Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease



Abstract
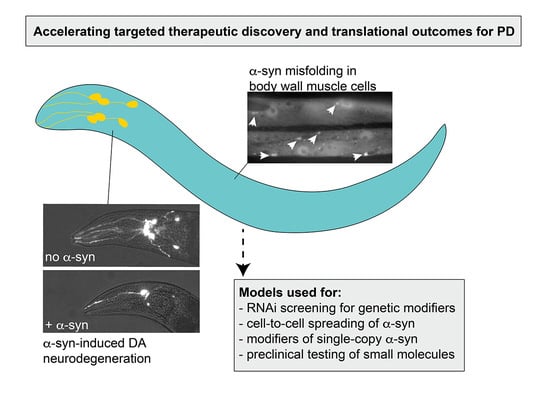
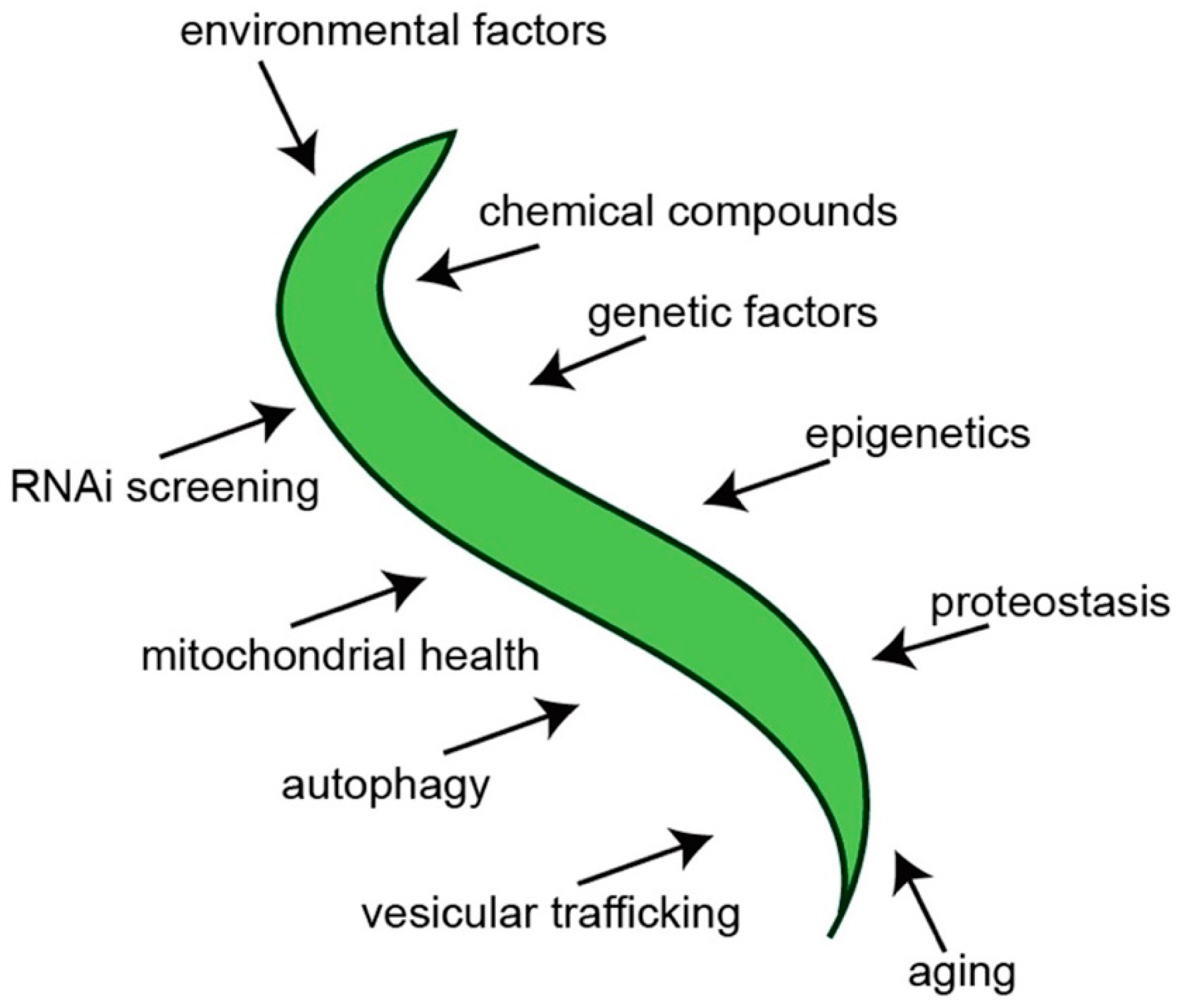
1. Introduction
“I once was lost, for now I’m found”-Amazing Grace, John Newton (1779)
2. C. elegans Models of PD
2.1. The First of Its Kind
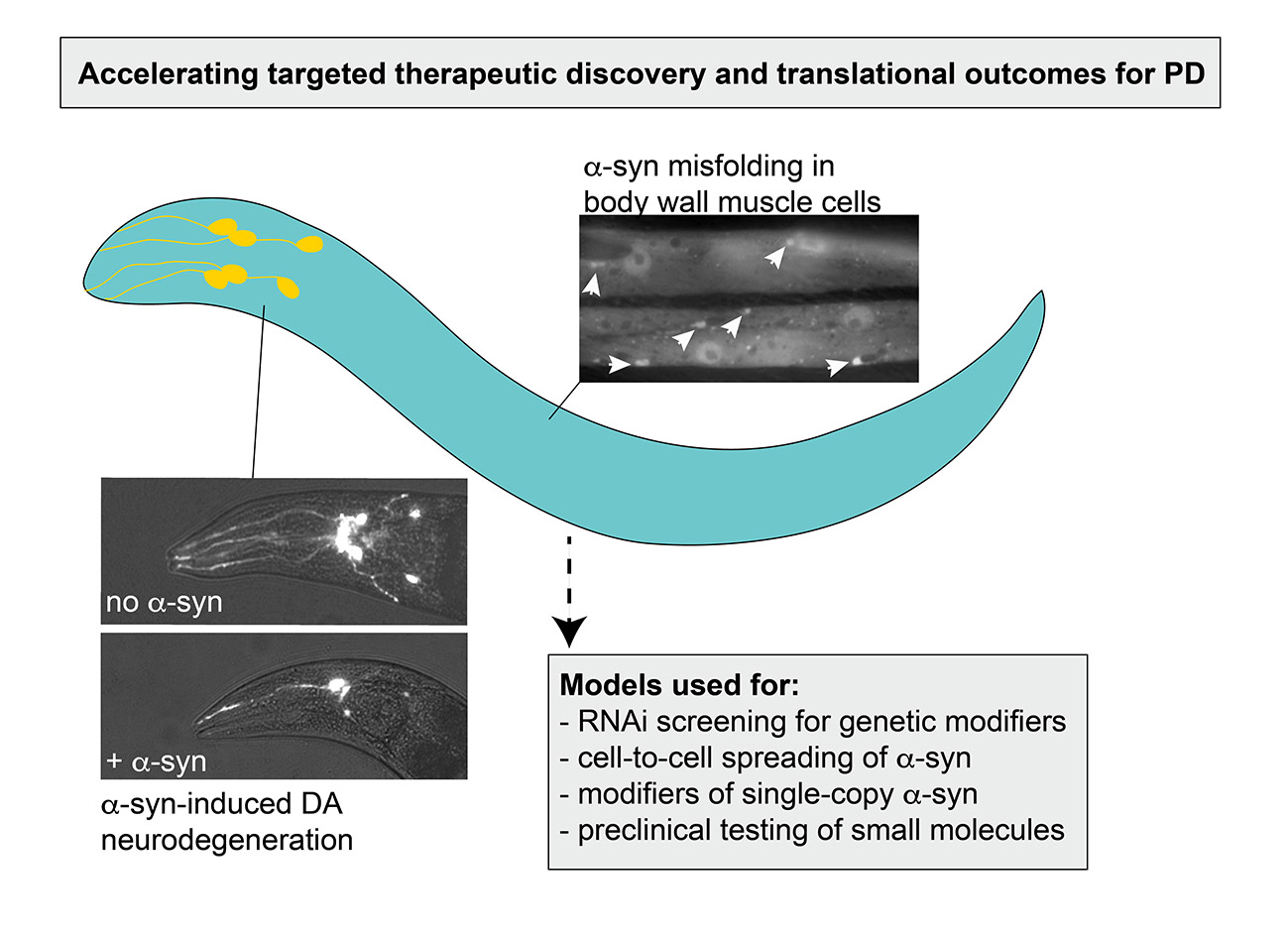
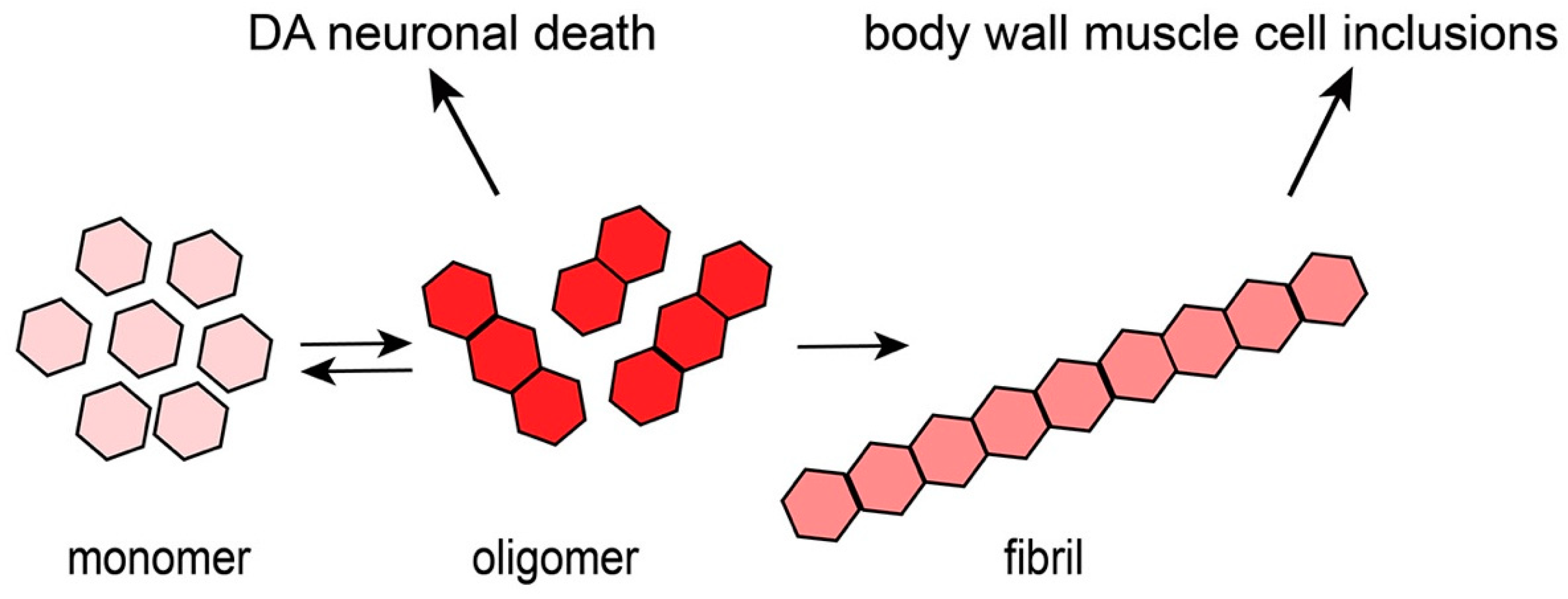
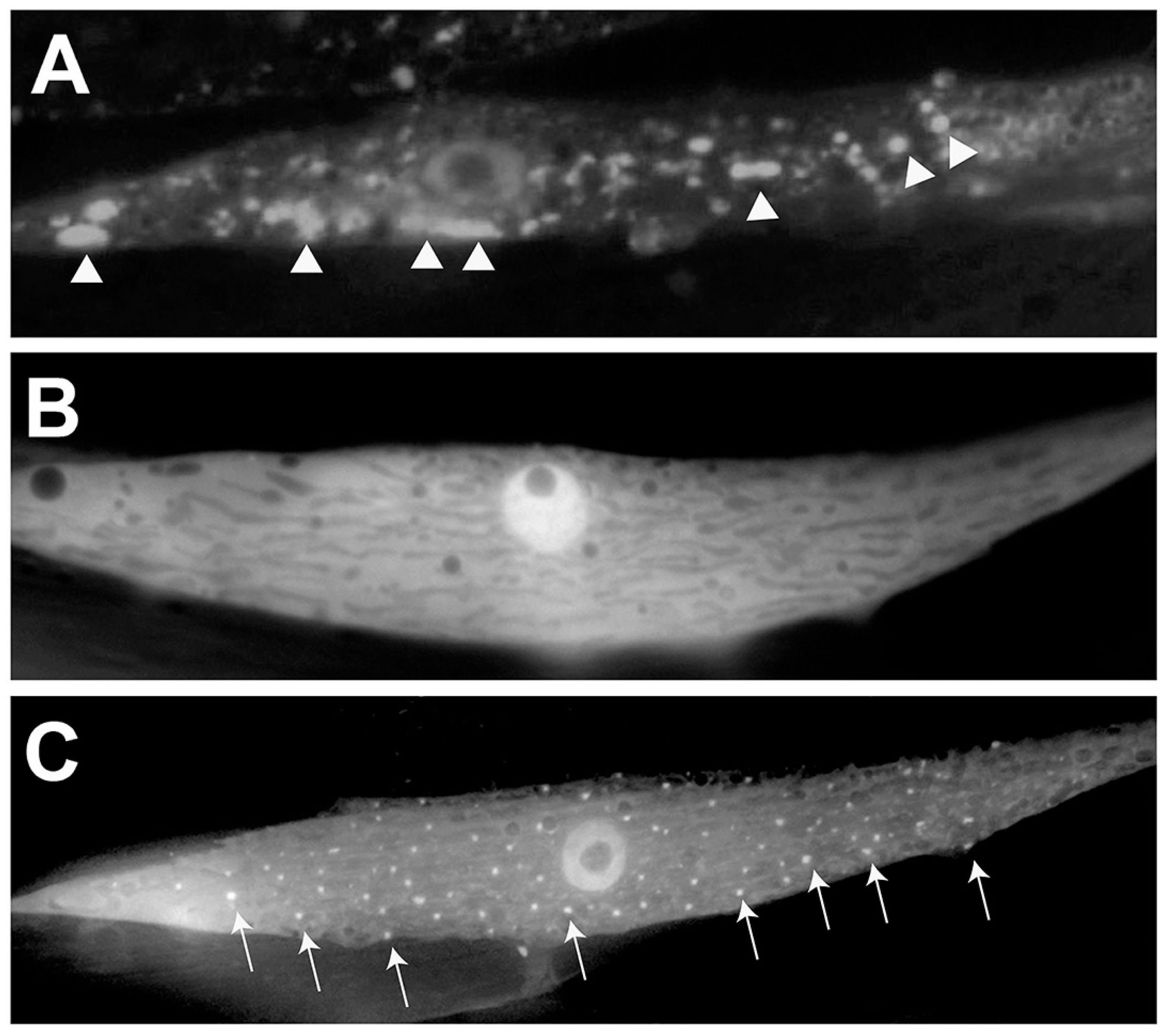
2.2. α-syn Aggregates in Muscle Cells
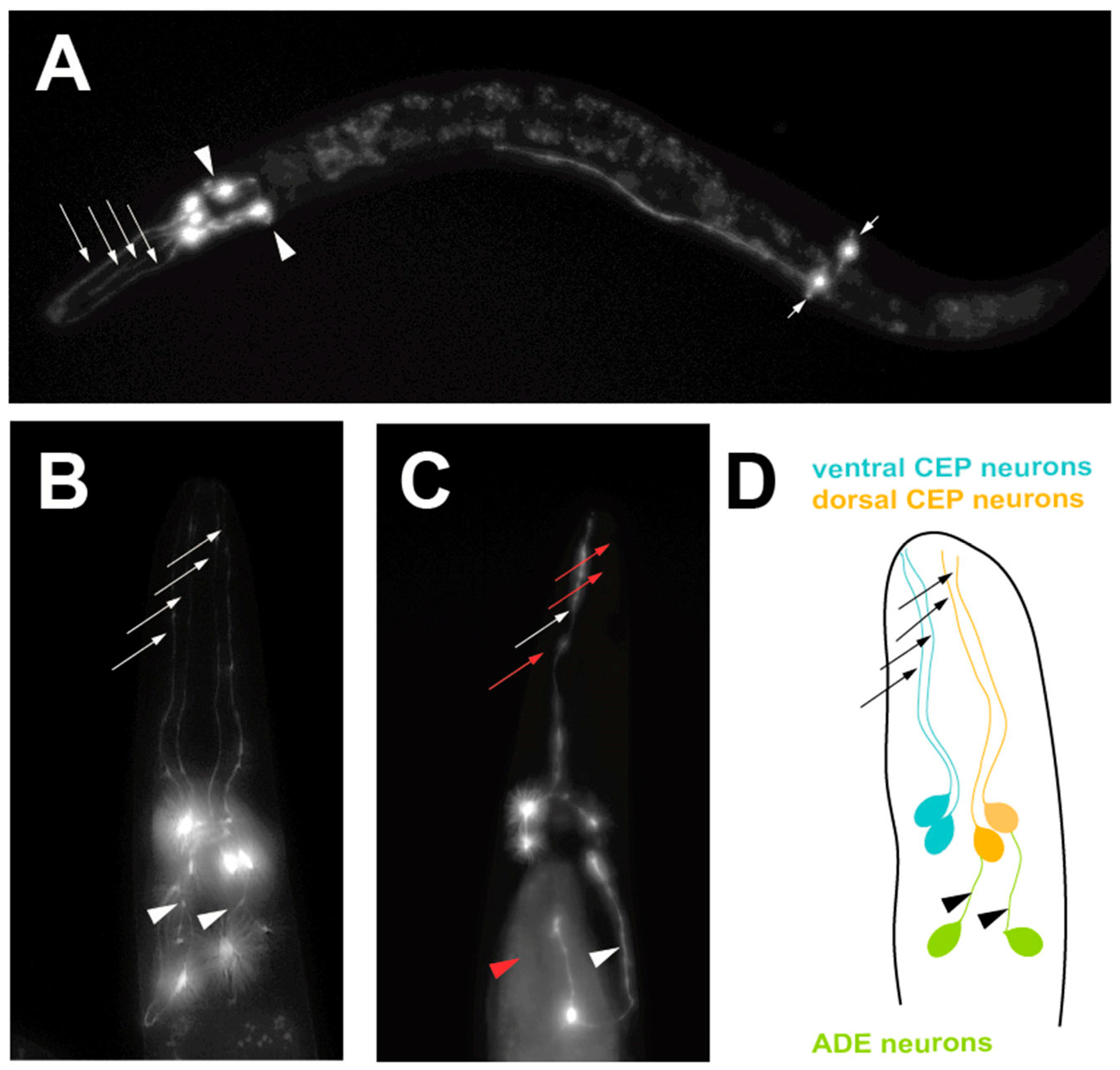
2.3. Modeling Progressive Dopaminergic Neurodegeneration
2.4. An Alternate Model for Investigation of α-syn Aggregation in Vivo
2.5. Single-Copy Transgenic Modeling of α-syn in C. elegans
2.6. A Model to Explore the “Prion-Like” Nature of α-syn
3. Use of C. elegans to Discern the Role of Dopamine in α-syn-Induced Neurotoxicity
4. C. elegans as a Tool to Uncover Epigenetic Mechanisms of PD Pathology
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Sämann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 Act Antagonistically in Stress Response and Neurite Outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joossee, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W.C. Identification of Novel Human Genes Evolutionarily Conserved in Caenorhabditis elegans by Comparative Proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef]
- Cooper, J.F.; Raamsdonk, M.V. Modeling Parkinson’s Disease in C. elegans. J. Parkinson’s Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef]
- Dexter, P.M.; Caldwell, K.A.; Caldwell, G.A. A Predictable Worm: Application of Caenorhabditis elegans for Mechanistic Investigation of Movement Disorders. Neurotherapeutics 2012, 9, 393–404. [Google Scholar] [CrossRef]
- Harrington, A.J.; Hamamichi, S.; Caldwell, G.A.; Caldwell, K.A. C. elegans as a Model Organism to Investigate Molecular Pathways Involved with Parkinson’s Disease. Dev. Dyn. 2010, 239, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.A.; Caldwell, K.A.; Caldwell, G.A. C. elegans as a model system to accelerate discovery for Parkinson disease. Curr. Opin. Genet. Dev. 2017, 44, 102–109. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M.Y. Lewy Body-like α-Synuclein Aggregates Resist Degradation and Impair Macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Growdon, W.; Gomez-Isla, T.; Newell, K.; George, J.M.; Clayton, D.F.; Hyman, B.T. Nigral and Cortical Lewy Bodies and Dystrophic Nigral Neurites in Parkinson’s Disease and Cortical Lewy Body Disease Contain alpha-synuclein Immunoreactivity. J. Neuropathol. Exp. Neurol. 1998, 57, 334–337. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreio, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Slow, E.J.; Graham, R.K.; Osmand, A.P.; Devon, R.S.; Lu, G.; Deng, Y.; Pearson, J.; Vaid, K.; Bissada, N.; Wetzel, R.; et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. USA 2005, 102, 11402–11407. [Google Scholar] [CrossRef]
- Yang, H.; Hu, H.U. Sequestration of cellular interacting partners by protein aggregates: Implication in a loss-of-function pathology. FEBS J. 2016, 283, 3705–3717. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Oligomeric Alpha-synuclein and Its Role in Neuronal Death. IUBMB Life 2010, 62, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.R.; Federoff, H.J.; Vicini, S.; Maguire-Zeiss, K.A. α-Synuclein mediates alterations in membrane conductance: A potential role for α-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 2010, 32, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human α-synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Koyama, A.; Gengyo-Ando, K.; Masuda, M.; Kowa, H.; Tsunoda, M.; Mitani, S.; Iwatsubo, T. Familial Parkinson Mutant α-Synuclein Causes Dopamine Neuron Dysfunction in Transgenic Caenorhabditis elegans. J. Biol. Chem. 2005, 281, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Koyama, A.; Koyama, S.; Yoshina, S.; Ren, C.H.; Kato, T.; Mitani, S.; Iwatsubo, T. A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in a-synuclein transgenic C. elegans. Hum. Mol. Genet. 2008, 17, 2997–3009. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.W.; Plasterk, R.H.A.; Nollen, E.A.A. C. elegans Model Identifies Genetic Modifiers of α-Synuclein Inclusion Formation During Aging. PLoS Genet. 2008, 4, e1000027. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Gelwix, C.S.; Caldwell, K.A.; Caldwell, G.A. Torsin-Mediated Protection from Cellular Stress in the Dopaminergic Neurons of Caenorhabditis elegans. J. Neurosci. 2005, 25, 3801–3812. [Google Scholar] [CrossRef]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef]
- Cooper, J.F.; Spielbauer, K.K.; Senchuk, M.M.; Nadarajan, S.; Colaiácovo, M.P.; Van Raamsdonk, J.M. α-synuclein expression from a single copy transgene increases sensitivity to stress and accelerates neuronal loss in genetic models of Parkinson’s disease. Exp. Neurol. 2018, 310, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Tyson, T.; Senchuk, M.; Cooper, J.F.; George, S.; Van Raamsdonk, J.M.; Brundin, P. Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci. Rep. 2017, 7, 7506. [Google Scholar] [CrossRef]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S.; Majounie, E.; et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 22. [Google Scholar] [CrossRef]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast Reveal a “Druggable” Rsp5/Nedd4 Network that Ameliorates α-Synuclein Toxicity in Neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and Rescue of α-Synuclein Toxicity in Parkinson Patient–Derived Neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.L.; Yan, X.; Hamamichi, S.; Ajjuri, R.R.; Mazzulli, J.R.; Zhang, M.W.; Daigle, J.G.; Zhang, S.; Borom, A.R.; Roberts, L.R.; et al. The Glycolytic Enzyme, GPI, Is a Functionally Conserved Modifier of Dopaminergic Neurodegeneration in Parkinson’s Models. Cell Metab. 2014, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Harrington, A.J.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol. Dis. 2010, 37, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Yacoubian, T.A.; Slone, S.R.; Caldwell, K.A.; Caldwell, G.A. Functional Analysis of VPS41-Mediated Neuroprotection in Caenorhabditis elegans and Mammalian Models of Parkinson’s Disease. J. Neurosci. 2012, 32, 2142–2153. [Google Scholar] [CrossRef] [PubMed]
- Griffin, E.F.; Yan, X.; Caldwell, K.A.; Caldwell, G.A. Distinct functional roles of Vps41-mediated neuroprotection in Alzheimer’s and Parkinson’s disease models of neurodegeneration. Hum. Mol. Genet. 2018, 27, 4176–4193. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 1644. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Calatayud, C.; Guha, S.; Fernández-Carasa, I.; Berkowitz, L.; Carballo-Carbajal, I.; Ezquerra, M.; Fernández-Santiago, R.; Kapahi, P.; Raya, Á.; Miranda-Vizuete, A.; et al. The Small GTPase RAC1/CED-10 Is Essential in Maintaining Dopaminergic Neuron Function and Survival Against α-Synuclein-Induced Toxicity. Mol. Neurobiol. 2018, 55, 7533–7552. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Dimitri, K. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1. [Google Scholar] [CrossRef]
- Yacoubian, T.A.; Slone, S.R.; Harrington, A.J.; Hamamichi, S.; Schieltz, J.M.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Cadlwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2008, 41, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Su, J.L.; Auluck, P.K.; Outeiro, T.F.; Yeger-Lotem, E.; Kritzer, J.A.; Tardiff, D.F.; Strathearn, K.E.; Liu, F.; Coa, S.; Hamamichi, S.; et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis. Model. Mech. 2010, 3, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Hewett, J.; Oxelius, L.J.; Ramesh, V.; McLean, P.J.; Breakefield, X.O.; Hyman, B.T. A Close Association of TorsinA and α-Synuclein in Lewy Bodies. Am. J. Pathol. 2001, 159, 339–344. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Angot, E.; Steiner, J.A.; Tomé, C.M.L.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-Synuclein Cell-to-Cell Transfer and Seeding in Grafted Dopaminergic Neurons In Vivo. PLoS ONE 2012, 7, e39465. [Google Scholar] [CrossRef]
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifi, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef]
- Seugnet, L.; Galvin, J.E.; Suzuki, Y.; Gottschalk, L.; Shaw, P.J. Persistent Short-Term Memory Defects Following Sleep Deprivation in a Drosophila Model of Parkinson Disease. Sleep 2009, 32, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.; Burkhart, K.; Gu, S.G.; Spracklin, G.; Kershner, A.; Fritz, H.; Kimble, J.; Fire, A.; Kennedy, S. A nuclear Argonaute promotes multi-generational epigenetic inheritance and germline immortality. Nature 2012, 489, 447–451. [Google Scholar] [CrossRef]
- Gu, S.G.; Pak, J.; Guang, S.; Maniar, J.M.; Kennedy, S.; Fire, A. A transgenerational impact of siRNA on chromatin: siRNA amplification in Caenorhabditis elegans generates a homology- targeted footprint of H3K9 methylated nucleosomes. Nat. Genet. 2013, 44, 157–164. [Google Scholar] [CrossRef]
- Devanapally, S.; Ravikumar, S.; Jose, A.M. Double-stranded RNA made in C. elegans neurons can enter the germline and cause transgenerational gene silencing. Proc. Natl. Acad. Sci. USA 2015, 112, 2133–2138. [Google Scholar] [CrossRef]
- Forthun, R.B.; SenGupta, T.; Skjeldam, H.K.; Lindvall, J.M.; McCormack, E.; Gjertsen, B.T.; Nilsen, H. Cross-Species Functional Genomic Analysis Identifies Resistance Genes of the Histone Deacetylase Inhibitor Valproic Acid. PLoS ONE 2012, 7, e48992. [Google Scholar] [CrossRef] [PubMed]
- Kautu, B.B.; Carrasquilla, A.; Hicks, M.L.; Caldwell, K.A.; Caldwell, G.A. Valproic acid ameliorates C. elegans dopaminergic neurodegeneration with implications for ERK-MAPK signaling. Neurosci. Lett. 2013, 541, 116–119. [Google Scholar] [CrossRef]
- Caldwell, K.A.; Thies, J.L.; Caldwell, G.A. No Country for Old Worms: A systematic review of the application of C. elegans to investigate a bacterial source of environmental neurotoxicity in Parkinson’s disease. Metabolites 2018, 8, 70. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description of Transgenic C. elegans α-syn Model(s) | Advantages | Disadvantages | Genotype of Model Strain |
|---|---|---|---|
| Non-progressive neurodegeneration, α-syn expressed both pan-neuronally and exclusively in DA neurons [22] | Exhibits dopaminergic neurodegeneration and deficits in locomotion | Dopaminergic neurodegeneration and deficits in locomotion do not worsen with age; Limited reports of experimental use in the literature | Wild-Type α-syn or A53T α-syn mutation
|
| Limited neurodegeneration, Wild-Type and rare mutant forms of α-syn expressed either pan-neuronally or exclusively in DA neurons from different promoters [23,24] |
| Dopaminergic neurodegeneration limited to neurites, not cell bodies and mainly observed with rare mutant forms of α-syn | Wild-type, A53T and A30P mutant forms of α-syn
|
| α-syn misfolding in body-wall muscle cells [25] | Exhibits visible inclusions of α-syn that increase with age; expression in large muscle cells facilitates biochemical analysis and fluorescent protein interaction studies | Employs an α-syn fusion protein with YFP and, therefore, may not represent native structural dynamics; Identified modulators of α-syn misfolding in this model have proven inconsistent with Drosophila and mammalian models of PD | NL5901 (pkIs2386 [Punc54::α-syn::YFP (pRP2386)unc-119(+)]) |
| Progressive neurodegeneration with Wild-Type α-syn expressed in DA neurons [26] | Exhibits dopaminergic neurodegeneration that worsens with age; established utility in discerning translational outcomes | α-syn is overexpressed; expression limited to the 8 DA neurons, thereby complicating biochemical analysis and precluding evaluation of non-cell autonomous effects of α-syn on DA neurodegeneration | UA44 (baIn11 [Pdat-1:: α-syn, Pdat-1::GFP]) |
| α-syn in body wall muscle cells in a background where misfolding is attenuated by chaperone co-expression [27] |
| α-syn is overexpressed as a fusion protein with GFP and thereby may not fully represent native structural dynamics; Presence of chaperone (TOR-2) in the strain background may complicate interpretation of α-syn-independent effects | Without TOR-2 co-expression: UA49 (baIn2 [Punc-54::α-syn::GFP, rol-6 (su1006)]) With TOR-2 co-expression: UA50 (baInl13 [Punc-54::α-syn::GFP, Punc-54::tor-2], rol-6 (su1006)]) |
| Single-copy transgene of α-syn expressed ubiquitously [28] |
| Does not exhibit dopaminergic degeneration or neuronal deficits on its own | JVR339 (jerIs004 [Peft-3::α-syn: TagRFP: let-858_unc-119(+)]; unc-119(−); vtIs7 [Pdat-1::GFP(pRB490)]) |
| “prion-like” α-syn spreading model [29] | Suitable model to investigate potential to lessen or worsen neuron-to-neuron α-syn transfer |
| JVR406 [(Pddr-2::BiFC1 (EGFH1-LINK-SYN); Ptph-1::BIFC2 (SYN-EGFH2); rol-6(su1006)] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaeta, A.L.; Caldwell, K.A.; Caldwell, G.A. Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sci. 2019, 9, 73. https://doi.org/10.3390/brainsci9040073
Gaeta AL, Caldwell KA, Caldwell GA. Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sciences. 2019; 9(4):73. https://doi.org/10.3390/brainsci9040073
Chicago/Turabian StyleGaeta, Anthony L., Kim A. Caldwell, and Guy A. Caldwell. 2019. "Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease" Brain Sciences 9, no. 4: 73. https://doi.org/10.3390/brainsci9040073
APA StyleGaeta, A. L., Caldwell, K. A., & Caldwell, G. A. (2019). Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sciences, 9(4), 73. https://doi.org/10.3390/brainsci9040073