Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells

 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. S100β Injections
2.3. Electrophysiology
2.4. Immunohistochemistry
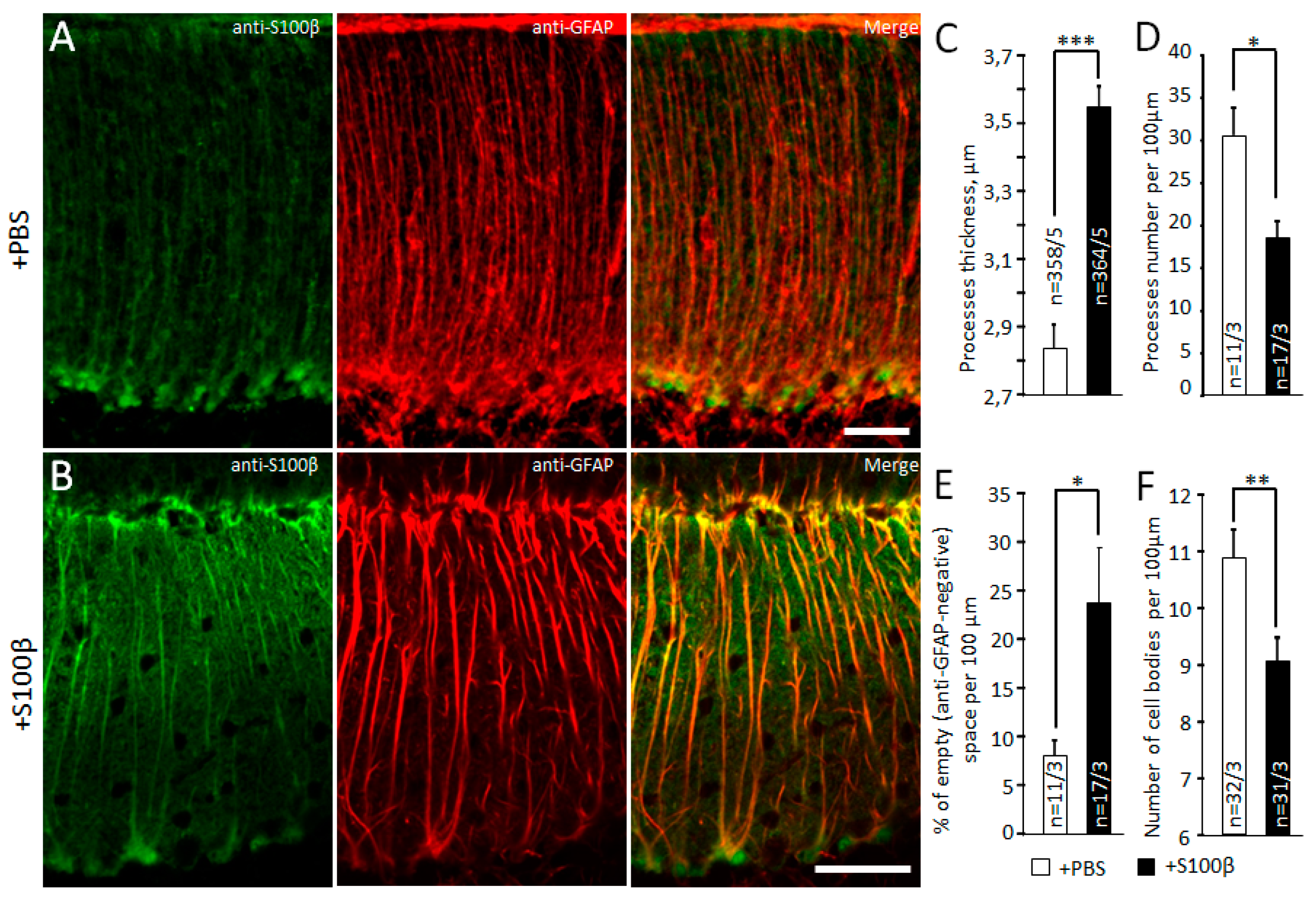
2.5. Confocal Microscopy and Morphometric Analysis
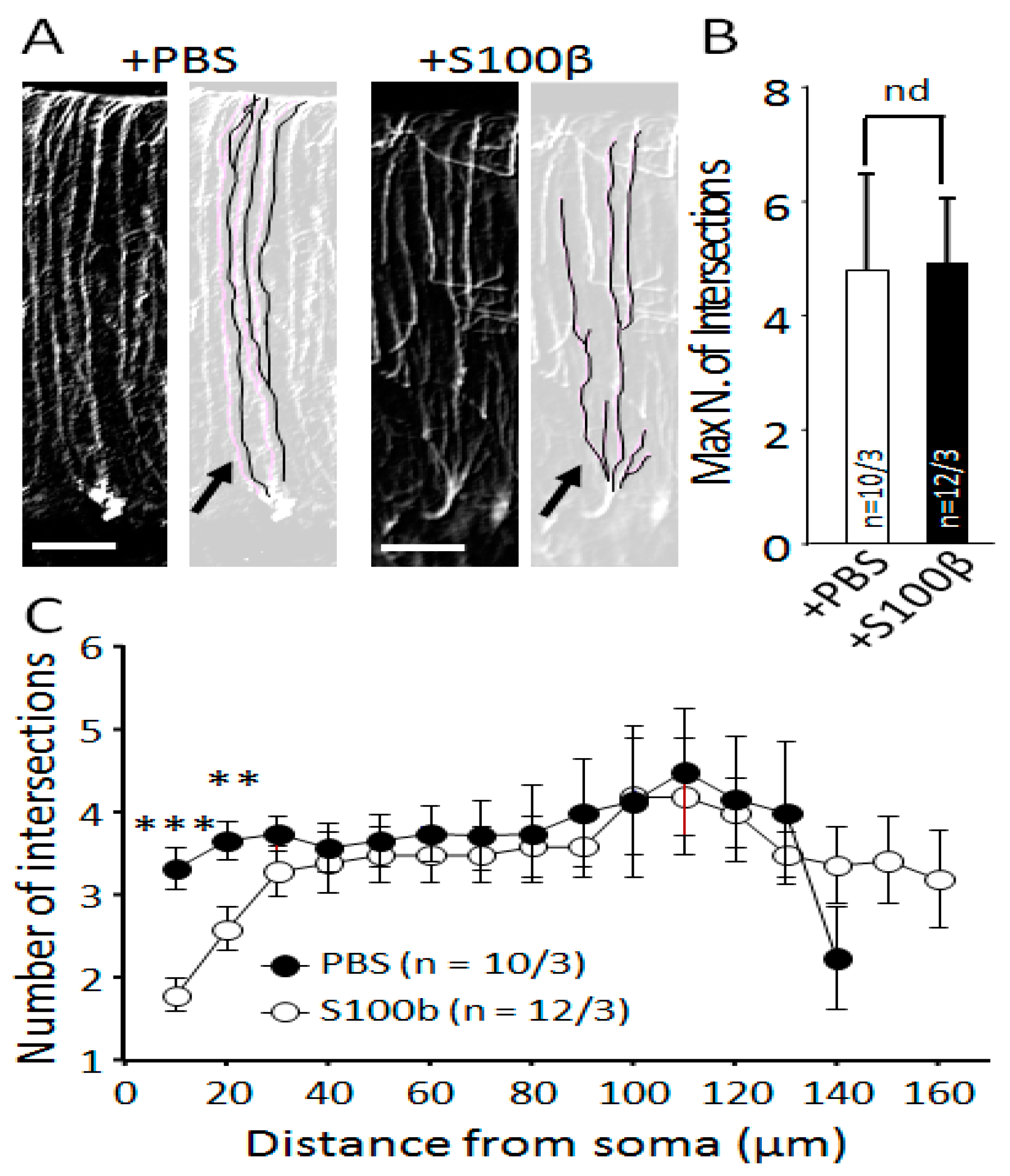
2.6. Sholl Analysis of Bergmann Glia Cells
2.7. Statistical and Mathematical Analysis
3. Results
3.1. Exogenous S100β Alters Morphology of Bergmann Glia
3.2. Extracellular S100β Alters Morphology of Purkinje Cells
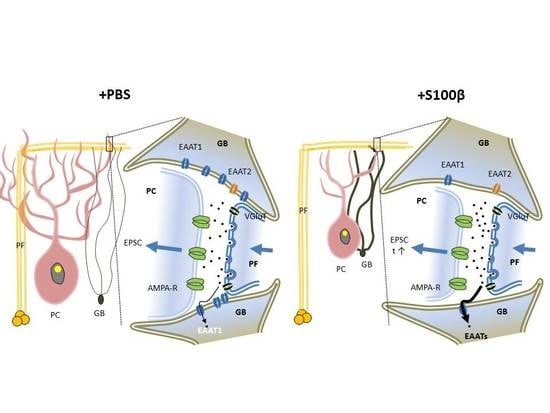
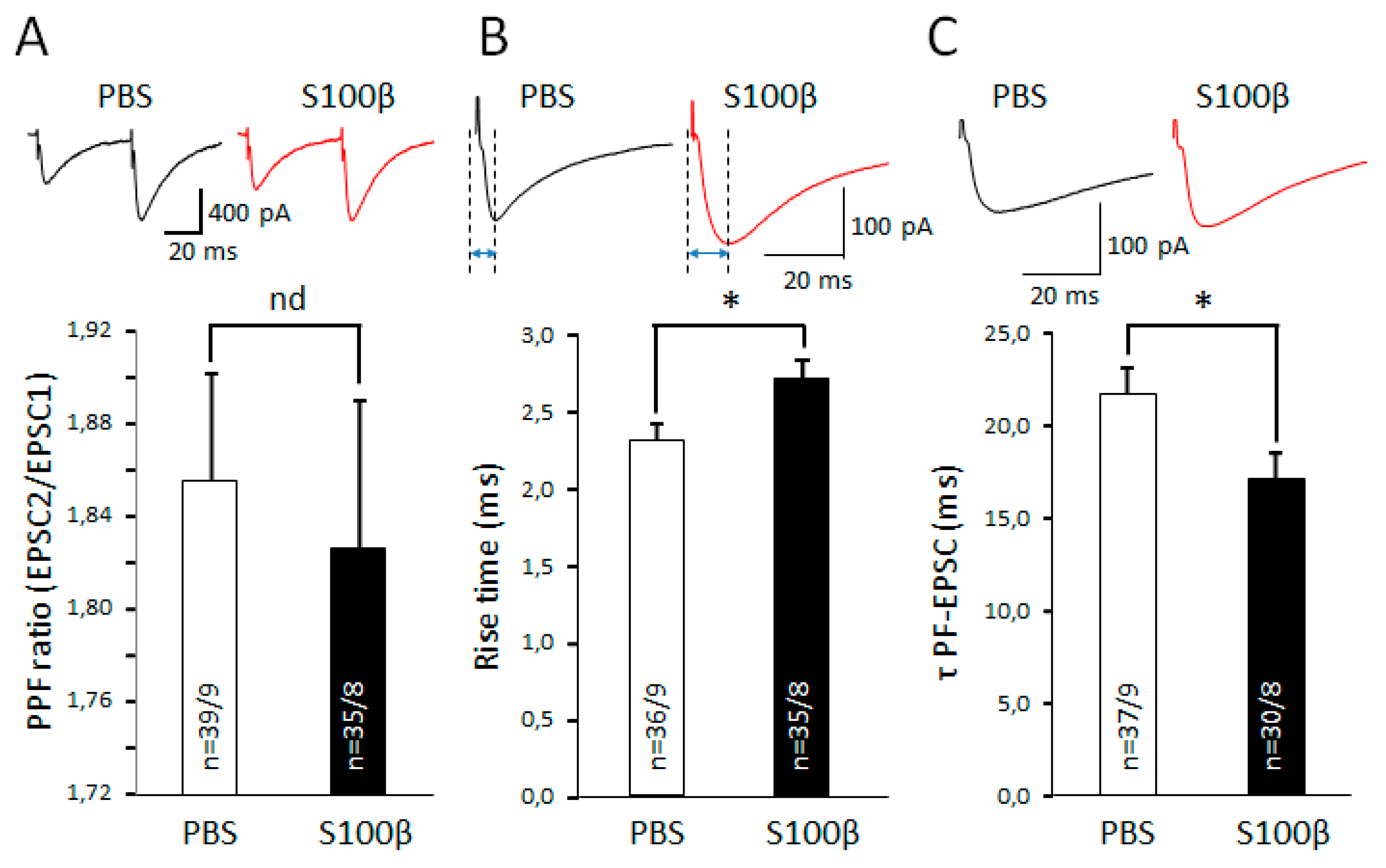
3.3. Extracellular S100β Alters Synaptic Transmission in PFs and PCs
3.4. Similarities in Changes in Synaptic Transmission in S100β-Injected Mice and Ataxin1 Mutant Animals
3.5. Extracellular S100β Alters Endocannabinoid-Dependent Short Term Plasticity in PF–PC Synapses
3.6. Effects of FC on PF–PC Transmission and Endocannabinoid Short Term Plasticity in PF–PC Synapses
3.7. Slowdown of Glutamate Uptake in Bergmann Glia by TBOA Leads to Alteration of PF-EPSC Kinetic, But Does Not Change Endocannabinoid Short Term Plasticity in PF-PC Synapses
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Sun, Y.-M.; Lu, C.; Wu, Z.-Y. Spinocerebellar ataxia: Relationship between phenotype and genotype—A review. Clin. Genet. 2016, 90, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, S.; Mariotti, C.; Taroni, F. Spinocerebellar ataxia type 1. Handb. Clin. Neurol. 2012, 103, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Tezenas du Montcel, S.; Durr, A.; Rakowicz, M.; Nanetti, L.; Charles, P.; Sulek, A.; Mariotti, C.; Rola, R.; Schols, L.; Bauer, P.; et al. Prediction of the age at onset in spinocerebellar ataxia type 1, 2, 3 and 6. J. Med. Genet. 2014, 51, 479–486. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide Repeat Disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Pathogenic Mechanisms of a Polyglutamine-mediated Neurodegenerative Disease, Spinocerebellar Ataxia Type 1. J. Biol. Chem. 2009, 284, 7425–7429. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K.; Globas, C.; Bösch, S.; Klockgether, T.; Zühlke, C.; Daum, I.; Dichgans, J. Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J. Neurol. 2003, 250, 207–211. [Google Scholar] [CrossRef]
- Lim, J.; Crespo-Barreto, J.; Jafar-Nejad, P.; Bowman, A.B.; Richman, R.; Hill, D.E.; Orr, H.T.; Zoghbi, H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452, 713–718. [Google Scholar] [CrossRef]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Duenas, A.M.; Goold, R.; Giunti, P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129, 1357–1370. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef]
- Cvetanovic, M.; Ingram, M.; Orr, H.; Opal, P. Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1. Neuroscience 2015, 289, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, R.; Maggio, N.; Rosaria Bianco, M.; Cavaliere, C.; Cirillo, G.; Lavitrano, M.; Papa, M. Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model. Neuron Glia Biol. 2007, 3, 335–351. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cvetanovic, M. Decreased Expression of Glutamate Transporter GLAST in Bergmann Glia Is Associated with the Loss of Purkinje Neurons in the Spinocerebellar Ataxia Type 1. Cerebellum 2015, 14, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Akassoglou, K.; Kassiotis, G.; Pasparakis, M.; Alexopoulou, L.; Kollias, G. TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J. Neuroimmunol. 1997, 72, 137–141. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Rothermundt, M.; Peters, M.; Prehn, J.H.M. S100B in Brain Damage and Neurodegeneration. Microsc. Res. Tech. 2003, 632, 614–632. [Google Scholar] [CrossRef]
- Vig, P.J.S.; Shao, Q.; Subramony, S.H.; Lopez, M.E.; Safaya, E. Bergmann Glial S100B Activates Myo-inositol Monophosphatase 1 and Co-localizes to Purkinje Cell Vacuoles in SCA1 Transgenic Mice. Cerebellum 2009, 8, 231–244. [Google Scholar] [CrossRef][Green Version]
- Heizmann, C.W.; Fritz, G.; Schäfer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.W.; McGregor, D. Chromatographic and electrophoretic fractionation of soluble proteins of brain and liver. J. Biol. Chem. 1965, 240, 1647–1653. [Google Scholar] [PubMed]
- Shuvaev, A.N.; Hosoi, N.; Sato, Y.; Yanagihara, D.; Hirai, H. Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J. Physiol. 2017, 595, 141–164. [Google Scholar] [CrossRef]
- Kreitzer, A.C.; Regehr, W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001, 29, 717–727. [Google Scholar] [CrossRef]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953, 87, 387–406. [Google Scholar]
- Major, G.; Evans, J.D.; Jack, J.J.B. Solutions for transients in arbitrarily branching cables: II. Voltage clamp theory. Biophys. J. 1993, 65, 450–468. [Google Scholar] [CrossRef]
- Sterratt, D.; Graham, B.; Gillies, A.; Willshaw, D. Principles of Computational Modelling in Neuroscience; Cambridge University Press: Cambridge, UK, 2011; 404p. [Google Scholar]
- Kaminsky, N.; Bihari, O.; Kanner, S.; Barzilai, A. Connecting Malfunctioning Glial Cells and Brain Degenerative Disorders. Genom. Proteom. Bioinform. 2016, 14, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170–1170.e1. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Manji, H.K.; Zarate, C.A. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology and Therapeutics of Mood Disorders. Neuroscientist 2010, 15, 525–539. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflugers Arch. 2014, 466, 3–24. [Google Scholar] [CrossRef]
- Swanson, R.A.; Graham, S.H. Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res. 1994, 664, 94–100. [Google Scholar] [CrossRef]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, D.N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Rasooli-Nejad, S.; Palygin, O.; Lalo, U.; Pankratov, Y. Cannabinoid receptors contribute to astroglial Ca2+-signalling and control of synaptic plasticity in the neocortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20140077. [Google Scholar] [CrossRef]
- Araque, A.; Li, N.; Doyle, R.T.; Haydon, P.G. SNARE protein-dependent glutamate release from astrocytes. J. Neurosci. 2000, 20, 666–673. [Google Scholar] [CrossRef]
- Beardsley, P.M.; Hauser, K.F. Glial modulators as potential treatments of psychostimulant abuse. Adv. Pharmacol. 2014, 69, 1–69. [Google Scholar] [CrossRef]
- Cisneros, I.E.; Ghorpade, A. Methamphetamine and HIV-1-induced neurotoxicity: Role of trace amine associated receptor 1 cAMP signaling in astrocytes. Neuropharmacology 2014, 85, 499–507. [Google Scholar] [CrossRef]
- Vig, P.J.S.; Lopez, M.E.; Wei, J.; D’Souza, D.R.; Subramony, S.H.; Henegar, J.; Fratkin, J.D. Glial S100B Positive Vacuoles in Purkinje Cells: Earliest Morphological Abnormality in SCA1 Transgenic Mice. J. Neurol. Sci. Turk. 2006, 23, 166–174. [Google Scholar] [CrossRef]
- Vig, P.J.S.; Hearst, S.; Shao, Q.; Lopez, M.E.; Murphy, H.A.; Safaya, E. Glial S100B protein modulates mutant ataxin-1 aggregation and toxicity: TRTK12 peptide, a potential candidate for SCA1 therapy. Cerebellum 2011, 0, 254–266. [Google Scholar] [CrossRef]
- Hoxha, E.; Gabriele, R.M.C.; Balbo, I.; Ravera, F.; Masante, L.; Zambelli, V.; Albergo, C.; Mitro, N.; Caruso, D.; Di Gregorio, E.; et al. Motor Deficits and Cerebellar Atrophy in Elovl5 Knock Out Mice. Front. Cell. Neurosci. 2017, 11, 343. [Google Scholar] [CrossRef]
- Morquette, P.; Verdier, D.; Kadala, A.; Féthière, J.; Philippe, A.G.; Robitaille, R.; Kolta, A. An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nat. Neurosci. 2015, 18, 844–854. [Google Scholar] [CrossRef]
- Llinás, R.R.; Sugimori, M. The Electrophysiology of the Cerebellar Purkinje Cell Revisited. Cereb. Rev. 1992, 1, 167–181. [Google Scholar]
- Shuvaev, A.N.; Hirai, H. Experience of mGluR1 gene therapy in transgenic models of SCA1 mice. Sovremennyetehnol. Med. 2016, 8, 141–154. [Google Scholar] [CrossRef][Green Version]
- Mitosek-Szewczyk, K.; Sulkowski, G.; Stelmasiak, Z.; Struzynska, L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neuroscience 2008, 155, 45–52. [Google Scholar] [CrossRef]
- Lin, C.L.G.; Kong, Q.; Cuny, G.D.; Glicksman, M.A. Glutamate transporter EAAT2: A new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012, 4, 1689–1700. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Navarrete, M.; Araque, A. Endocannabinoids mediate neuron-astrocyte communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belozor, O.S.; Yakovleva, D.A.; Potapenko, I.V.; Shuvaev, A.N.; Smolnikova, M.V.; Vasilev, A.; Pozhilenkova, E.A.; Shuvaev, A.N. Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells. Brain Sci. 2019, 9, 80. https://doi.org/10.3390/brainsci9040080
Belozor OS, Yakovleva DA, Potapenko IV, Shuvaev AN, Smolnikova MV, Vasilev A, Pozhilenkova EA, Shuvaev AN. Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells. Brain Sciences. 2019; 9(4):80. https://doi.org/10.3390/brainsci9040080
Chicago/Turabian StyleBelozor, Olga S., Dariya A. Yakovleva, Ilya V. Potapenko, Andrey N. Shuvaev, Marina V. Smolnikova, Alex Vasilev, Elena A. Pozhilenkova, and Anton N. Shuvaev. 2019. "Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells" Brain Sciences 9, no. 4: 80. https://doi.org/10.3390/brainsci9040080
APA StyleBelozor, O. S., Yakovleva, D. A., Potapenko, I. V., Shuvaev, A. N., Smolnikova, M. V., Vasilev, A., Pozhilenkova, E. A., & Shuvaev, A. N. (2019). Extracellular S100β Disrupts Bergman Glia Morphology and Synaptic Transmission in Cerebellar Purkinje Cells. Brain Sciences, 9(4), 80. https://doi.org/10.3390/brainsci9040080