Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome


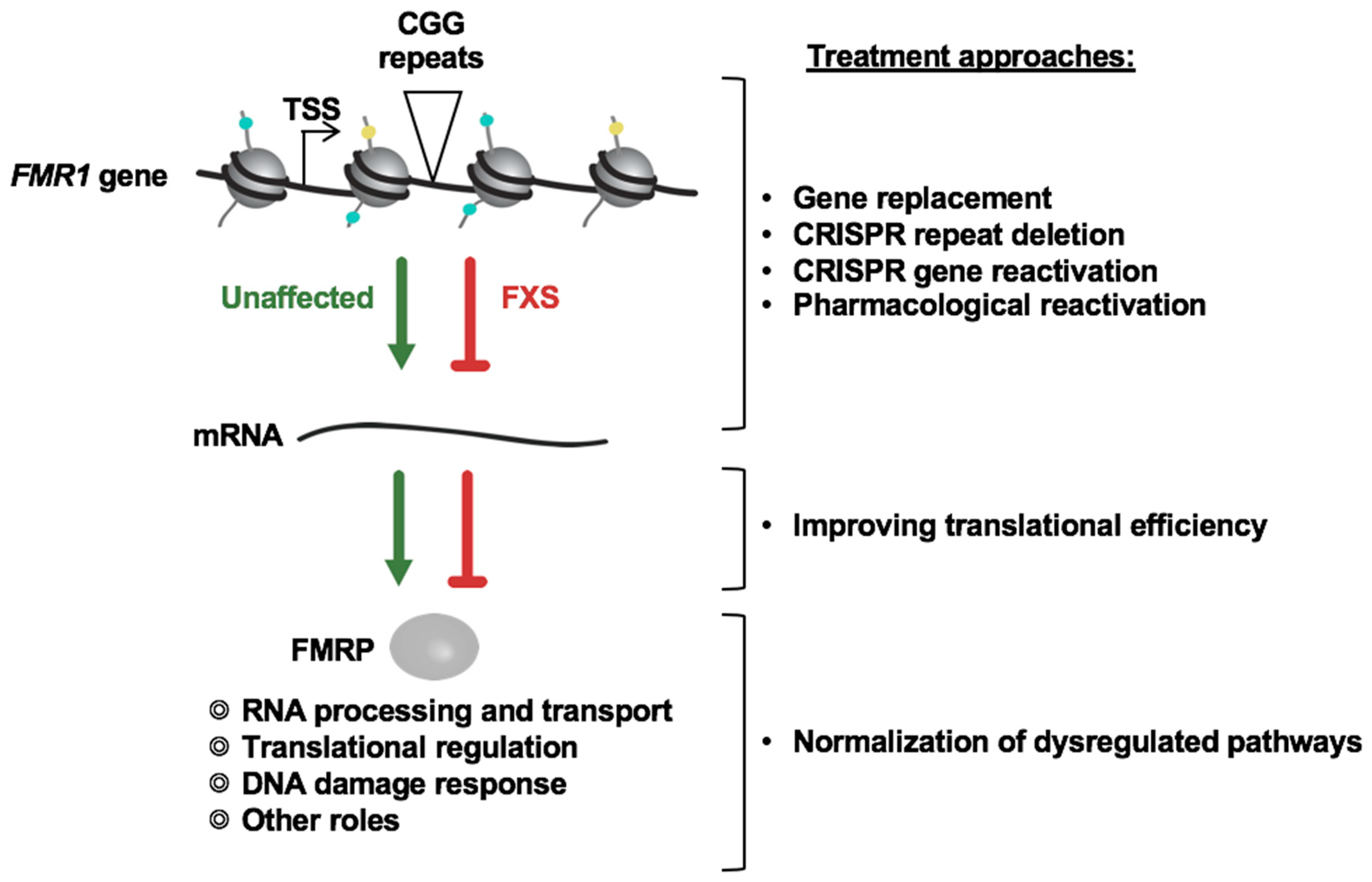{kind=link}
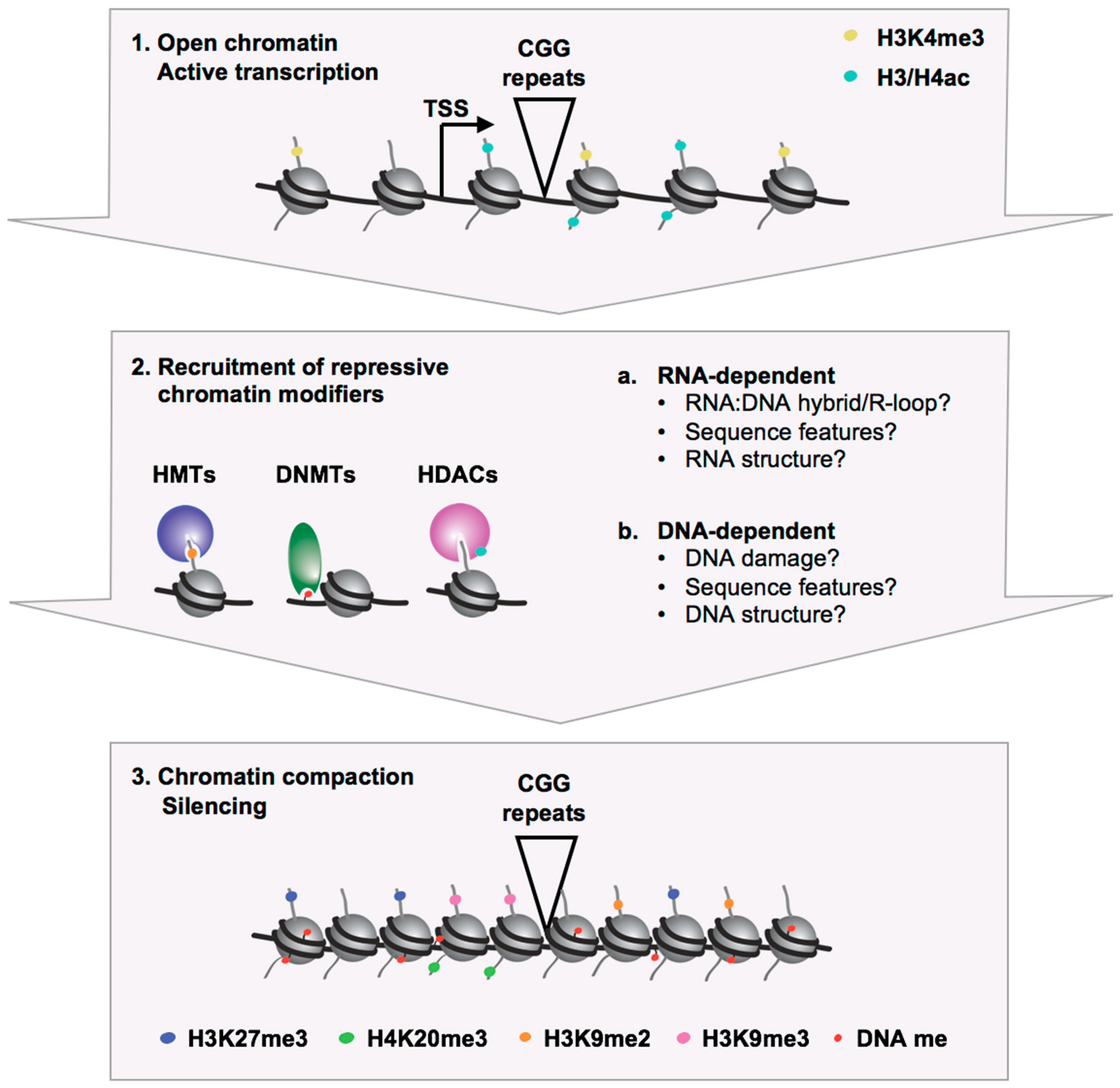{kind=link}
Abstract
1. Introduction
2. Targeting Specific Pathways and Proteins Involved in FMR1 Gene Silencing in FXS
2.1. Epigenetic Marks Associated with the Silenced FMR1 Gene in FXS
2.2. Models for FMR1 Gene Silencing
2.3. Targeting the Activity of Repressive Chromatin Modifiers for Gene Reactivation
2.4. Targeting the Recruitment of Chromatin Modifiers for Gene Reactivation
3. Unbiased High-Throughput Screens to Identify Compounds that Reverse FMR1 Gene Silencing
3.1. HTS Based on Measuring Endogenous FMRP Levels
3.2. HTS Using Knock-In Reporter Cell Lines
4. Limitations and Challenges of Using FMR1 Gene Reactivation as a Treatment Approach for FXS
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iong, K.P.; Tong, T.H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012, 4, 100. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef]
- Monaghan, K.G.; Lyon, E.; Spector, E.B.; erican College of Medical Genetics and Genomics. ACMG Standards and Guidelines for fragile X testing: A revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet. Med. 2013, 15, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- De Boulle, K.; Verkerk, A.J.; Reyniers, E.; Vits, L.; Hendrickx, J.; Van Roy, B.; Van den Bos, F.; de Graaff, E.; Oostra, B.A.; Willems, P.J. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 1993, 3, 31–35. [Google Scholar] [CrossRef]
- Lugenbeel, K.A.; Peier, A.M.; Carson, N.L.; Chudley, A.E.; Nelson, D.L. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat. Genet. 1995, 10, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.C.; Bray, S.M.; Suhl, J.A.; Cutler, D.J.; Coffee, B.; Zwick, M.E.; Warren, S.T. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am. J. Med. Genet. A 2010, 152A, 2512–2520. [Google Scholar] [CrossRef]
- Suhl, J.A.; Warren, S.T. Single-Nucleotide Mutations in FMR1 Reveal Novel Functions and Regulatory Mechanisms of the Fragile X Syndrome Protein FMRP. J. Exp. Neurosci. 2015, 9, 35–41. [Google Scholar] [CrossRef] [PubMed]
- McConkie-Rosell, A.; Lachiewicz, A.M.; Spiridigliozzi, G.A.; Tarleton, J.; Schoenwald, S.; Phelan, M.C.; Goonewardena, P.; Ding, X.; Brown, W.T. Evidence that methylation of the FMR-I locus is responsible for variable phenotypic expression of the fragile X syndrome. Am. J. Hum. Genet. 1993, 53, 800–809. [Google Scholar] [PubMed]
- Merenstein, S.A.; Shyu, V.; Sobesky, W.E.; Staley, L.; Berry-Kravis, E.; Nelson, D.L.; Lugenbeel, K.A.; Taylor, A.K.; Pennington, B.F.; Hagerman, R.J. Fragile X syndrome in a normal IQ male with learning and emotional problems. J. Am. Acad. Child Adolesc. Psychiatry 1994, 33, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Robb, L.J.; Rouillard, P.; Der Kaloustian, V.M. No mental retardation in a man with 40% abnormal methylation at the FMR-1 locus and transmission of sperm cell mutations as premutations. Hum. Mol. Genet. 1994, 3, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Smeets, H.J.; Smits, A.P.; Verheij, C.E.; Theelen, J.P.; Willemsen, R.; van de Burgt, I.; Hoogeveen, A.T.; Oosterwijk, J.C.; Oostra, B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995, 4, 2103–2108. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Spiridigliozzi, G.A.; McConkie-Rosell, A.; Burgess, D.; Feng, Y.; Warren, S.T.; Tarleton, J. A fragile X male with a broad smear on Southern blot analysis representing 100–500 CGG repeats and no methylation at the EagI site of the FMR-1 gene. Am. J. Med. Genet. 1996, 64, 278–282. [Google Scholar] [CrossRef]
- Wang, Z.; Taylor, A.K.; Bridge, J.A. FMR1 fully expanded mutation with minimal methylation in a high functioning fragile X male. J. Med. Genet. 1996, 33, 376–378. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Ikle, D.N.; Dyer, P.N.; Lampe, M.; Willemsen, R.; Oostra, B.A.; Taylor, A.K. FMRP expression as a potential prognostic indicator in fragile X syndrome. Am. J. Med. Genet. 1999, 84, 250–261. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Hatton, D.D.; Sideris, J.; Skinner, M.; Mankowski, J.; Bailey, D.B., Jr.; Roberts, J.; Mirrett, P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. A 2006, 140A, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, D.E.; Malter, H.E.; Feng, Y.; Warren, S.T. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996, 5, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
- Kao, D.I.; Aldridge, G.M.; Weiler, I.J.; Greenough, W.T. Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc. Natl. Acad. Sci. USA 2010, 107, 15601–15606. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B. Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 2017, 170, 1209.e20–1223.e20. [Google Scholar] [CrossRef]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef]
- De Rubeis, S.; Bagni, C. Fragile X mental retardation protein control of neuronal mRNA metabolism: Insights into mRNA stability. Mol. Cell Neurosci. 2010, 43, 43–50. [Google Scholar] [CrossRef]
- Jeon, S.J.; Seo, J.E.; Yang, S.I.; Choi, J.W.; Wells, D.; Shin, C.Y.; Ko, K.H. Cellular stress-induced up-regulation of FMRP promotes cell survival by modulating PI3K-Akt phosphorylation cascades. J. Biomed. Sci. 2011, 18, 17. [Google Scholar] [CrossRef]
- Luca, R.; Averna, M.; Zalfa, F.; Vecchi, M.; Bianchi, F.; La Fata, G.; Del Nonno, F.; Nardacci, R.; Bianchi, M.; Nuciforo, P.; et al. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol. Med. 2013, 5, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stutzer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cheng, Y.; Li, Y.; Chen, Z.; Jin, P.; Chen, D. A feed-forward mechanism involving Drosophila fragile X mental retardation protein triggers a replication stress-induced DNA damage response. Hum. Mol. Genet. 2014, 23, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Ye, S.H.; Yang, H.X.; Zhou, Y.T.; Zhao, Q.H.; Sun, W.W.; Gao, M.M.; Yi, Y.H.; Long, Y.S. A novel role of fragile X mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience 2017, 349, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Shamay-Ramot, A.; Khermesh, K.; Porath, H.T.; Barak, M.; Pinto, Y.; Wachtel, C.; Zilberberg, A.; Lerer-Goldshtein, T.; Efroni, S.; Levanon, E.Y.; et al. Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish. PLoS Genet. 2015, 11, e1005702. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the Fragile X Mental Retardation Protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Lindemann, L.; Jonch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Des Portes, V.; Hossain, F.; et al. Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 2018, 17, 280–299. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PLoS ONE 2016, 11, e0165499. [Google Scholar] [CrossRef]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Haenfler, J.M.; Skariah, G.; Rodriguez, C.M.; Monteiro da Rocha, A.; Parent, J.M.; Smith, G.D.; Todd, P.K. Targeted Reactivation of FMR1 Transcription in Fragile X Syndrome Embryonic Stem Cells. Front. Mol. Neurosci. 2018, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Zeier, Z.; Kumar, A.; Bodhinathan, K.; Feller, J.A.; Foster, T.C.; Bloom, D.C. Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther. 2009, 16, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, S.; Arsenault, J.; Xuan, I.C.; Pacey, L.K.; Hampson, D.R. Reduced phenotypic severity following adeno-associated virus-mediated Fmr1 gene delivery in fragile X mice. Neuropsychopharmacology 2014, 39, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Willemsen, R.; Oostra, B.A.; Neri, G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 1998, 7, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Biacsi, R.; Kumari, D.; Usdin, K. SIRT1 inhibition alleviates gene silencing in Fragile X mental retardation syndrome. PLoS Genet. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016, 25, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. The myelodysplastic syndrome as a prototypical epigenetic disease. Blood 2013, 121, 3811–3817. [Google Scholar] [CrossRef] [PubMed]
- Sripathy, S.; Leko, V.; Adrianse, R.L.; Loe, T.; Foss, E.J.; Dalrymple, E.; Lao, U.; Gatbonton-Schwager, T.; Carter, K.T.; Payer, B.; et al. Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-beta superfamily as a regulator of XIST expression. Proc. Natl. Acad. Sci. USA 2017, 114, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Carrette, L.L.G.; Wang, C.Y.; Wei, C.; Press, W.; Ma, W.; Kelleher, R.J., 3rd; Lee, J.T. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. USA 2018, 115, E668–E675. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Allen, J.A.; Mabb, A.M.; King, I.F.; Miriyala, J.; Taylor-Blake, B.; Sciaky, N.; Dutton, J.W., Jr.; Lee, H.M.; Chen, X.; et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef] [PubMed]
- She, A.; Kurtser, I.; Reis, S.A.; Hennig, K.; Lai, J.; Lang, A.; Zhao, W.N.; Mazitschek, R.; Dickerson, B.C.; Herz, J.; et al. Selectivity and Kinetic Requirements of HDAC Inhibitors as Progranulin Enhancers for Treating Frontotemporal Dementia. Cell Chem. Biol. 2017, 24, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Soragni, E.; Gottesfeld, J.M. Translating HDAC inhibitors in Friedreich’s ataxia. Expert Opin Orphan Drugs 2016, 4, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Halder-Sinha, S.; Clement, J.P.; Kundu, T.K. Epigenetic modulation by small molecule compounds for neurodegenerative disorders. Pharmacol. Res. 2018, 132, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Lee, Y.M.; Lee, S.C.; Wang, T.R. In vitro DNA methylation inhibits FMR-1 promoter. Biochem. Biophys. Res. Commun. 1993, 193, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Interaction of the transcription factors USF1, USF2, and alpha-Pal/Nrf-1 with the FMR1 promoter. Implications for Fragile X mental retardation syndrome. J. Biol. Chem. 2001, 276, 4357–4364. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Zhang, F.; Ceman, S.; Warren, S.T.; Reines, D. Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 2002, 71, 923–932. [Google Scholar] [CrossRef]
- Tabolacci, E.; Moscato, U.; Zalfa, F.; Bagni, C.; Chiurazzi, P.; Neri, G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008, 16, 1487–1498. [Google Scholar] [CrossRef]
- Kumari, D.; Usdin, K. The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum. Mol. Genet. 2010, 19, 4634–4642. [Google Scholar] [CrossRef]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Hum. Mol. Genet. 2014, 23, 6575–6583. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pray-Grant, M.G.; Grant, P.A. Effector proteins for methylated histones: An expanding family. Cell Cycle 2005, 4, 919–926. [Google Scholar] [CrossRef]
- Smallwood, A.; Esteve, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Appanah, R.; Lee, S.; Lam, L.L.; Goyal, P.; Lorincz, M.C. Targeting of EZH2 to a defined genomic site is sufficient for recruitment of Dnmt3a but not de novo DNA methylation. Epigenetics 2009, 4, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Naumann, A.; Hochstein, N.; Weber, S.; Fanning, E.; Doerfler, W. A distinct DNA-methylation boundary in the 5′-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am. J. Hum. Genet. 2009, 85, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224.e15–238.e15. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef]
- Lanni, S.; Goracci, M.; Borrelli, L.; Mancano, G.; Chiurazzi, P.; Moscato, U.; Ferre, F.; Helmer-Citterich, M.; Tabolacci, E.; Neri, G. Role of CTCF protein in regulating FMR1 locus transcription. PLoS Genet. 2013, 9, e1003601. [Google Scholar] [CrossRef]
- Usdin, K.; Hayward, B.E.; Kumari, D.; Lokanga, R.A.; Sciascia, N.; Zhao, X.N. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet. 2014, 5, 226. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Loeb, L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. USA 1994, 91, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mariappan, S.V.; Catasti, P.; Ratliff, R.; Moyzis, R.K.; Laayoun, A.; Smith, S.S.; Bradbury, E.M.; Gupta, G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: Structure and biological implications. Proc. Natl. Acad. Sci. USA 1995, 92, 5199–5203. [Google Scholar] [CrossRef] [PubMed]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef] [PubMed]
- Mitas, M.; Yu, A.; Dill, J.; Haworth, I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 1995, 34, 12803–12811. [Google Scholar] [CrossRef] [PubMed]
- Nadel, Y.; Weisman-Shomer, P.; Fry, M. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J. Biol. Chem. 1995, 270, 28970–28977. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef]
- Patel, P.K.; Bhavesh, N.S.; Hosur, R.V. Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of Fragile X Syndrome: NMR observations. Biochem. Biophys. Res. Commun. 2000, 278, 833–838. [Google Scholar] [CrossRef]
- Handa, V.; Saha, T.; Usdin, K. The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 2003, 31, 6243–6248. [Google Scholar] [CrossRef]
- Zumwalt, M.; Ludwig, A.; Hagerman, P.J.; Dieckmann, T. Secondary structure and dynamics of the r(CGG) repeat in the mRNA of the fragile X mental retardation 1 (FMR1) gene. RNA Biol. 2007, 4, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Sanz, L.A.; Chedin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Malgowska, M.; Gudanis, D.; Kierzek, R.; Wyszko, E.; Gabelica, V.; Gdaniec, Z. Distinctive structural motifs of RNA G-quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res. 2014, 42, 10196–10207. [Google Scholar] [CrossRef] [PubMed]
- Abu Diab, M.; Mor-Shaked, H.; Cohen, E.; Cohen-Hadad, Y.; Ram, O.; Epsztejn-Litman, S.; Eiges, R. The G-rich Repeats in FMR1 and C9orf72 Loci Are Hotspots for Local Unpairing of DNA. Genetics 2018, 210, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.S.; Laayoun, A.; Lingeman, R.G.; Baker, D.J.; Riley, J. Hypermethylation of telomere-like foldbacks at codon 12 of the human c-Ha-ras gene and the trinucleotide repeat of the FMR-1 gene of fragile X. J. Mol. Biol. 1994, 243, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; Perrera, V.; Scaranaro, M.; de la Rosa-Velazquez, I.A.; van de Nobelen, S.; Shukeir, N.; Popow, J.; Gerle, B.; Opravil, S.; Pagani, M.; et al. A transcription factor-based mechanism for mouse heterochromatin formation. Nat. Struct. Mol. Biol. 2012, 19, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Saksouk, N.; Simboeck, E.; Dejardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 2015, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Peschansky, V.J.; Barbouth, D.; Mehta, A.; Silva, J.P.; Wahlestedt, C. Comprehensive analysis of the transcriptional landscape of the human FMR1 gene reveals two new long noncoding RNAs differentially expressed in Fragile X syndrome and Fragile X-associated tremor/ataxia syndrome. Hum. Genet. 2014, 133, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Tabib, A.; Kahan, T.; Orlanski, S.; Gropp, M.; Tabach, Y.; Yanuka, O.; Benvenisty, N.; Keshet, I.; Cedar, H. Epigenetic mechanism of FMR1 inactivation in Fragile X syndrome. Int. J. Dev. Biol. 2017, 61, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Fine, N.A.; Fujisawa, T.; Gorovsky, M.A. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell 2002, 110, 689–699. [Google Scholar] [CrossRef]
- Taverna, S.D.; Coyne, R.S.; Allis, C.D. Methylation of histone h3 at lysine 9 targets programmed DNA elimination in tetrahymena. Cell 2002, 110, 701–711. [Google Scholar] [CrossRef]
- Pal-Bhadra, M.; Bhadra, U.; Birchler, J.A. RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Mol. Cell 2002, 9, 315–327. [Google Scholar] [CrossRef]
- Morris, K.V.; Chan, S.W.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef]
- Kim, D.H.; Villeneuve, L.M.; Morris, K.V.; Rossi, J.J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar] [CrossRef]
- Sarshad, A.A.; Juan, A.H.; Muler, A.I.C.; Anastasakis, D.G.; Wang, X.; Genzor, P.; Feng, X.; Tsai, P.F.; Sun, H.W.; Haase, A.D.; et al. Argonaute-miRNA Complexes Silence Target mRNAs in the Nucleus of Mammalian Stem Cells. Mol. Cell 2018, 71, 1040.e8–1050.e8. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef]
- Nagano, T.; Mitchell, J.A.; Sanz, L.A.; Pauler, F.M.; Ferguson-Smith, A.C.; Feil, R.; Fraser, P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008, 322, 1717–1720. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Beckedorff, F.C.; Ayupe, A.C.; Crocci-Souza, R.; Amaral, M.S.; Nakaya, H.I.; Soltys, D.T.; Menck, C.F.; Reis, E.M.; Verjovski-Almeida, S. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 2013, 9, e1003705. [Google Scholar] [CrossRef] [PubMed]
- Velazquez Camacho, O.; Galan, C.; Swist-Rosowska, K.; Ching, R.; Gamalinda, M.; Karabiber, F.; De La Rosa-Velazquez, I.; Engist, B.; Koschorz, B.; Shukeir, N.; et al. Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. Elife 2017, 6. [Google Scholar] [CrossRef]
- Wang, X.; Goodrich, K.J.; Gooding, A.R.; Naeem, H.; Archer, S.; Paucek, R.D.; Youmans, D.T.; Cech, T.R.; Davidovich, C. Targeting of Polycomb Repressive Complex 2 to RNA by Short Repeats of Consecutive Guanines. Mol. Cell 2017, 65, 1056.e5–1067.e5. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Porro, A.; Feuerhahn, S.; Delafontaine, J.; Riethman, H.; Rougemont, J.; Lingner, J. Functional characterization of the TERRA transcriptome at damaged telomeres. Nat. Commun. 2014, 5, 5379. [Google Scholar] [CrossRef]
- Johnson, W.L.; Yewdell, W.T.; Bell, J.C.; McNulty, S.M.; Duda, Z.; O’Neill, R.J.; Sullivan, B.A.; Straight, A.F. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 2017, 6. [Google Scholar] [CrossRef]
- Shirai, A.; Kawaguchi, T.; Shimojo, H.; Muramatsu, D.; Ishida-Yonetani, M.; Nishimura, Y.; Kimura, H.; Nakayama, J.I.; Shinkai, Y. Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. Elife 2017, 6. [Google Scholar] [CrossRef]
- Kanhere, A.; Viiri, K.; Araujo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.T.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells. Stem Cells 2017, 35, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Detich, N.; Bovenzi, V.; Szyf, M. Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 2003, 278, 27586–27592. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; De Pascalis, I.; Accadia, M.; Terracciano, A.; Moscato, U.; Chiurazzi, P.; Neri, G. Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet. Genomics 2008, 18, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Nesher, G.; Moore, T.L.; Dorner, R.W. In vitro effects of methotrexate on peripheral blood monocytes: Modulation by folinic acid and S-adenosylmethionine. Ann. Rheum. Dis. 1991, 50, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Tanaka, Y.; Ueda, K. Evidence for hypomethylation in two children with acute lymphoblastic leukemia and leukoencephalopathy. Cancer 2000, 89, 925–931. [Google Scholar] [CrossRef]
- Brendel, C.; Mielke, B.; Hillebrand, M.; Gartner, J.; Huppke, P. Methotrexate treatment of FraX fibroblasts results in FMR1 transcription but not in detectable FMR1 protein levels. J. Neurodev. Disord. 2013, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Nakao, M.; Fujita, N.; Sado, T.; Tanaka, M.; Taguchi, H.; Okumura, K. 5-Aza-2′-deoxycytidine induces histone hyperacetylation of mouse centromeric heterochromatin by a mechanism independent of DNA demethylation. Biochem. Biophys. Res. Commun. 2001, 288, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002, 62, 6456–6461. [Google Scholar] [PubMed]
- Chiurazzi, P.; Pomponi, M.G.; Pietrobono, R.; Bakker, C.E.; Neri, G.; Oostra, B.A. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999, 8, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Dolskiy, A.A.; Pustylnyak, V.O.; Yarushkin, A.A.; Lemskaya, N.A.; Yudkin, D.V. Inhibitors of Histone Deacetylases Are Weak Activators of the FMR1 Gene in Fragile X Syndrome Cell Lines. Biomed. Res. Int. 2017, 2017, 3582601. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Gabrielson, E.; Chen, W.; Anbazhagan, R.; van Engeland, M.; Weijenberg, M.P.; Herman, J.G.; Baylin, S.B. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002, 31, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Mancano, G.; Lanni, S.; Palumbo, F.; Goracci, M.; Ferre, F.; Helmer-Citterich, M.; Neri, G. Genome-wide methylation analysis demonstrates that 5-aza-2-deoxycytidine treatment does not cause random DNA demethylation in fragile X syndrome cells. Epigenetics Chromatin 2016, 9, 12. [Google Scholar] [CrossRef]
- Palii, S.S.; Van Emburgh, B.O.; Sankpal, U.T.; Brown, K.D.; Robertson, K.D. DNA methylation inhibitor 5-Aza-2’-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell Biol. 2008, 28, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Orta, M.L.; Calderon-Montano, J.M.; Dominguez, I.; Pastor, N.; Burgos-Moron, E.; Lopez-Lazaro, M.; Cortes, F.; Mateos, S.; Helleday, T. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 2013, 41, 5827–5836. [Google Scholar] [CrossRef]
- Disney, M.D.; Liu, B.; Yang, W.Y.; Sellier, C.; Tran, T.; Charlet-Berguerand, N.; Childs-Disney, J.L. A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem. Biol. 2012, 7, 1711–1718. [Google Scholar] [CrossRef]
- Tran, T.; Childs-Disney, J.L.; Liu, B.; Guan, L.; Rzuczek, S.; Disney, M.D. Targeting the r(CGG) repeats that cause FXTAS with modularly assembled small molecules and oligonucleotides. ACS Chem. Biol. 2014, 9, 904–912. [Google Scholar] [CrossRef]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014, 83, 1043–1050. [Google Scholar] [CrossRef]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Bontekoe, C.J.; Severijnen, L.A.; Oostra, B.A. Timing of the absence of FMR1 expression in full mutation chorionic villi. Hum. Genet. 2002, 110, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism. 2016, 7, 42. [Google Scholar] [CrossRef]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef]
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 2002, 8, 1482–1488. [Google Scholar] [PubMed]
- Brouwer, J.R.; Mientjes, E.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; Severijnen, L.A.; Van der Linde, H.C.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp. Cell Res. 2007, 313, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Hundscheid, R.D.; Braat, D.D.; Kiemeney, L.A.; Smits, A.P.; Thomas, C.M. Increased serum FSH in female fragile X premutation carriers with either regular menstrual cycles or on oral contraceptives. Hum. Reprod. 2001, 16, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Leavitt, B.R.; Farzin, F.; Jacquemont, S.; Greco, C.M.; Brunberg, J.A.; Tassone, F.; Hessl, D.; Harris, S.W.; Zhang, L.; et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am. J. Hum. Genet. 2004, 74, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Welt, C.K.; Smith, P.C.; Taylor, A.E. Evidence of early ovarian aging in fragile X premutation carriers. J. Clin. Endocrinol. Metab. 2004, 89, 4569–4574. [Google Scholar] [CrossRef]
- Allen, E.G.; Sullivan, A.K.; Marcus, M.; Small, C.; Dominguez, C.; Epstein, M.P.; Charen, K.; He, W.; Taylor, K.C.; Sherman, S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 2007, 22, 2142–2152. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Sherwell, S.; Kinsella, G.; Tassone, F.; Taylor, A.; Amor, D.; Sung, S.; Evans, A. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 2012, 82, 88–92. [Google Scholar] [CrossRef]
- Santa Maria, L.; Pugin, A.; Alliende, M.A.; Aliaga, S.; Curotto, B.; Aravena, T.; Tang, H.T.; Mendoza-Morales, G.; Hagerman, R.; Tassone, F. FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin. Genet. 2014, 86, 378–382. [Google Scholar] [CrossRef]
- Basuta, K.; Schneider, A.; Gane, L.; Polussa, J.; Woodruff, B.; Pretto, D.; Hagerman, R.; Tassone, F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A 2015, 167A, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- LaFauci, G.; Adayev, T.; Kascsak, R.; Kascsak, R.; Nolin, S.; Mehta, P.; Brown, W.T.; Dobkin, C. Fragile X screening by quantification of FMRP in dried blood spots by a Luminex immunoassay. J. Mol. Diagn. 2013, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.K.; Safanda, J.F.; Fall, M.Z.; Quince, C.; Lang, K.A.; Hull, C.E.; Carpenter, I.; Staley, L.W.; Hagerman, R.J. Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA 1994, 271, 507–514. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Wiegers, A.M.; Smits, A.P.; Mohkamsing, S.; Duivenvoorden, H.J.; Fryns, J.P.; Curfs, L.M.; Halley, D.J.; Oostra, B.A.; van den Ouweland, A.M.; et al. Mental status of females with an FMR1 gene full mutation. Am. J. Hum. Genet. 1996, 58, 1025–1032. [Google Scholar] [PubMed]
- Kieffer, E.; Nicod, J.C.; Gardes, N.; Kastner, C.; Becker, N.; Celebi, C.; Pirrello, O.; Rongieres, C.; Koscinski, I.; Gosset, P.; et al. Improving preimplantation genetic diagnosis for Fragile X syndrome: Two new powerful single-round multiplex indirect and direct tests. Eur. J. Hum. Genet. 2016, 24, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Raspa, M.; Bishop, E.; Holiday, D. No change in the age of diagnosis for fragile x syndrome: Findings from a national parent survey. Pediatrics 2009, 124, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Allan, A.M.; Zong, R.; Zhang, L.; Johnson, E.B.; Schaller, E.G.; Murthy, A.C.; Goggin, S.L.; Eisch, A.J.; Oostra, B.A.; et al. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat. Med. 2011, 17, 559–565. [Google Scholar] [CrossRef]
- Siegel, J.J.; Chitwood, R.A.; Ding, J.M.; Payne, C.; Taylor, W.; Gray, R.; Zemelman, B.V.; Johnston, D. Prefrontal Cortex Dysfunction in Fragile X Mice Depends on the Continued Absence of Fragile X Mental Retardation Protein in the Adult Brain. J. Neurosci. 2017, 37, 7305–7317. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, D.; Gazy, I.; Usdin, K. Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome. Brain Sci. 2019, 9, 39. https://doi.org/10.3390/brainsci9020039
Kumari D, Gazy I, Usdin K. Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome. Brain Sciences. 2019; 9(2):39. https://doi.org/10.3390/brainsci9020039
Chicago/Turabian StyleKumari, Daman, Inbal Gazy, and Karen Usdin. 2019. "Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome" Brain Sciences 9, no. 2: 39. https://doi.org/10.3390/brainsci9020039
APA StyleKumari, D., Gazy, I., & Usdin, K. (2019). Pharmacological Reactivation of the Silenced FMR1 Gene as a Targeted Therapeutic Approach for Fragile X Syndrome. Brain Sciences, 9(2), 39. https://doi.org/10.3390/brainsci9020039