α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Differentiation
2.2. Cell Treatment with Aβ1-42 and α-Tocopherol
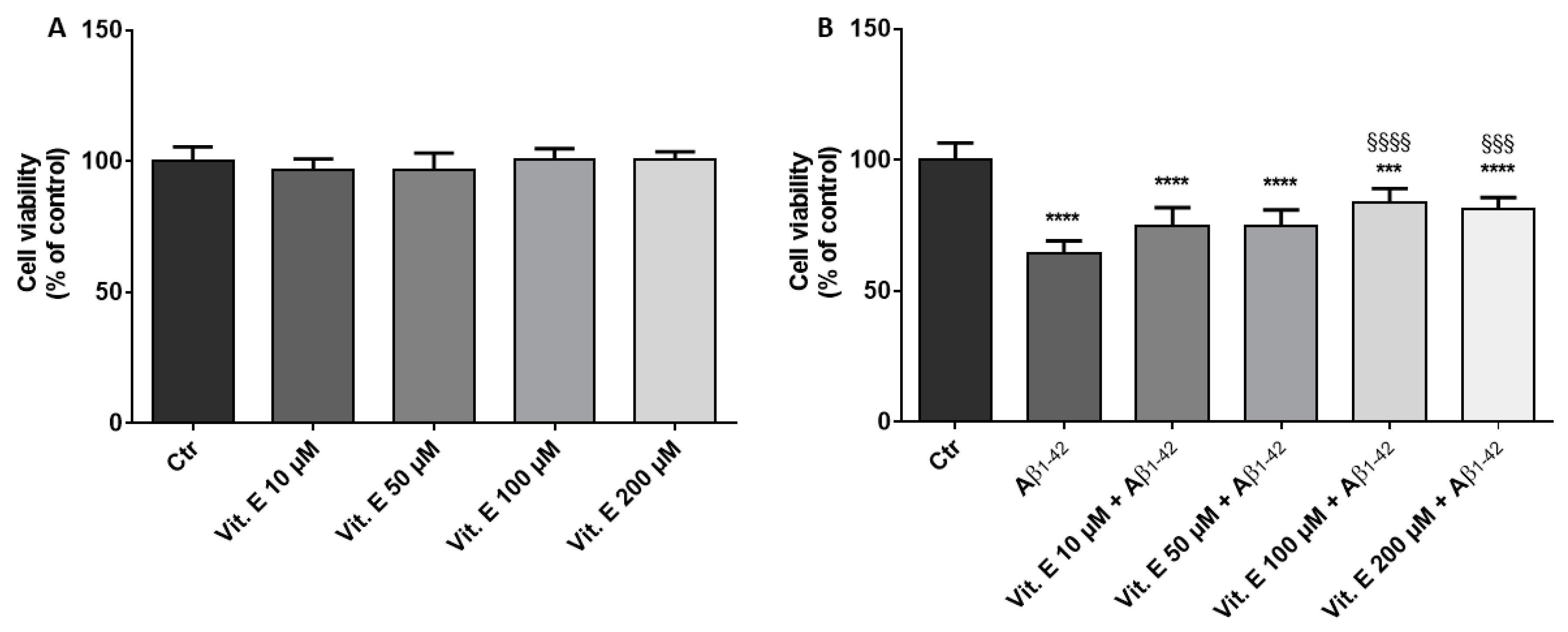
2.3. Cell Viability
2.4. Extraction of Total RNA and cDNA Library Preparation
2.5. RNA-Seq Data Analysis and Gene Evaluation
2.6. Immunocytochemistry
2.7. Statistical Analysis
3. Results
3.1. Cell Viability
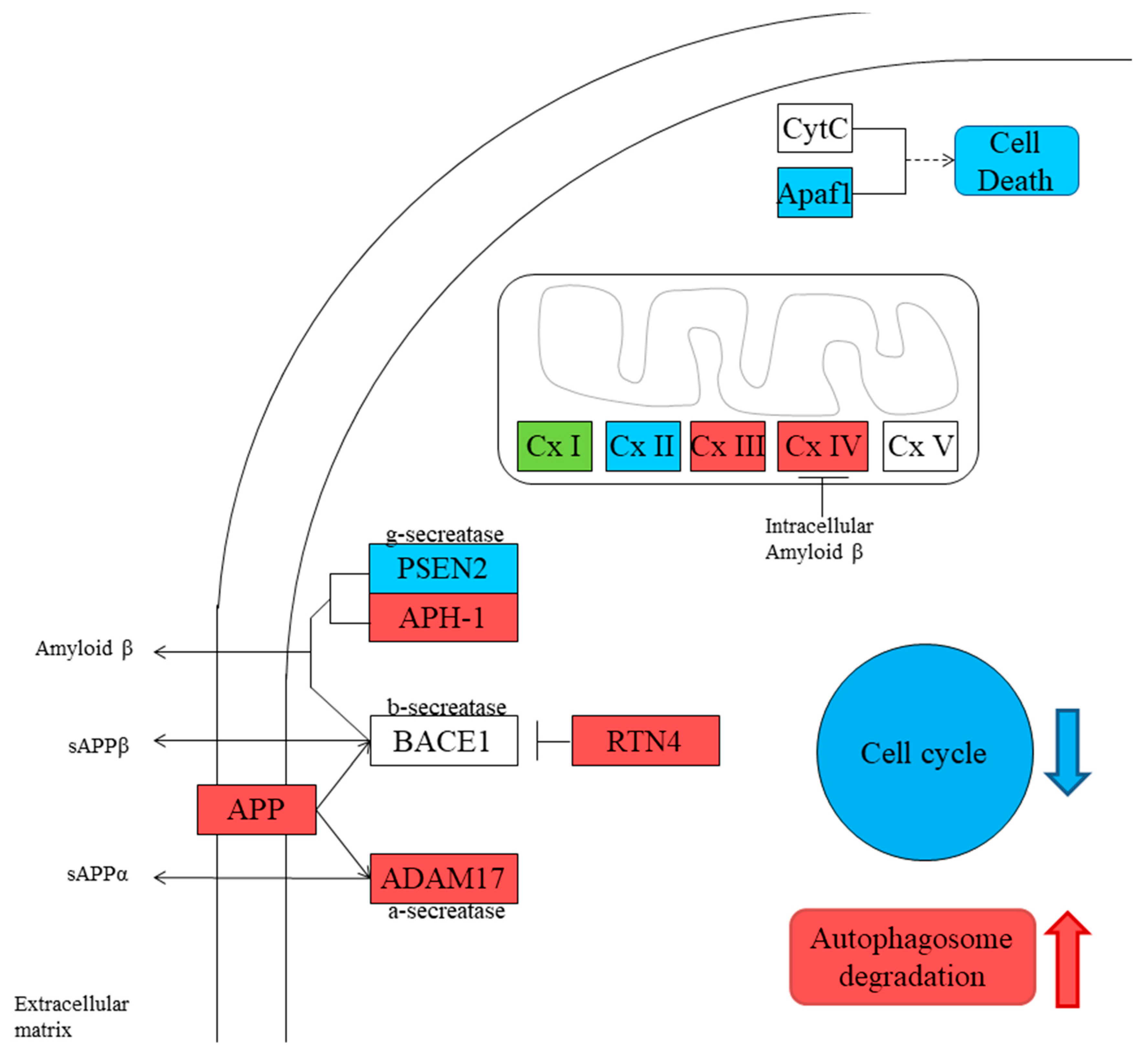
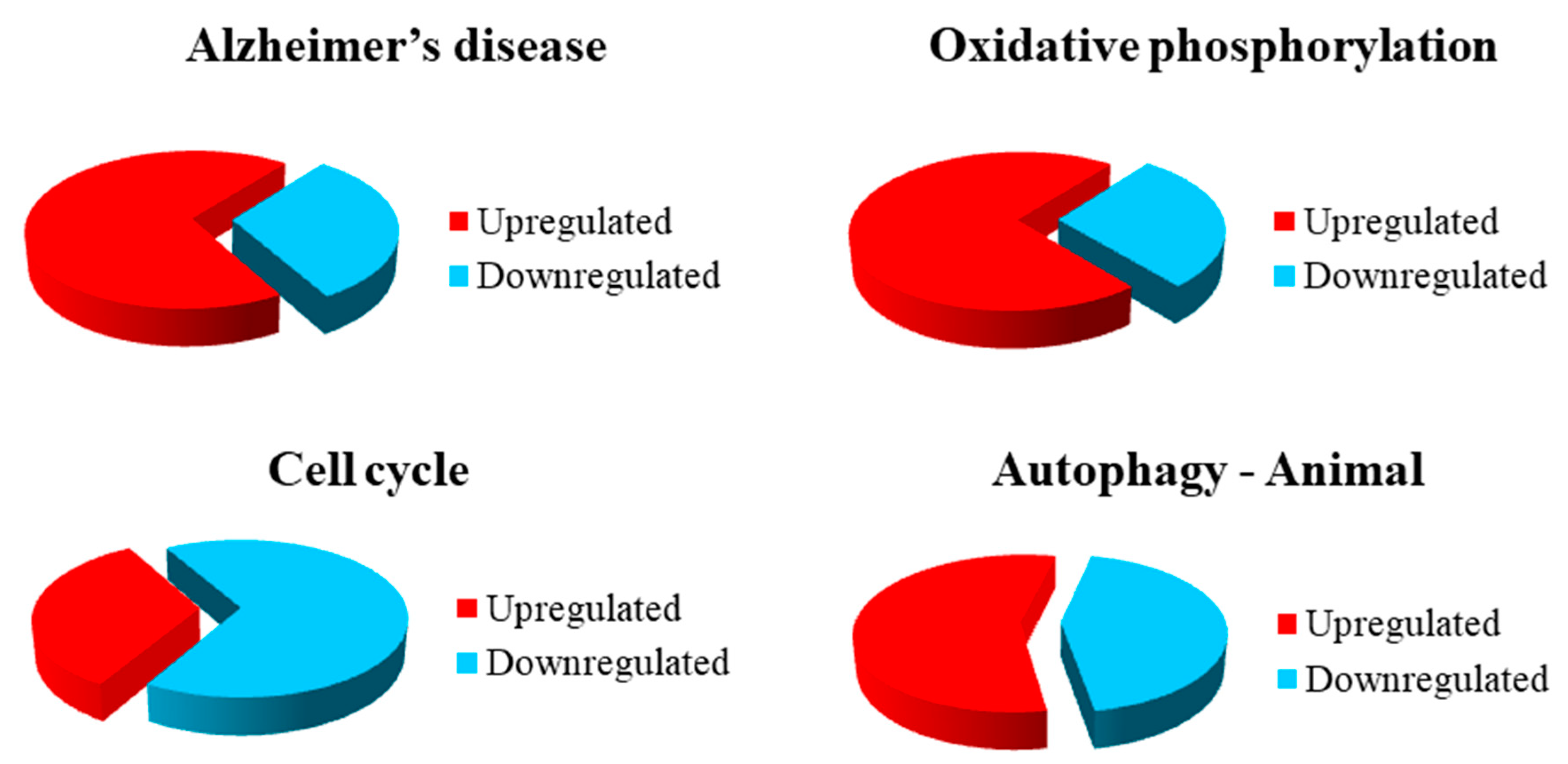
3.2. Gene Inspection
3.3. Immunocytochemistry for iNOS and Nrf2
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Zverova, M. Clinical aspects of Alzheimer’s disease. Clin. Biochem. 2019. [Google Scholar] [CrossRef]
- Fiscon, G.; Weitschek, E.; Cialini, A.; Felici, G.; Bertolazzi, P.; De Salvo, S.; Bramanti, A.; Bramanti, P.; De Cola, M.C. Combining eeg signal processing with supervised methods for Alzheimer’s patients classification. BMC Med. Inform. Decis. Mak. 2018, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Lo Buono, V.; Bonanno, L.; Corallo, F.; Foti, M.; Palmeri, R.; Marra, A.; Di Lorenzo, G.; Todaro, A.; Bramanti, P.; Bramanti, A.; et al. Qualitative analysis of mini mental state examination pentagon in vascular dementia and alzheimer’s disease: A longitudinal explorative study. J. Stroke Cerebrovasc. Dis. 2018, 27, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Allone, C.; Lo Buono, V.; Corallo, F.; Bonanno, L.; Palmeri, R.; Di Lorenzo, G.; Marra, A.; Bramanti, P.; Marino, S. Cognitive impairment in Parkinson’s disease, Alzheimer’s dementia, and vascular dementia: The role of the clock-drawing test. Psychogeriatrics 2018, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Tacal, O. Trafficking and proteolytic processing of amyloid precursor protein and secretases in alzheimer’s disease development: An up-to-date review. Eur. J. Pharmacol. 2019, 856, 172415. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Liu, Q.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Abeta42 and abeta40: Similarities and differences. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2015, 21, 522–529. [Google Scholar]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Penke, B.; Bogar, F.; Fulop, L. Beta-amyloid and the pathomechanisms of alzheimer’s disease: A comprehensive view. Molecules 2017, 22, 1692. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzeriat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef]
- Bostanciklioglu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef]
- Meng, T.; Lin, S.; Zhuang, H.; Huang, H.; He, Z.; Hu, Y.; Gong, Q.; Feng, D. Recent progress in the role of autophagy in neurological diseases. Cell Stress 2019, 3, 141–161. [Google Scholar] [CrossRef]
- Ntsapi, C.; Lumkwana, D.; Swart, C.; du Toit, A.; Loos, B. New insights into autophagy dysfunction related to amyloid beta toxicity and neuropathology in alzheimer’s disease. Int. Rev. Cell Mol. Biol. 2018, 336, 321–361. [Google Scholar]
- Dong, Y.; Chen, X.; Liu, Y.; Shu, Y.; Chen, T.; Xu, L.; Li, M.; Guan, X. Do low-serum vitamin e levels increase the risk of alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int. J. Geriatr. Psychiatry 2018, 33, 257–263. [Google Scholar] [CrossRef]
- Jimenez-Jimenez, F.J.; de Bustos, F.; Molina, J.A.; Benito-Leon, J.; Tallon-Barranco, A.; Gasalla, T.; Orti-Pareja, M.; Guillamon, F.; Rubio, J.C.; Arenas, J.; et al. Cerebrospinal fluid levels of alpha-tocopherol (vitamin e) in Alzheimer’s disease. J. Neural Transm. 1997, 104, 703–710. [Google Scholar] [CrossRef]
- Mangialasche, F.; Kivipelto, M.; Mecocci, P.; Rizzuto, D.; Palmer, K.; Winblad, B.; Fratiglioni, L. High plasma levels of vitamin e forms and reduced Aalzheimer’s disease risk in advanced age. J. Alzheimer’s Dis. JAD 2010, 20, 1029–1037. [Google Scholar] [CrossRef]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Ozer, N.K. Vitamin e: Emerging aspects and new directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef]
- Zingg, J.M. Vitamin e: Regulatory role on signal transduction. IUBMB Life 2019, 71, 456–478. [Google Scholar] [CrossRef]
- Niki, E. Role of vitamin e as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Ulatowski, L.; Parker, R.; Warrier, G.; Sultana, R.; Butterfield, D.A.; Manor, D. Vitamin E is essential for purkinje neuron integrity. Neuroscience 2014, 260, 120–129. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Role of vitamin e in the treatment of Alzheimer’s disease: Evidence from animal models. Int. J. Mol. Sci. 2017, 18, 2504. [Google Scholar] [CrossRef]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The effectiveness of vitamin E treatment in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef]
- Yang, S.G.; Wang, W.Y.; Ling, T.J.; Feng, Y.; Du, X.T.; Zhang, X.; Sun, X.X.; Zhao, M.; Xue, D.; Yang, Y.; et al. Alpha-tocopherol quinone inhibits beta-amyloid aggregation and cytotoxicity, disaggregates preformed fibrils and decreases the production of reactive oxygen species, no and inflammatory cytokines. Neurochem. Int. 2010, 57, 914–922. [Google Scholar] [CrossRef]
- Castillo, W.O.; Aristizabal-Pachon, A.F.; de Lima Montaldi, A.P.; Sakamoto-Hojo, E.T.; Takahashi, C.S. Galanthamine decreases genotoxicity and cell death induced by beta-amyloid peptide in sh-sy5y cell line. Neurotoxicology 2016, 57, 291–297. [Google Scholar] [CrossRef]
- Castillo, W.O.; Aristizabal-Pachon, A.F.; Sakamoto-Hojo, E.; Gasca, C.A.; Cabezas-Fajardo, F.A.; Takahashi, C. Caliphruria subedentata (amaryllidaceae) decreases genotoxicity and cell death induced by beta-amyloid peptide in sh-sy5y cell line. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2018, 836, 54–61. [Google Scholar] [CrossRef]
- Mei, Z.; Yan, P.; Situ, B.; Mou, Y.; Liu, P. Cryptotanshinione inhibits beta-amyloid aggregation and protects damage from beta-amyloid in sh-sy5y cells. Neurochem. Res. 2012, 37, 622–628. [Google Scholar] [CrossRef]
- Seino, S.; Kimoto, T.; Yoshida, H.; Tanji, K.; Matsumiya, T.; Hayakari, R.; Seya, K.; Kawaguchi, S.; Tsuruga, K.; Tanaka, H.; et al. Gnetin c, a resveratrol dimer, reduces amyloid-beta 1-42 (abeta42) production and ameliorates abeta42-lowered cell viability in cultured sh-sy5y human neuroblastoma cells. Biomed. Res. 2018, 39, 105–115. [Google Scholar] [CrossRef]
- Chiricosta, L.; Diomede, F.; Trubiani, O.; Bramanti, P.; Mazzon, E. Physiological expression of ion channel receptors in human periodontal ligament stem cells. Cells 2019, 8, 219. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- University of California Santa Cruz (Ucsc). Available online: http://labshare.cshl.edu/shares/gingeraslab/www-data/dobin/STAR/STARgenomes/ENSEMBL/homo_sapiens/ENSEMBL.homo_sapiens.release-75/Homo_sapiens.GRCh37.75.dna.primary_assembly.fa (accessed on 1 June 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with rna-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kyoto encyclopedia of genes and genomes (kegg). Available online: https://www.genome.jp/kegg/pathway.html (accessed on 20 June 2019).
- Cervantes, B.; Ulatowski, L.M. Vitamin E and Alzheimer’s disease-is it time for personalized medicine? Antioxidants 2017, 6, 45. [Google Scholar] [CrossRef]
- La Fata, G.; Weber, P.; Mohajeri, M.H. Effects of vitamin e on cognitive performance during ageing and in alzheimer’s disease. Nutrients 2014, 6, 5453–5472. [Google Scholar] [CrossRef]
- Meng, P.; Yoshida, H.; Matsumiya, T.; Imaizumi, T.; Tanji, K.; Xing, F.; Hayakari, R.; Dempoya, J.; Tatsuta, T.; Aizawa-Yashiro, T.; et al. Carnosic acid suppresses the production of amyloid-beta 1-42 by inducing the metalloprotease gene tace/adam17 in sh-sy5y human neuroblastoma cells. Neurosci. Res. 2013, 75, 94–102. [Google Scholar] [CrossRef]
- Murayama, K.S.; Kametani, F.; Saito, S.; Kume, H.; Akiyama, H.; Araki, W. Reticulons rtn3 and rtn4-b/c interact with bace1 and inhibit its ability to produce amyloid beta-protein. Eur. J. Neurosci. 2006, 24, 1237–1244. [Google Scholar] [CrossRef]
- He, W.; Lu, Y.; Qahwash, I.; Hu, X.Y.; Chang, A.; Yan, R. Reticulon family members modulate bace1 activity and amyloid-beta peptide generation. Nat. Med. 2004, 10, 959–965. [Google Scholar] [CrossRef]
- Checler, F.; Dunys, J.; Pardossi-Piquard, R.; Alves da Costa, C. P53 is regulated by and regulates members of the gamma-secretase complex. Neuro Degener. Dis. 2010, 7, 50–55. [Google Scholar] [CrossRef]
- Dunys, J.; Kawarai, T.; Sevalle, J.; Dolcini, V.; George-Hyslop, P.S.; Da Costa, C.A.; Checler, F. P53-dependent aph-1 and pen-2 anti-apoptotic phenotype requires the integrity of the gamma-secretase complex but is independent of its activity. J. Biol. Chem. 2007, 282, 10516–10525. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Chan, C.H.; Ma, Q.H.; Xu, X.H.; Xiao, Z.C.; Tan, E.K. The roles of amyloid precursor protein (app) in neurogenesis: Implications to pathogenesis and therapy of Alzheimer disease. Cell Adhes. Migr. 2011, 5, 280–292. [Google Scholar] [CrossRef]
- Ryan, M.M.; Morris, G.P.; Mockett, B.G.; Bourne, K.; Abraham, W.C.; Tate, W.P.; Williams, J.M. Time-dependent changes in gene expression induced by secreted amyloid precursor protein-alpha in the rat hippocampus. BMC Genom. 2013, 14, 376. [Google Scholar] [CrossRef][Green Version]
- Chasseigneaux, S.; Allinquant, B. Functions of abeta, sappalpha and sappbeta: Similarities and differences. J. Neurochem. 2012, 120, 99–108. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Qu, D.H.; Wang, K. Therapeutic approaches to Alzheimer’s disease through stimulating of non-amyloidogenic processing of amyloid precursor protein. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 2389–2403. [Google Scholar]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of a beta accumulation in alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Proteinopathies and oxphos dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar atpase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-the emerging role of v-atpase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic pathways: Ten minutes to dead. Cell 2005, 121, 671–674. [Google Scholar] [CrossRef]
- Bonda, D.J.; Lee, H.P.; Kudo, W.; Zhu, X.; Smith, M.A.; Lee, H.G. Pathological implications of cell cycle re-entry in Alzheimer disease. Expert Rev. Mol. Med. 2010, 12, 19. [Google Scholar] [CrossRef]
- Frasca, G.; Chiechio, S.; Vancheri, C.; Nicoletti, F.; Copani, A.; Angela Sortino, M. Beta-amyloid-activated cell cycle in sh-sy5y neuroblastoma cells: Correlation with the map kinase pathway. J. Mol. Neurosci. MN 2004, 22, 231–236. [Google Scholar] [CrossRef]
- Folch, J.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pizarro, J.G.; Beas-Zarate, C.; Pallas, M.; Camins, A. Role of cell cycle re-entry in neurons: A common apoptotic mechanism of neuronal cell death. Neurotox. Res. 2012, 22, 195–207. [Google Scholar] [CrossRef]
- Colicino, E.G.; Hehnly, H. Regulating a key mitotic regulator, polo-like kinase 1 (plk1). Cytoskeleton 2018, 75, 481–494. [Google Scholar] [CrossRef]
- Ishimi, Y. Regulation of mcm2-7 function. Genes Genet. Syst. 2018, 93, 125–133. [Google Scholar] [CrossRef]
- Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. CB 2015, 25, 1002–1018. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Kernan, J.; Bonacci, T.; Emanuele, M.J. Who guards the guardian? Mechanisms that restrain apc/c during the cell cycle. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1924–1933. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Lloret, A.; Vina, J. New functions of apc/c ubiquitin ligase in the nervous system and its role in alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1057. [Google Scholar] [CrossRef]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating cdc25 activity in the cell cycle: Clinical implications of cdc25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef]
- Gardino, A.K.; Yaffe, M.B. 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 2011, 22, 688–695. [Google Scholar] [CrossRef]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of g2/m transition by inhibition of wee1 and pkmyt1 kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef]
- Zheng, N.; Zhou, Q.; Wang, Z.; Wei, W. Recent advances in scf ubiquitin ligase complex: Clinical implications. Biochim. Biophys. Acta 2016, 1866, 12–22. [Google Scholar] [CrossRef]
- Gui, M.C.; Chen, B.; Yu, S.S.; Bu, B.T. Effects of suppressed autophagy on mitochondrial dynamics and cell cycle of n2a cells. J. Huazhong Univ. Sci. Technol. Med 2014, 34, 157–160. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ulk1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Viret, C.; Faure, M. Regulation of syntaxin 17 during autophagosome maturation. Trends Cell Biol. 2019, 29, 1–3. [Google Scholar] [CrossRef]
- Vergne, I.; Deretic, V. The role of pi3p phosphatases in the regulation of autophagy. FEBS Lett. 2010, 584, 1313–1318. [Google Scholar] [CrossRef]
- Grimmel, M.; Backhaus, C.; Proikas-Cezanne, T. Wipi-mediated autophagy and longevity. Cells 2015, 4, 202–217. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. Lc3/gabarap family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian atg8/lc3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Maruyama, T.; Noda, N.N. Autophagy-regulating protease atg4: Structure, function, regulation and inhibition. J. Antibiot. 2017, 71, 72. [Google Scholar] [CrossRef]
- Kaminskyy, V.; Zhivotovsky, B. Proteases in autophagy. Biochim. Biophys. Acta 2012, 1824, 44–50. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Expression Level SH-SY5Y-Aβ | Expression Level SH-SY5Y-Aβ-αT | Fold Change SH-SY5Y-Aβ vs SH-SY5Y-Aβ-αT | KEGG |
|---|---|---|---|---|
| APP | 790.97 | 831.23 | 0.07 | APP |
| ADAM17 | 43.75 | 51.53 | 0.24 | ADAM17 |
| RTN4 | 145.96 | 172.05 | 0.24 | RTN4 |
| PSEN2 | 36.60 | 27.17 | −0.43 | PSEN |
| APH1B | 0.71 | 4.27 | 2.59 | APH-1 |
| NDUFB1 | 315.96 | 386.93 | 0.29 | Cx I |
| NDUFB3 | 33.35 | 45.25 | 0.44 | Cx I |
| NDUFC2 | 125.39 | 157.85 | 0.33 | Cx I |
| NDUFS4 | 110.35 | 134.55 | 0.29 | Cx I |
| NDUFA1 | 140.33 | 116.04 | −0.27 | Cx I |
| NDUFA6 | 44.59 | 27.70 | −0.69 | Cx I |
| SDHA | 90.76 | 72.85 | −0.32 | Cx II |
| UQCRB | 319.48 | 465.93 | 0.54 | Cx III |
| UQCRH | 409.51 | 459.12 | 0.17 | Cx III |
| COX7A2 | 317.64 | 368.03 | 0.21 | Cx IV |
| APAF1 | 32.15 | 23.13 | −0.48 | Apaf1 |
| Gene | Expression Level SH-SY5Y-Aβ | Expression Level SH-SY5Y-Aβ-αT | Fold Change SH-SY5Y-Aβ vs SH-SY5Y-Aβ-αT | KEGG |
|---|---|---|---|---|
| NDUFB1 | 315.96 | 386.93 | 0.29 | Cx I |
| NDUFB3 | 33.35 | 45.25 | 0.44 | Cx I |
| NDUFC2 | 125.39 | 157.85 | 0.33 | Cx I |
| NDUFS4 | 110.35 | 134.55 | 0.29 | Cx I |
| NDUFA1 | 140.33 | 116.04 | −0.27 | Cx I |
| NDUFA6 | 44.59 | 27.70 | −0.69 | Cx I |
| SDHA | 90.76 | 72.85 | −0.32 | Cx II |
| UQCRB | 319.48 | 465.93 | 0.54 | Cx III |
| UQCRH | 409.51 | 459.12 | 0.17 | Cx III |
| COX7A2 | 317.64 | 368.03 | 0.21 | Cx IV |
| COX10 | 21.36 | 30.59 | 0.52 | Cx IV |
| ATP6V0A2 | 107.86 | 83.99 | −0.36 | V-type ATPase |
| ATP6V1C1 | 31.00 | 47.08 | 0.60 | V-type ATPase |
| ATP6V1H | 17.21 | 28.31 | 0.72 | V-type ATPase |
| Gene | Expression Level SH-SY5Y-Aβ | Expression Level SH-SY5Y-Aβ-αT | Fold Change SH-SY5Y-Aβ vs SH-SY5Y-Aβ-αT | KEGG |
|---|---|---|---|---|
| SMC1A | 308.34 | 265.09 | −0.22 | Smc1 |
| PTTG1 | 238.77 | 195.25 | −0.29 | PTTG |
| ANAPC1 | 27.67 | 21.81 | −0.34 | APC/C |
| CDC27 | 72.41 | 96.18 | 0.41 | APC/C |
| CDC16 | 47.73 | 56.45 | 0.24 | APC/C |
| MAD2L1 | 41.47 | 20.97 | −0.98 | Mad2 |
| BUB1B | 192.20 | 162.39 | −0.24 | BubR1 |
| BUB3 | 108.93 | 85.13 | −0.36 | Bub3 |
| YWHAE | 839.28 | 920.19 | 0.13 | 14-3-3 |
| YWHAQ | 528.10 | 617.89 | 0.23 | 14-3-3 |
| YWHAZ | 337.73 | 382.05 | 0.18 | 14-3-3 |
| YWHAG | 54.03 | 59.33 | 0.14 | 14-3-3 |
| CDC25B | 22.56 | 16.98 | −0.41 | Cdc25B |
| PLK1 | 75.39 | 44.80 | −0.75 | Plk1 |
| CCNB1 | 115.68 | 82.62 | −0.49 | CycB |
| CCNB2 | 32.32 | 24.88 | −0.38 | CycB |
| CDK1 | 55.86 | 40.01 | −0.48 | CDK1 |
| CCNA2 | 28.88 | 24.31 | −0.25 | CycA |
| WEE1 | 54.85 | 69.70 | 0.35 | Wee |
| MCM4 | 424.40 | 327.06 | −0.38 | MCM |
| MCM6 | 42.88 | 36.64 | −0.23 | MCM |
| ABL1 | 26.96 | 22.39 | −0.27 | Abl |
| CCND1 | 326.55 | 269.68 | −0.28 | CycD |
| SKP1 | 505.53 | 563.13 | 0.16 | SCF |
| Gene | Expression Level SH-SY5Y-Aβ | Expression Level SH-SY5Y-Aβ-αT | Fold Change SH-SY5Y-Aβ vs SH-SY5Y-Aβ-αT | KEGG |
|---|---|---|---|---|
| CTSB | 29.90 | 46.25 | 0.63 | CTSB |
| CTSL | 25.55 | 17.53 | −0.54 | CTSL |
| CTSD | 39.26 | 51.95 | 0.40 | CTSD |
| STX17 | 34.35 | 17.14 | −1.00 | STX17 |
| GABARAP | 322.29 | 387.87 | 0.27 | LC3 |
| GABARAPL1 | 10.63 | 19.36 | 0.87 | LC3 |
| ATG3 | 98.92 | 126.93 | 0.36 | ATG3 |
| ATG4A | 8.14 | 3.51 | −1.21 | ATG4 |
| ATG4B | 65.88 | 43.33 | −0.61 | ATG4 |
| MTMR3 | 27.34 | 38.55 | 0.50 | MRMR3 |
| UVRAG | 9.34 | 16.50 | 0.82 | UVRAG |
| AMBRA1 | 13.36 | 17.45 | 0.39 | AMBRA1 |
| VMP1 | 72.46 | 57.04 | −0.35 | VMP1 |
| WIPI1 | 9.11 | 15.35 | 0.75 | WIPI |
| WIPI2 | 58.48 | 45.25 | −0.37 | WIPI |
| ULK2 | 58.01 | 37.97 | −0.61 | ULK2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gugliandolo, A.; Chiricosta, L.; Silvestro, S.; Bramanti, P.; Mazzon, E. α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study. Brain Sci. 2019, 9, 196. https://doi.org/10.3390/brainsci9080196
Gugliandolo A, Chiricosta L, Silvestro S, Bramanti P, Mazzon E. α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study. Brain Sciences. 2019; 9(8):196. https://doi.org/10.3390/brainsci9080196
Chicago/Turabian StyleGugliandolo, Agnese, Luigi Chiricosta, Serena Silvestro, Placido Bramanti, and Emanuela Mazzon. 2019. "α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study" Brain Sciences 9, no. 8: 196. https://doi.org/10.3390/brainsci9080196
APA StyleGugliandolo, A., Chiricosta, L., Silvestro, S., Bramanti, P., & Mazzon, E. (2019). α-Tocopherol Modulates Non-Amyloidogenic Pathway and Autophagy in an In Vitro Model of Alzheimer’s Disease: A Transcriptional Study. Brain Sciences, 9(8), 196. https://doi.org/10.3390/brainsci9080196