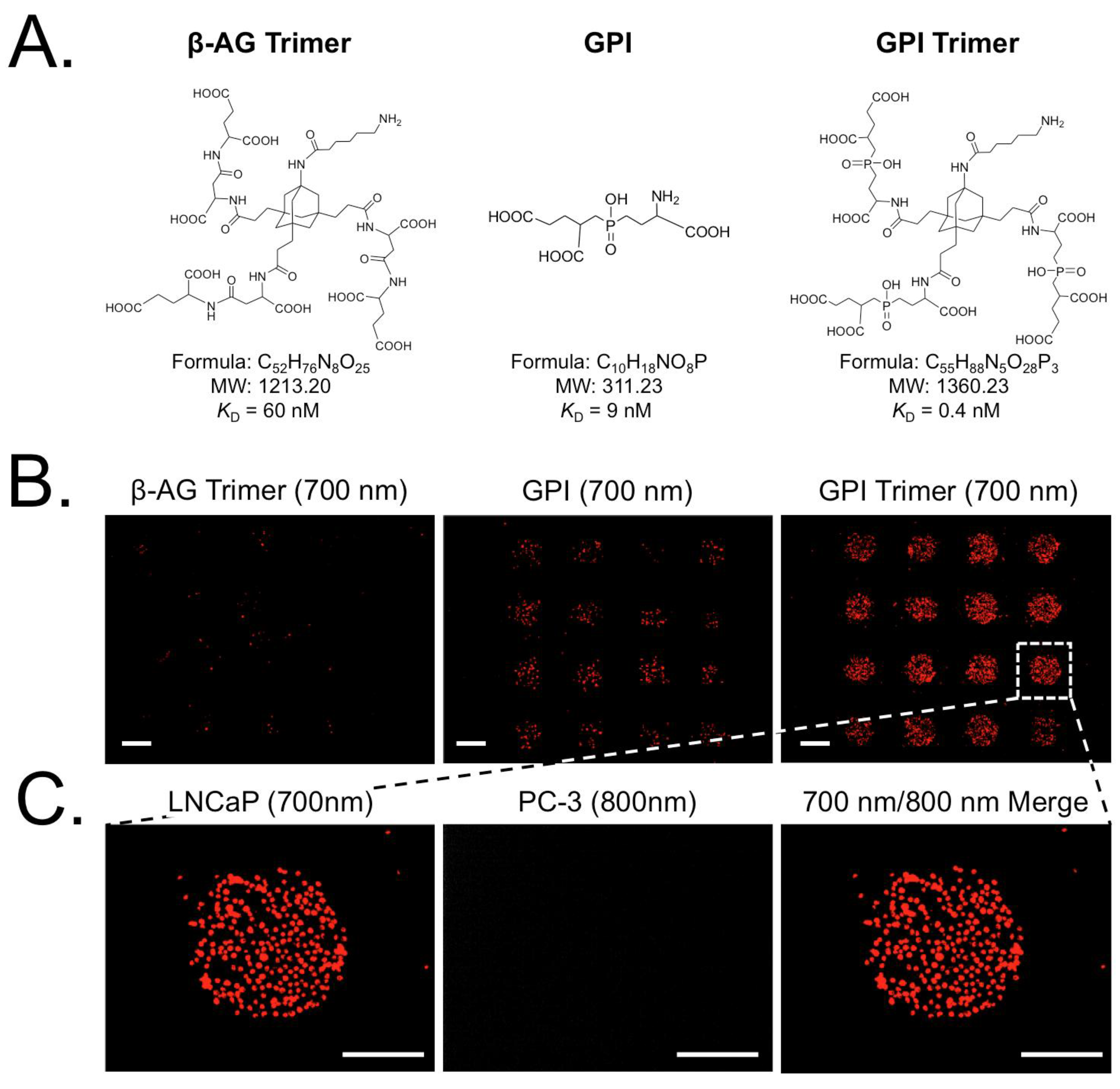
| β-AG trimer | 1213.20 | 60 nM | PSMA | LNCaP/PC3 | [22,23] |
| GPI | 311.23 | 9 nM | PSMA | LNCaP/PC3 | [22,23] |
| GPI trimer | 1360.23 | 0.7 nM | PSMA | LNCaP/PC3 | [22,23] |
| KUE | 319.31 | 15 nM | PSMA | LNCaP/PC3 | [28] |
| cRGDyK | 619.67 | 50 nM | Integrin αvβ3 | M21/M21-L | [30] |
| α-MSH | 1664.88 | 0.4 nM | MC1R | B16/LNCaP | [29] |
| PAAm | ~70,000 | N.A. | Nonspecific | All Cells | [21] |
3.4. Exploring the Relationship between Ligand Affinity and Bmax
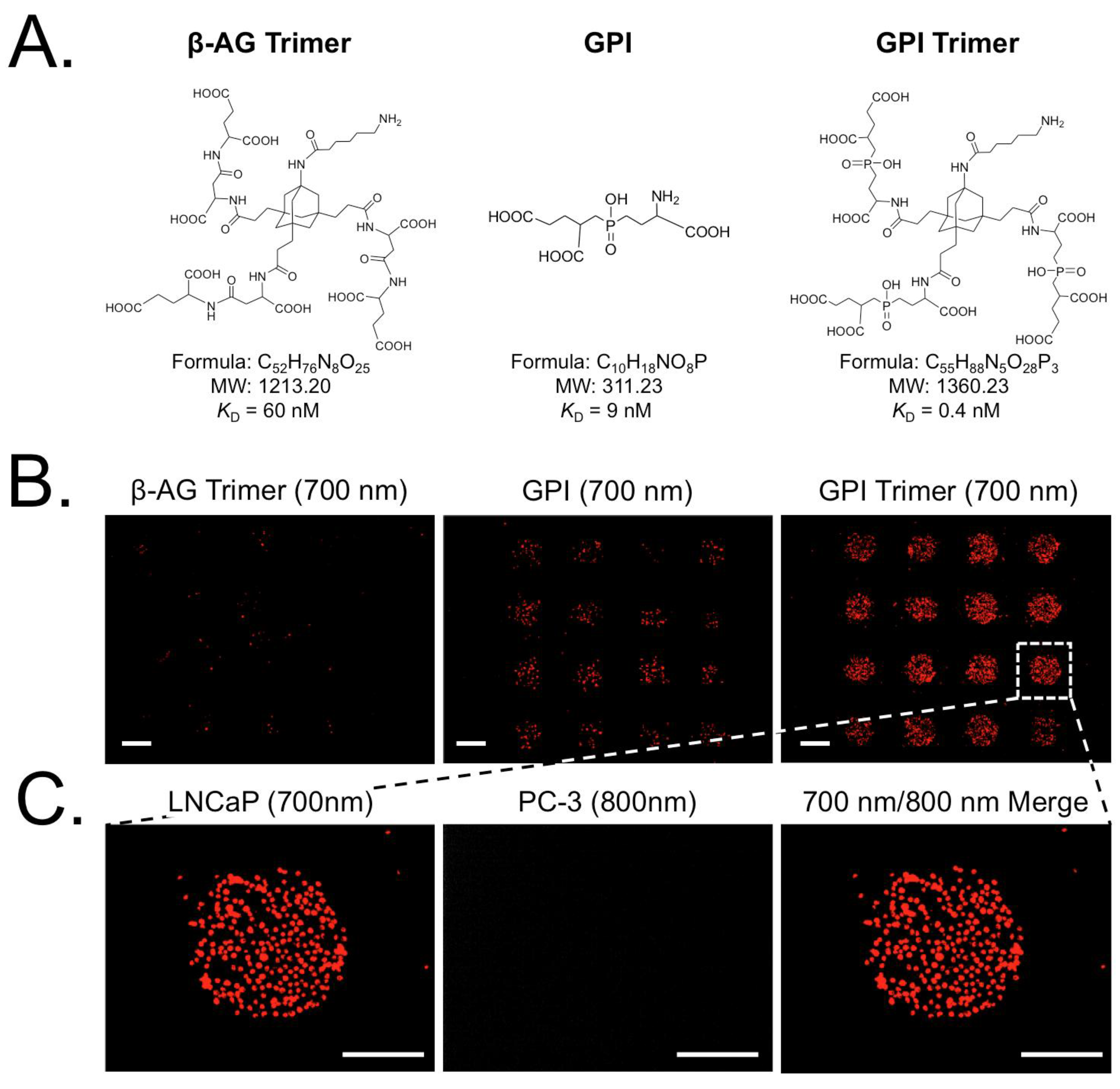
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Geysen, H.M.; Schoenen, F.; Wagner, D.; Wagner, R. Combinatorial compound libraries for drug discovery: An ongoing challenge. Nat. Rev. Drug Discov. 2003, 2, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.V.; Godon, E.M. Applications of small-molecule combinatorial chemistry to drug discovery. Drug Discov. Today 1996, 1, 134–144. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Uttamchandani, M.; Walsh, D.P.; Yao, S.Q.; Chang, Y.T. Small molecule microarrays: Recent advances and applications. Curr. Opin. Chem. Biol. 2005, 9, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Horiuchi, K.Y. Chemical microarray: A new tool for drug screening and discovery. Drug Discov. Today 2006, 11, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Barnes-Seeman, D.; Park, S.B.; Koehler, A.N.; Schreiber, S.L. Expanding the functional group compatibility of small-molecule microarrays: Discovery of novel calmodulin ligands. Angew Chem. Int. Ed. Engl. 2003, 42, 2376–2379. [Google Scholar] [CrossRef] [PubMed]
- Uttamchandani, M.; Walsh, D.P.; Khersonsky, S.M.; Huang, X.; Yao, S.Q.; Chang, Y.T. Microarrays of tagged combinatorial triazine libraries in the discovery of small-molecule ligands of human IgG. J. Combin. Chem. 2004, 6, 862–868. [Google Scholar] [CrossRef]
- Wu, H.; Ge, J.; Uttamchandani, M.; Yao, S.Q. Small molecule microarrays: The first decade and beyond. Chem. Comm. 2011, 47, 5664–5670. [Google Scholar] [PubMed]
- Bradner, J.E.; McPherson, O.M.; Mazitschek, R.; Barnes-Seeman, D.; Shen, J.P.; Dhaliwal, J.; Stevenson, K.E.; Duffner, J.L.; Park, S.B.; Neuberg, D.S.; et al. A robust small-molecule microarray platform for screening cell lysates. Chem. Biol. 2006, 13, 493–504. [Google Scholar]
- Pop, M.S.; Wassaf, D.; Koehler, A.N. Probing small-molecule microarrays with tagged proteins in cell lysates. Curr. Protoc. Chem. Biol. 2014, 6, 209–220. [Google Scholar] [PubMed]
- Shi, H.; Uttamchandani, M.; Yao, S.Q. Applying small molecule microarrays and resulting affinity probe cocktails for proteome profiling of mammalian cell lysates. Chem. Asian J. 2011, 6, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- El-Ali, J.; Sorger, P.K.; Jensen, K.F. Cells on chips. Nature 2006, 442, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Rantala, J.B.; Kwon, S.; Korkola, J.; Gray, J.W. Expanding the diversity of imaging-based rnai screen applications using cell spot microarrays. Microarrays 2013, 2, 97–114. [Google Scholar] [CrossRef]
- Kim, J.; Taylor, D.; Agrawal, N.; Wang, H.; Kim, H.; Han, A.; Rege, K.; Jayaraman, A. A programmable microfluidic cell array for combinatorial drug screening. Lab Chip 2012, 12, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Davis, M.M. Molecular and functional analysis using live cell microarrays. Curr. Opin. Chem. Biol. 2006, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, H.S.; Nasr, K.A.; Ha, M.; Kim, Y.; Frangioni, J.V. High-throughput small molecule identification using maldi-tof and a nanolayered substrate. Anal. Chem. 2011, 83, 5283–5289. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hyun, H.; Cross, C.J.; Henary, M.; Nasr, K.A.; Oketokoun, R.; Choi, H.S.; Frangioni, J.V. Rapid and facile microwave-assisted surface chemistry for functionalized microarray slides. Adv. Funct. Mater. 2012, 22, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.; Hyun, H.; Bordo, M.W.; Oketokoun, R.; Nasr, K.A.; Frangioni, J.V.; Choi, H.S. High-throughput screening of small molecule ligands targeted to live bacteria surface. Anal. Chem. 2013, 85, 3508–3514. [Google Scholar] [CrossRef] [PubMed]
- Humblet, V.; Misra, P.; Bhushan, K.R.; Nasr, K.; Ko, Y.S.; Tsukamoto, T.; Pannier, N.; Frangioni, J.V.; Maison, W. Multivalent scaffolds for affinity maturation of small molecule cell surface binders and their application to prostate tumor targeting. J. Med. Chem. 2009, 52, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase ii: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, G.; Hyun, H.; Lee, J.H.; Ashitate, Y.; Choi, J.; Hong, G.H.; Owens, E.A.; Henary, M.; Choi, H.S. Near-infrared lipophilic fluorophores for tracing tissue growth. Biomed. Mater. 2013, 8, 014110. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ashitate, Y.; Lee, J.H.; Kim, S.H.; Matsui, A.; Insin, N.; Bawendi, M.G.; Semmler-Behnke, M.; Frangioni, J.V.; Tsuda, A. Rapid translocation of nanoparticles from the lung airspaces to the body. Nat. Biotechnol. 2010, 28, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Gibbs, S.L.; Lee, J.H.; Kim, S.H.; Ashitate, Y.; Liu, F.; Hyun, H.; Park, G.; Xie, Y.; Bae, S.; et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nat. Biotechnol. 2013, 31, 148–153. [Google Scholar]
- Cressman, S.; Dobson, I.; Lee, J.B.; Tam, Y.Y.; Cullis, P.R. Synthesis of a labeled rgd-lipid, its incorporation into liposomal nanoparticles, and their trafficking in cultured endothelial cells. Bioconjugate Chem. 2009, 20, 1404–1411. [Google Scholar] [CrossRef]
- Hillier, S.M.; Maresca, K.P.; Femia, F.J.; Marquis, J.C.; Foss, C.A.; Nguyen, N.; Zimmerman, C.N.; Barrett, J.A.; Eckelman, W.C.; Pomper, M.G.; et al. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69, 6932–6940. [Google Scholar]
- Chen, J.; Cheng, Z.; Hoffman, T.J.; Jurisson, S.S.; Quinn, T.P. Melanoma-targeting properties of (99m)technetium-labeled cyclic alpha-melanocyte-stimulating hormone peptide analogues. Cancer Res. 2000, 60, 5649–5658. [Google Scholar] [PubMed]
- Chen, X.; Park, R.; Tohme, M.; Shahinian, A.H.; Bading, J.R.; Conti, P.S. Micropet and autoradiographic imaging of breast cancer alpha v-integrin expression using 18f- and 64cu-labeled rgd peptide. Bioconjugate Chem. 2004, 15, 41–49. [Google Scholar] [CrossRef]
- Misra, P.; Humblet, V.; Pannier, N.; Maison, W.; Frangioni, J.V. Production of multimeric prostate-specific membrane antigen small-molecule radiotracers using a solid-phase 99mtc preloading strategy. J. Nucl. Med. 2007, 48, 1379–1389. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Bao, K.; Frangioni, J.V.; Choi, H.S. Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells. Microarrays 2015, 4, 53-63. https://doi.org/10.3390/microarrays4010053
Lee JH, Bao K, Frangioni JV, Choi HS. Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells. Microarrays. 2015; 4(1):53-63. https://doi.org/10.3390/microarrays4010053
Chicago/Turabian StyleLee, Jeong Heon, Kai Bao, John V. Frangioni, and Hak Soo Choi. 2015. "Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells" Microarrays 4, no. 1: 53-63. https://doi.org/10.3390/microarrays4010053
APA StyleLee, J. H., Bao, K., Frangioni, J. V., & Choi, H. S. (2015). Screening of Small Molecule Microarrays for Ligands Targeted to the Extracellular Epitopes of Living Cells. Microarrays, 4(1), 53-63. https://doi.org/10.3390/microarrays4010053