Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Instability in Humans and Mice May Share a Common Molecular Basis
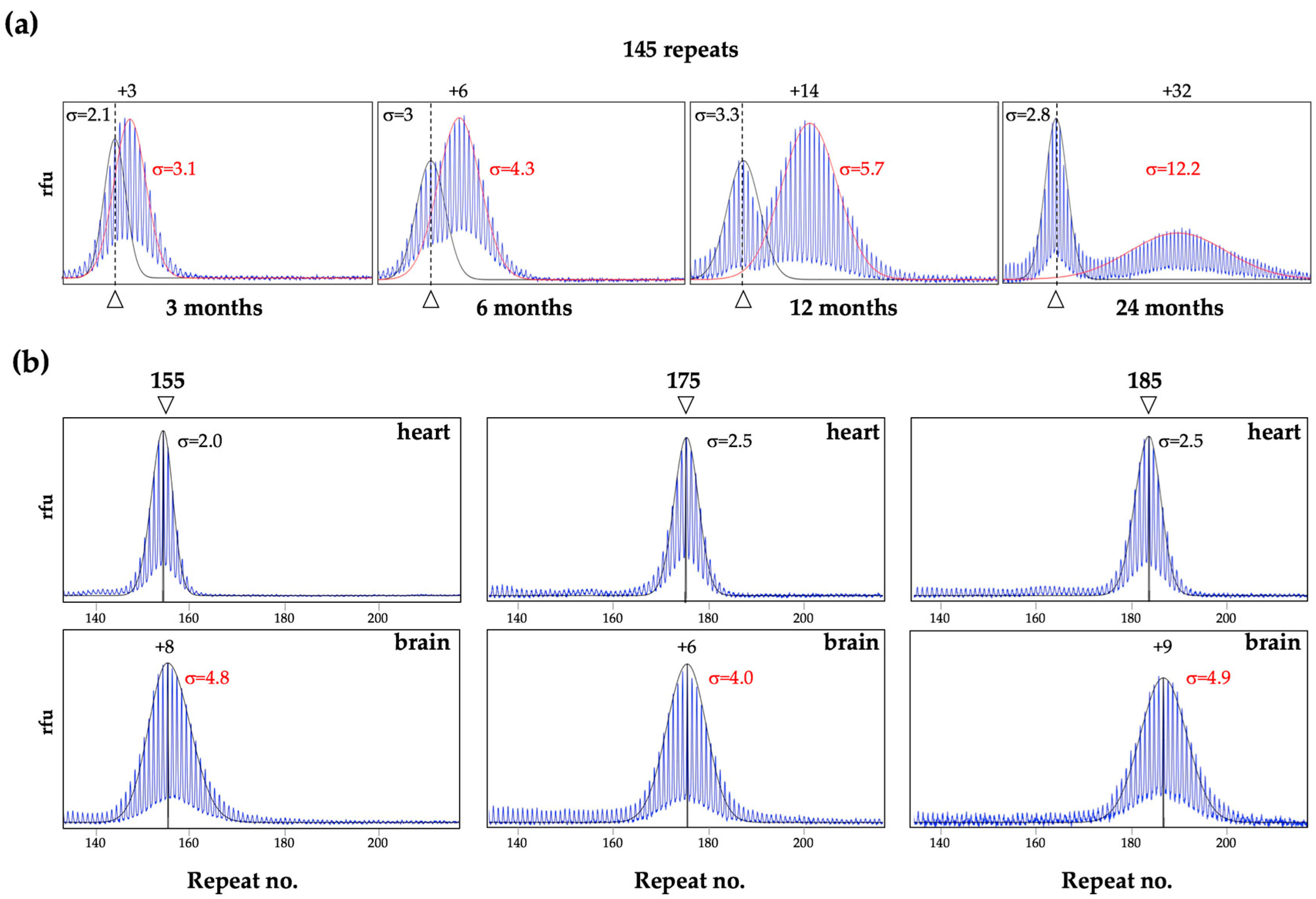
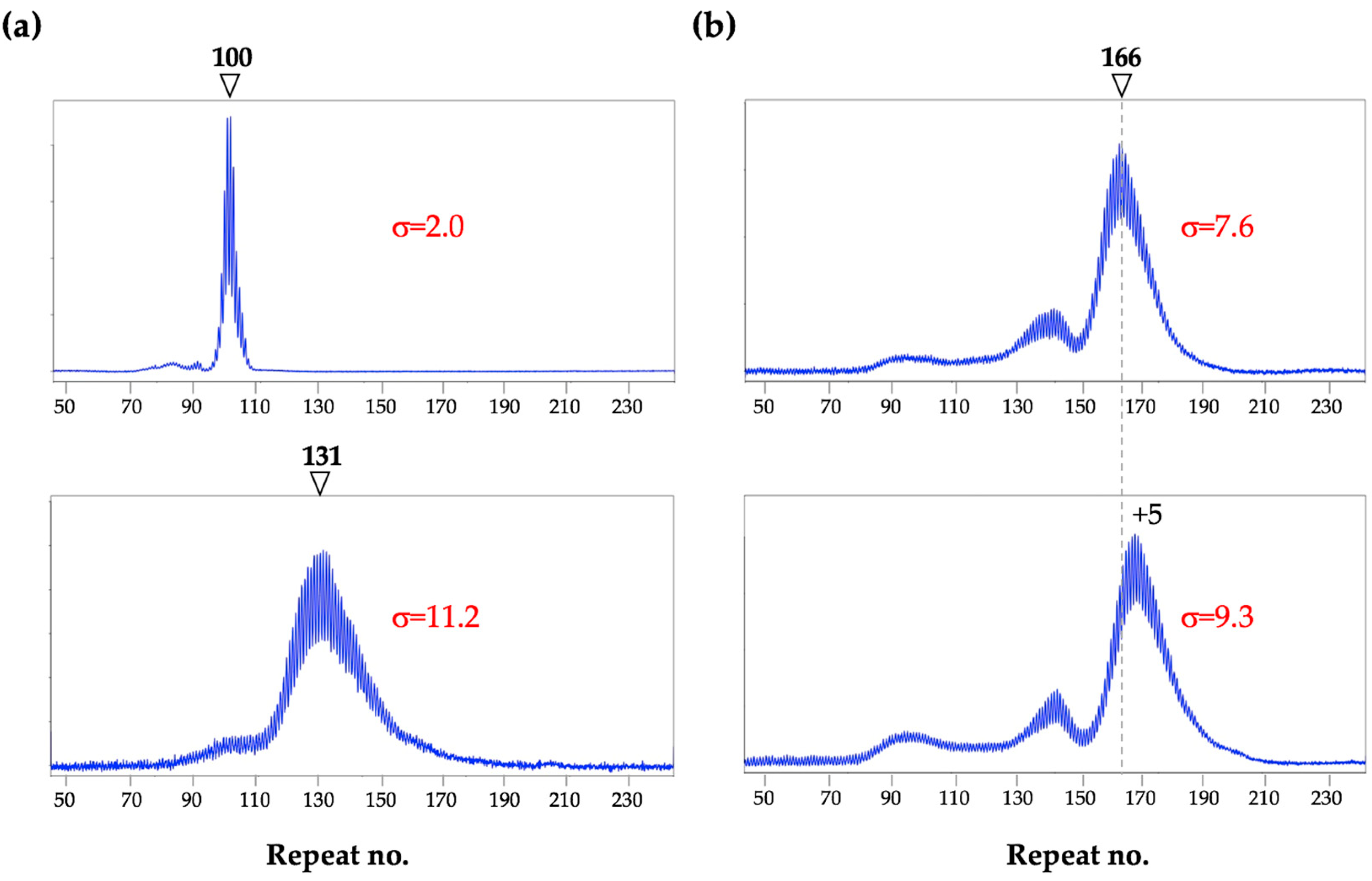
3. Different Cell Types Show Different Propensities to Expand in Mice
4. Expansions in Females Only Occurs on The Active X Chromosome
5. Expansion in the Male and Female Germline
6. Genetic and Environmental Factors Affecting Instability
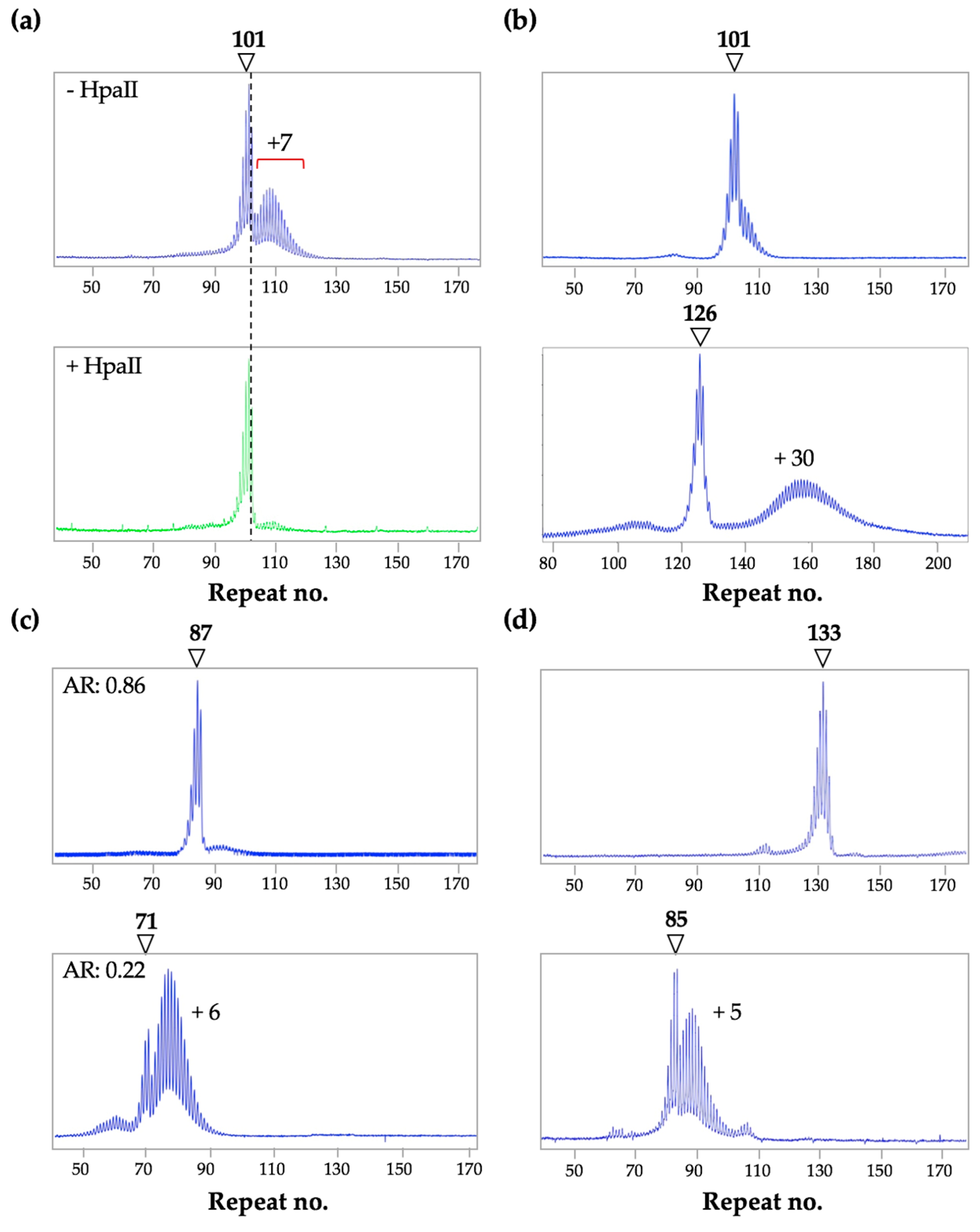
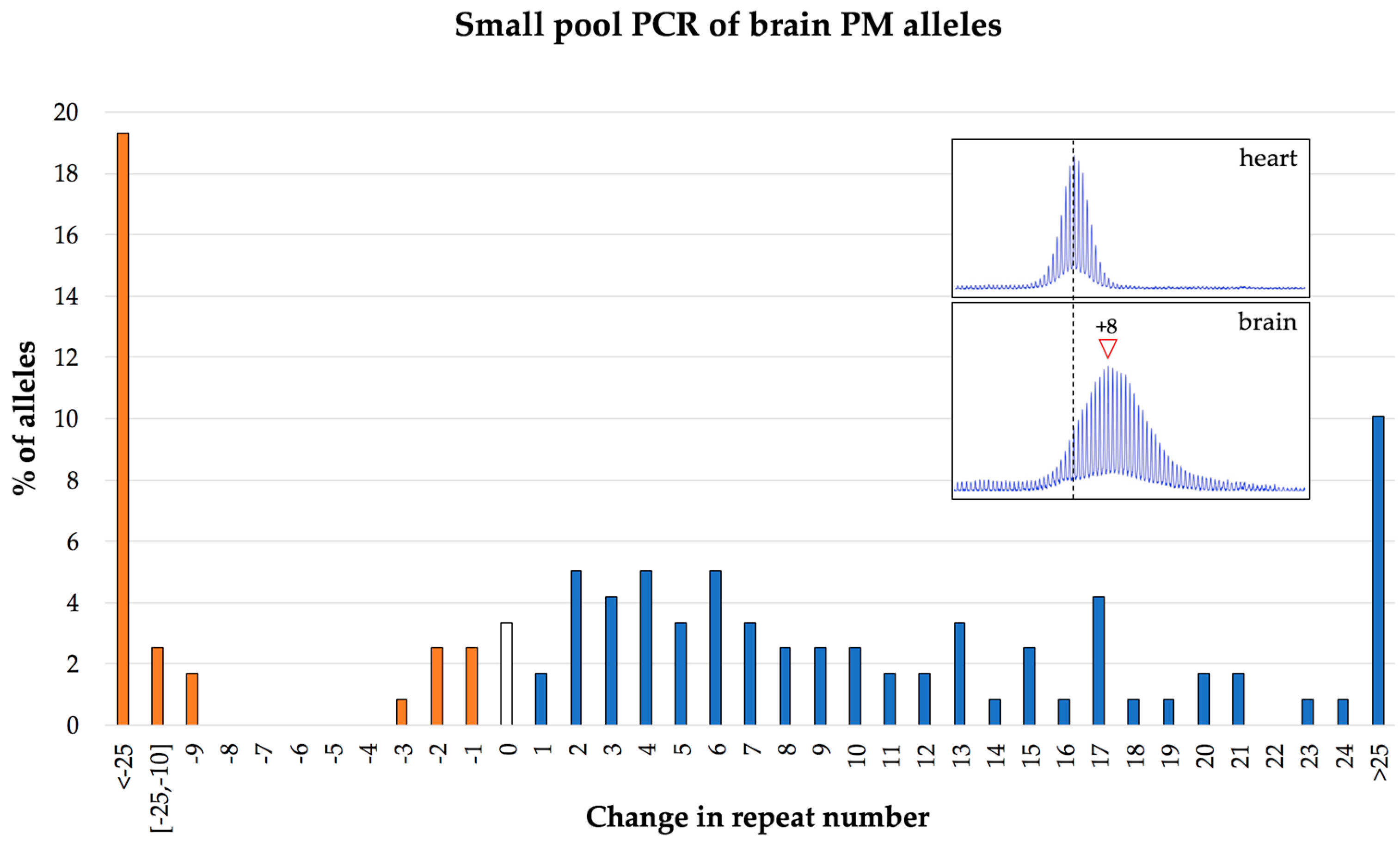
7. The Frequency of Large Contractions and Expansions can be Underestimated
8. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lozano, R.; Rosero, C.A.; Hagerman, R.J. Fragile X spectrum disorders. Intractable Rare Dis. Res. 2014, 3, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Houck, G.E., Jr.; Brown, W.T.; Dobkin, C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Basuta, K.; Schneider, A.; Gane, L.; Polussa, J.; Woodruff, B.; Pretto, D.; Hagerman, R.; Tassone, F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A 2015, 167, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.T.; Aliaga, S.M.; Arpone, M.; Francis, D.; Li, X.; Chong, B.; Slater, H.R.; Rogers, C.; Bretherton, L.; Hunter, M.; et al. Partially methylated alleles, microdeletion, and tissue mosaicism in a fragile X male with tremor and ataxia at 30 years of age: A case report. Am. J. Med. Genet. A 2016, 170, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Wohrle, D.; Salat, U.; Hameister, H.; Vogel, W.; Steinbach, P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am. J. Hum. Genet. 2001, 69, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Zaninovic, N.; Zhan, Q.; Madireddy, A.; Nolin, S.L.; Ersalesi, N.; Yan, Z.; Rosenwaks, Z.; Schildkraut, C.L. Cis-acting DNA sequence at a replication origin promotes repeat expansion to fragile X full mutation. J. Cell Biol. 2014, 206, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Brykczynska, U.; Pecho-Vrieseling, E.; Thiemeyer, A.; Klein, J.; Fruh, I.; Doll, T.; Manneville, C.; Fuchs, S.; Iazeolla, M.; Beibel, M.; et al. CGG repeat-Induced FMR1 silencing depends on the expansion size in human iPSCs and neurons carrying unmethylated full mutations. Stem Cell Rep. 2016, 7, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, C.J.; de Graaff, E.; Nieuwenhuizen, I.M.; Willemsen, R.; Oostra, B.A. FMR1 premutation allele (CGG)81 is stable in mice. Eur. J. Hum. Genet. 1997, 5, 293–298. [Google Scholar] [PubMed]
- Lavedan, C.; Grabczyk, E.; Usdin, K.; Nussbaum, R.L. Long uninterrupted CGG repeats within the first exon of the human FMR1 gene are not intrinsically unstable in transgenic mice. Genomics 1998, 50, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Hoogeveen-Westerveld, M.; Reis, S.; Holstege, J.; Severijnen, L.A.; Nieuwenhuizen, I.M.; Schrier, M.; van Unen, L.; Tassone, F.; Hoogeveen, A.T.; et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum. Mol. Genet. 2003, 12, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Glaser, D.; Wohrle, D.; Salat, U.; Vogel, W.; Steinbach, P. Mitotic behavior of expanded CGG repeats studied on cultured cells: further evidence for methylation-mediated triplet repeat stability in fragile X syndrome. Am. J. Med. Genet. 1999, 84, 226–228. [Google Scholar] [CrossRef]
- Lokanga, R.; Zhao, X.N.; Entezam, A.; Usdin, K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum. Mol. Genet. 2014, 23, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. Timing of expansion of fragile X premutation alleles during intergenerational transmission in a mouse model of the fragile X-related disorders. Front. Genet. 2018, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Satirapod, C.; Ohguchi, Y.; Park, E.S.; Woods, D.C.; Tilly, J.L. Genetic studies in mice directly link oocytes produced during adulthood to ovarian function and natural fertility. Sci. Rep. 2017, 7, 10011. [Google Scholar] [CrossRef] [PubMed]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M.; et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Hensman Moss, D.J.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD Investigators; REGISTRY Investigators; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. The Repeat Expansion Diseases: The dark side of DNA repair. DNA Repair (Amst.) 2015, 32, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. Ups and downs: mechanisms of repeat Instability in the fragile X-related disorders. Genes 2016, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Kumari, D.; Gupta, S.; Wu, D.; Evanitsky, M.; Yang, W.; Usdin, K. Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 2015, 24, 7087–7096. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Lokanga, R.; Allette, K.; Gazy, I.; Wu, D.; Usdin, K. A MutSbeta-dependent contribution of MutSalpha to repeat expansions in fragile X premutation mice? PLoS Genet. 2016, 12, e1006190. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Entezam, A.; Kumari, D.; Yudkin, D.; Qin, M.; Smith, C.B.; Usdin, K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013, 34, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Gazy, I.; Hayward, B.; Potapova, S.; Zhao, X.; Usdin, K. Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair (Amst.) 2019, 74, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Senejani, A.G.; Sweasy, J.B.; Usdin, K. Heterozygosity for a hypomorphic Polbeta mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genet. 2015, 11, e1005181. [Google Scholar] [CrossRef] [PubMed]
- Mollersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Wilkins, K.; Edelmann, W.; Usdin, K. MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 2018, 14, e1007719. [Google Scholar] [CrossRef] [PubMed]
- Hayward, B.E.; Zhou, Y.; Kumari, D.; Usdin, K. A set of assays for the comprehensive analysis of FMR1 alleles in the fragile X-related disorders. J. Mol. Diagn. 2016, 18, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 1996, 64, 296–301. [Google Scholar] [CrossRef]
- Maddalena, A.; Yadvish, K.N.; Spence, W.C.; Howard-Peebles, P.N. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am. J. Med. Genet. 1996, 64, 309–312. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- MacKenzie, J.J.; Sumargo, I.; Taylor, S.A. A cryptic full mutation in a male with a classical fragile X phenotype. Clin. Genet. 2006, 70, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014, 51, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with fragile X syndrome. Expert Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Gennaro, E.; Pirozzi, F.; Baldo, C.; Forzano, F.; Turolla, L.; Faravelli, F.; Gastaldo, D.; Coviello, D.; Grasso, M.; et al. FXS-like phenotype in two unrelated patients carrying a methylated premutation of the FMR1 gene. Front. Genet. 2018, 9, 442. [Google Scholar] [CrossRef] [PubMed]
- Mailick, M.R.; Movaghar, A.; Hong, J.; Greenberg, J.S.; DaWalt, L.S.; Zhou, L.; Jackson, J.; Rathouz, P.J.; Baker, M.W.; Brilliant, M.; et al. Health profiles of mosaic versus non-mosaic FMR1 premutation carrier mothers of children with fragile X syndrome. Front. Genet. 2018, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Houck, G.E., Jr.; Gargano, A.D.; Blumstein, H.; Dobkin, C.S.; Brown, W.T. FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am. J. Hum. Genet. 1999, 65, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Guitart, M.; Rodriguez-Revenga, L.; Madrigal, I.; Gabau, E.; Mila, M. Paternal transmission of a FMR1 full mutation allele. Am. J. Med. Genet. A 2017, 173, 2795–2797. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Quizoz, M.E.; Jesus, S.; Ramos, I.; Garcia, A.E.; Martinez, R.; Mir, P.; Pintado, E. Tissue-specific size and methylation analysis in two fragile X families: Contribution to the clinical phenotype. J. Mol. Genet. Med. 2016, 10. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 2014, 35, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.K.; Crawford, D.C.; Scott, E.H.; Leslie, M.L.; Sherman, S.L. Paternally transmitted FMR1 alleles are less stable than maternally transmitted alleles in the common and intermediate size range. Am. J. Hum. Genet. 2002, 70, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Loeb, L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. USA 1994, 91, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Mitas, M.; Yu, A.; Dill, J.; Haworth, I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 1995, 34, 12803–12811. [Google Scholar] [CrossRef] [PubMed]
- Nadel, Y.; Weisman-Shomer, P.; Fry, M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 1995, 270, 28970–28977. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, D.; Hayward, B.E.; Aladjem, M.I.; Kumari, D.; Usdin, K. Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 2014, 23, 2940–2952. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 2016, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.B.; Lee, W.R. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen. 1995, 25 (Suppl. 26), 48–64. [Google Scholar] [CrossRef]
- Crow, J.F. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 2000, 1, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Malter, H.E.; Iber, J.C.; Willemsen, R.; de Graaff, E.; Tarleton, J.C.; Leisti, J.; Warren, S.T.; Oostra, B.A. Characterization of the full fragile X syndrome mutation in fetal gametes. Nat. Genet. 1997, 15, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A.J.; Jorens, H.Z.; Darby, J.K.; Oostra, B.; et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 1993, 4, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Robb, L.J.; Rouillard, P.; Der Kaloustian, V.M. No mental retardation in a man with 40% abnormal methylation at the FMR-1 locus and transmission of sperm cell mutations as premutations. Hum. Mol. Genet. 1994, 3, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Lokanga, A.R.; Le, W.; Hoffman, G.; Usdin, K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2010, 31, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair. (Amst.) 2018, 69, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mariappan, S.V.; Catasti, P.; Ratliff, R.; Moyzis, R.K.; Laayoun, A.; Smith, S.S.; Bradbury, E.M.; Gupta, G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: structure and biological implications. Proc. Natl. Acad. Sci. USA 1995, 92, 5199–5203. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, S.V.; Catasti, P.; Chen, X.; Ratliff, R.; Moyzis, R.K.; Bradbury, E.M.; Gupta, G. Solution structures of the individual single strands of the fragile X DNA triplets (GCC)n.(GGC)n. Nucleic Acids Res. 1996, 24, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. The transcription-coupled repair protein ERCC6/CSB also protects against repeat expansion in a mouse model of the fragile X premutation. Hum. Mutat. 2015, 36, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; Degtyareva, N.P.; Koloteva, N.N.; Sugino, A.; Masumoto, H.; Gordenin, D.A.; Resnick, M.A. Replication slippage between distant short repeats in Saccharomyces cerevisiae depends on the direction of replication and the RAD50 and RAD52 genes. Mol. Cell. Biol. 1995, 15, 5607–5617. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.C.; White, P.J. Cloned human FMR1 trinucleotide repeats exhibit a length- and orientation-dependent instability suggestive of in vivo lagging strand secondary structure. Nucleic. Acids Res. 1998, 26, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Bichara, M.; Wagner, J.; Lambert, I.B. Mechanisms of tandem repeat instability in bacteria. Mutat. Res. 2006, 598, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J. DNA inverted repeats and human disease. Front. Biosci. 1998, 3, 408–418. [Google Scholar] [CrossRef]
- Lovett, S.T. Encoded errors: Mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol. Microbiol. 2004, 52, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Cho, J.E.; Li, Y.C.; Jinks-Robertson, S. RNA/DNA hybrids initiate quasi-palindrome-associated mutations in highly transcribed yeast DNA. PLoS Genet 2013, 9, e1003924. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Sah, S.; Glicksman, A.; Sherman, S.L.; Allen, E.; Berry-Kravis, E.; Tassone, F.; Yrigollen, C.; Cronister, A.; Jodah, M.; et al. Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am. J. Med. Genet. A 2013, 161A, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Pearson, C.E.; Coutinho, P.; Provost, S.; Amorim, A.; Dube, M.P.; Sequeiros, J.; Rouleau, G.A. Modifiers of (CAG)(n) instability in Machado-Joseph disease (MJD/SCA3) transmissions: an association study with DNA replication, repair and recombination genes. Hum. Genet. 2014, 133, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Vasquez, M.; Santamaria, C.; Cuenca, P.; Corrales, E.; Monckton, D.G. A polymorphism in the MSH3 mismatch repair gene is associated with the levels of somatic instability of the expanded CTG repeat in the blood DNA of myotonic dystrophy type 1 patients. DNA Repair (Amst.) 2016, 40, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Usdin, K. ATR protects the genome against CGG.CCG-repeat expansion in fragile X premutation mice. Nucleic Acids Res. 2008, 36, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Usdin, K. ATM and ATR protect the genome against two different types of tandem repeat instability in fragile X premutation mice. Nucleic Acids Res. 2009, 37, 6371–6377. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; Lebel, L.A.; Vrbanac, V.; Teed, A.; te Riele, H.; MacDonald, M.E. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum. Mol. Genet. 2003, 12, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Couto, J.M.; Higham, C.F.; Hogg, G.; Cuenca, P.; Braida, C.; Wilson, R.H.; Adam, B.; del Valle, G.; Brian, R.; et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 2012, 21, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Pena, S.; McMurray, C.T. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. 2015, 11, e1005267. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Gazy, I.; Hayward, B.; Pintado, E.; Hwang, Y.H.; Tassone, F.; Usdin, K. Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model. Brain Sci. 2019, 9, 52. https://doi.org/10.3390/brainsci9030052
Zhao X, Gazy I, Hayward B, Pintado E, Hwang YH, Tassone F, Usdin K. Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model. Brain Sciences. 2019; 9(3):52. https://doi.org/10.3390/brainsci9030052
Chicago/Turabian StyleZhao, Xiaonan, Inbal Gazy, Bruce Hayward, Elizabeth Pintado, Ye Hyun Hwang, Flora Tassone, and Karen Usdin. 2019. "Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model" Brain Sciences 9, no. 3: 52. https://doi.org/10.3390/brainsci9030052
APA StyleZhao, X., Gazy, I., Hayward, B., Pintado, E., Hwang, Y. H., Tassone, F., & Usdin, K. (2019). Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model. Brain Sciences, 9(3), 52. https://doi.org/10.3390/brainsci9030052