Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy

{kind=link}
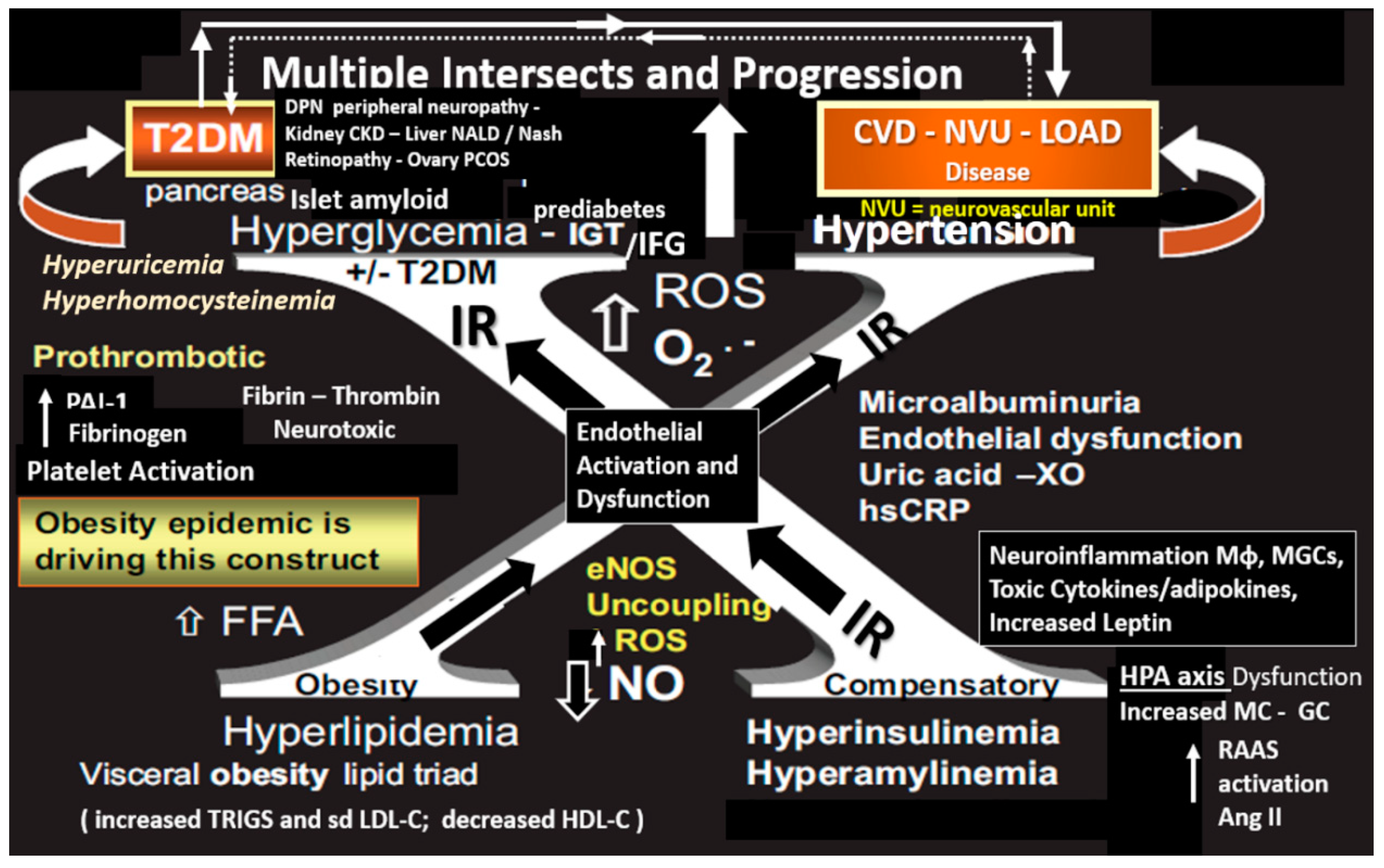{kind=link}
{kind=link}
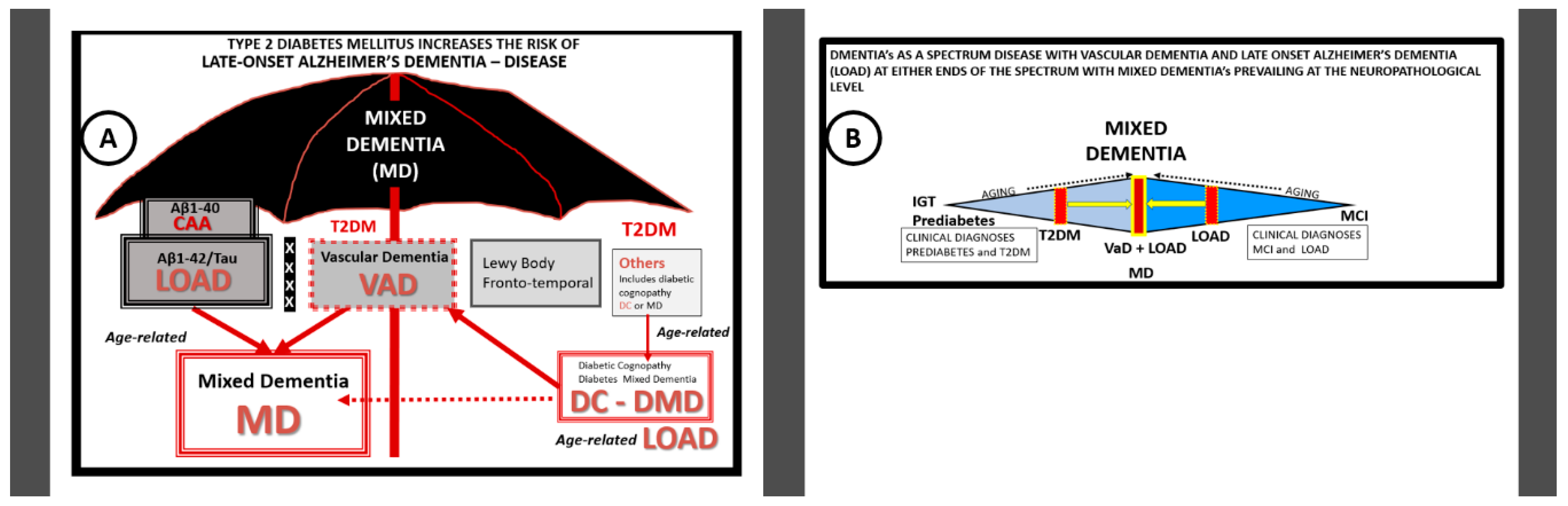{kind=link}
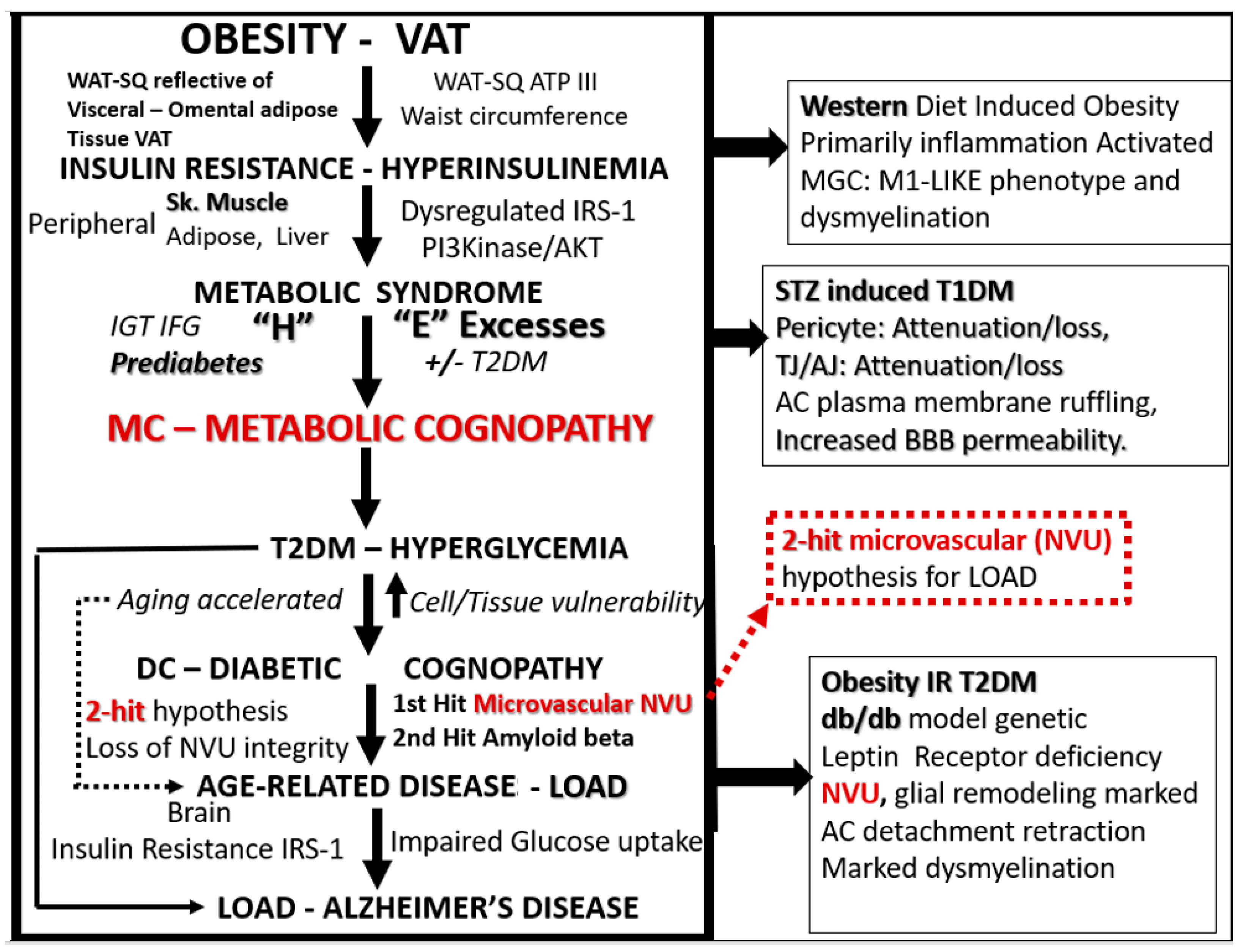{kind=link}
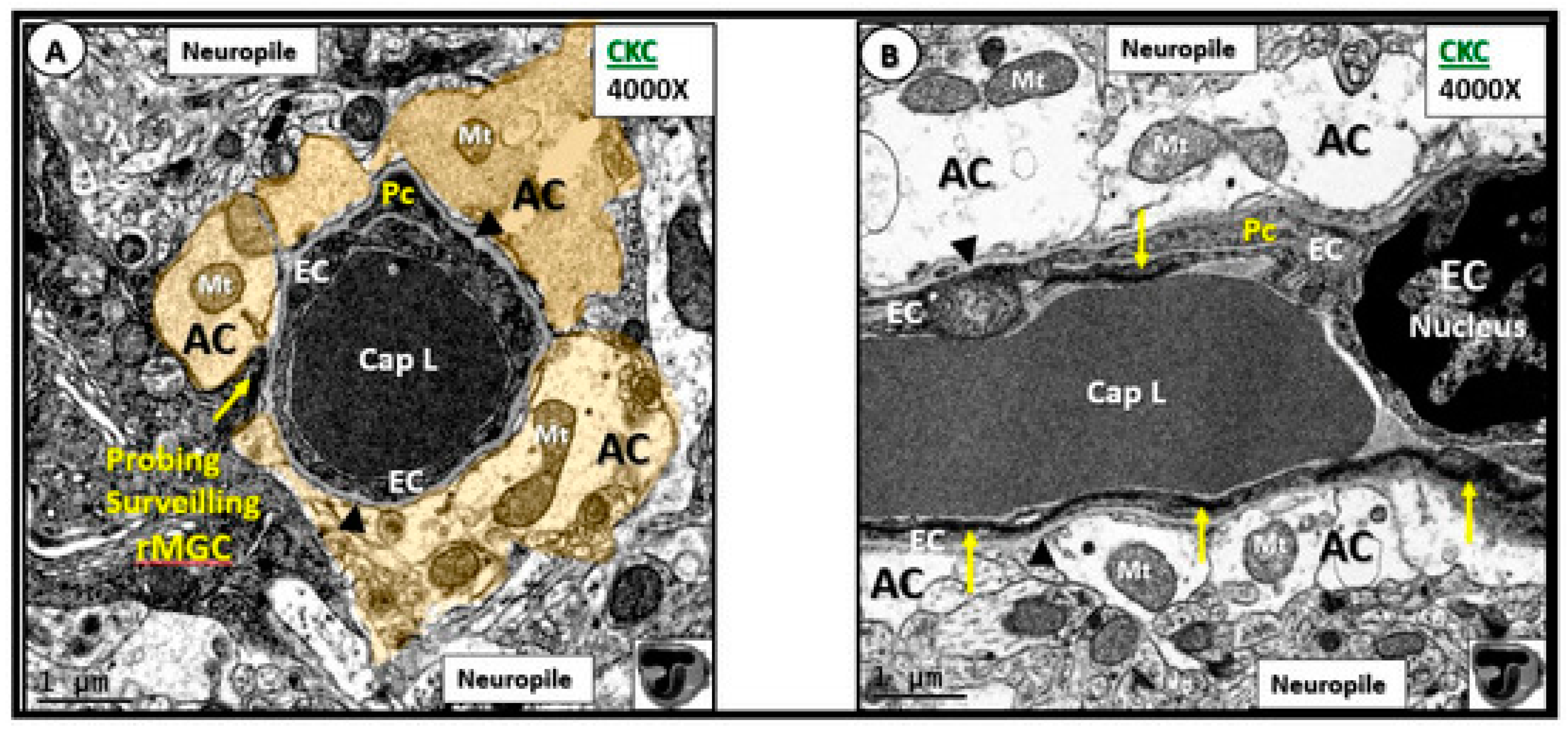{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. T2DM, Late-Onset Alzheimer’s Disease–Dementia (LOAD) and Societal Aging
1.2. Aging, Obesity, Insulin Resistance, Metabolic Syndrome (MetS)
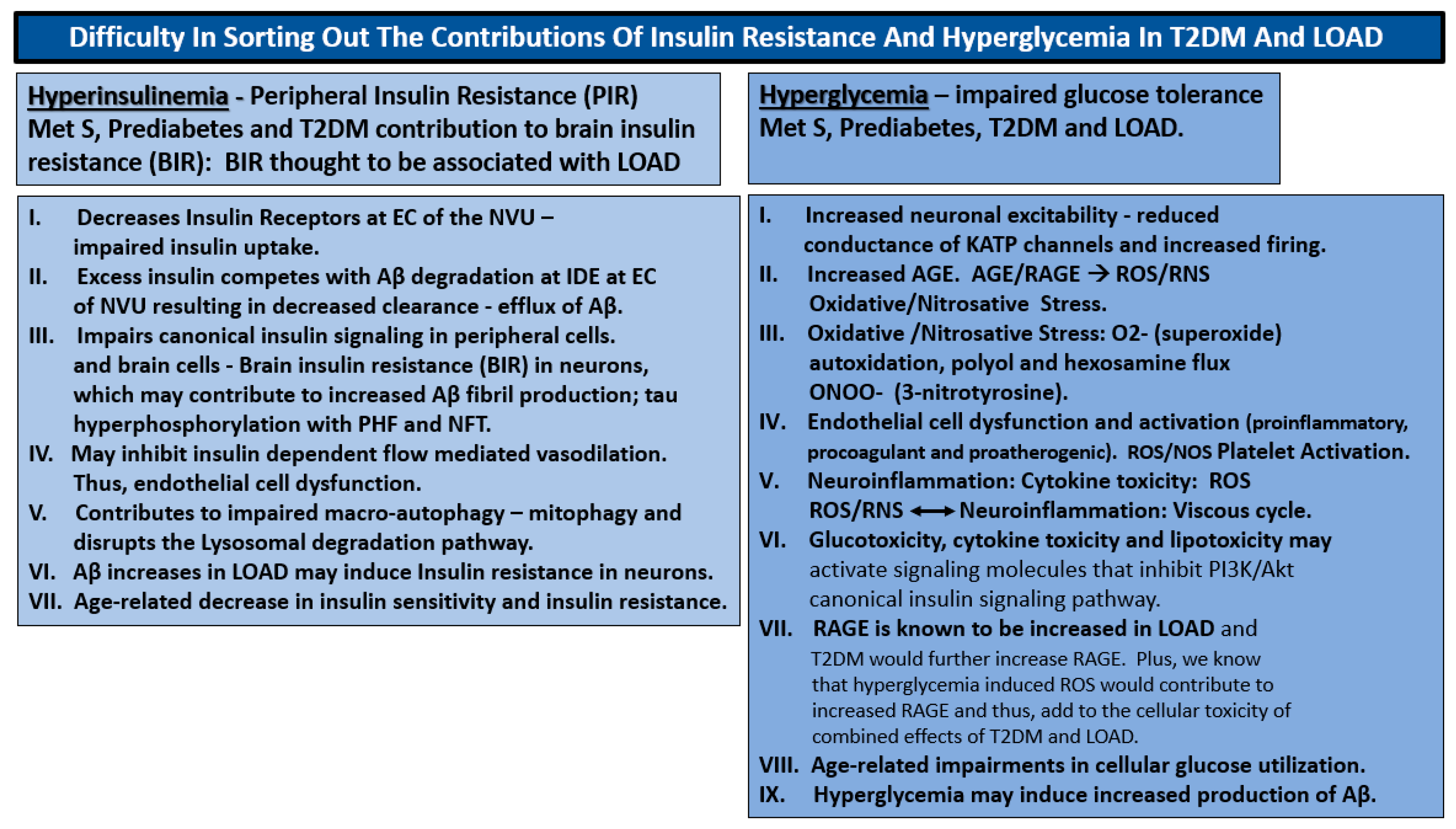
1.3. Insulin Resistance: Peripheral and Central-Brain Insulin Resistance
1.4. Hypothalamic–Pituitary–Adrenal (HPA) Axis Dysfunction in the Metabolic Syndrome (MetS)
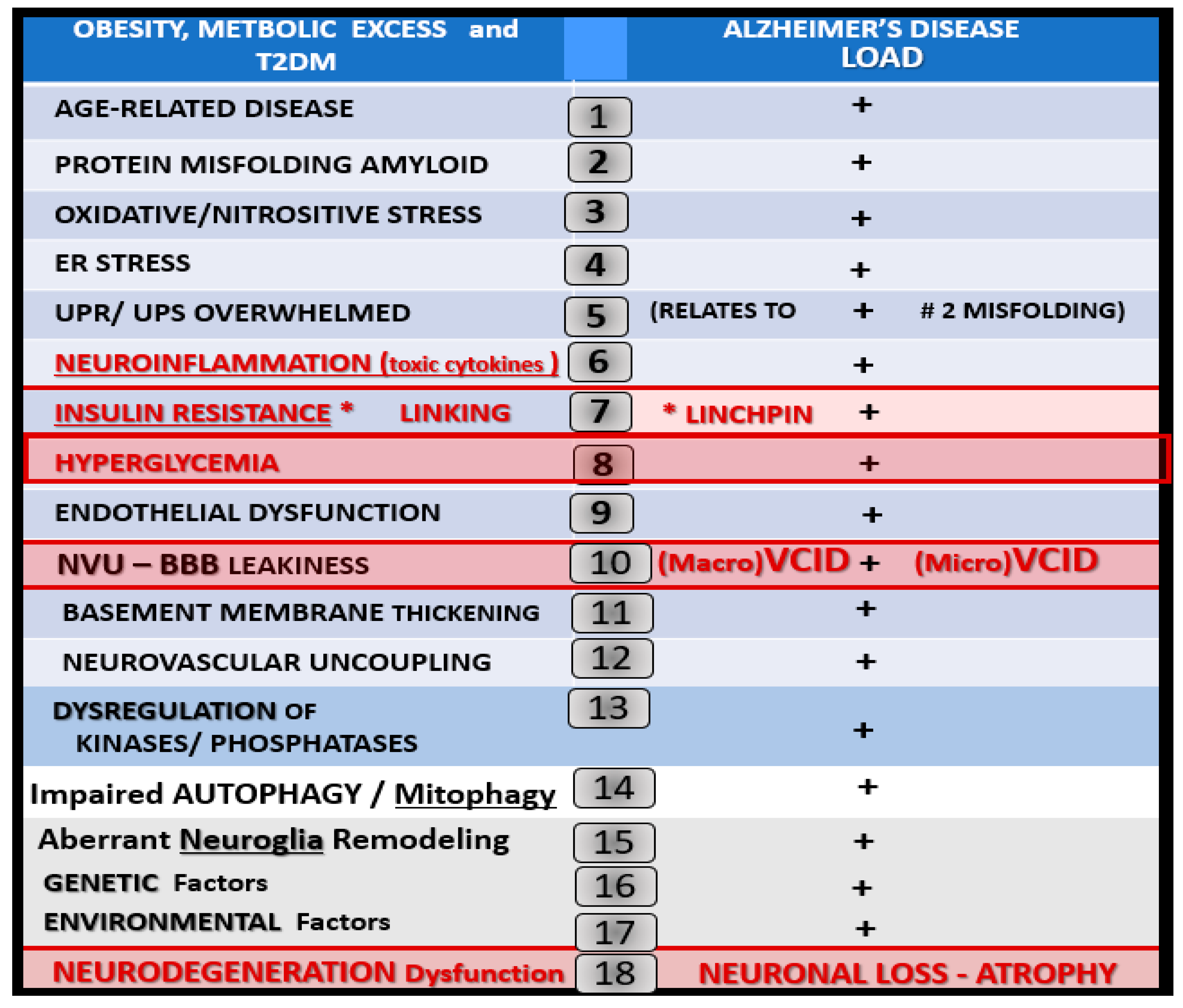
1.5. Type 2 Diabetes Mellitus, Late-Onset Alzheimer’s Disease and Mixed Dementias Intersections
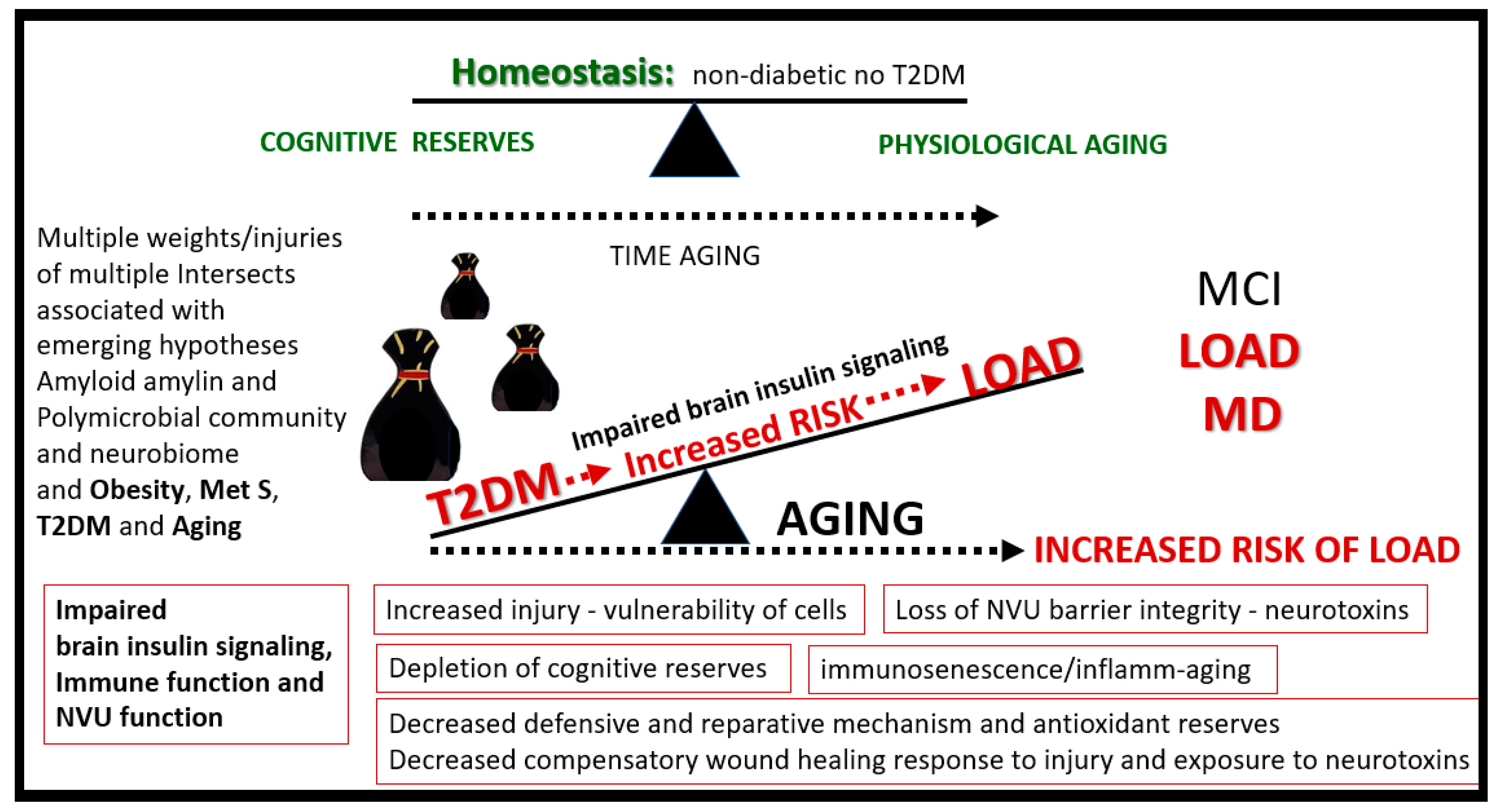
1.6. Continuum of Progression: From Obesity, Insulin Resistance to T2DM to LOAD
2. Capillary Neurovascular Unit (NVU)
2.1. Endothelial Cell(s) (EC)—Endothelium
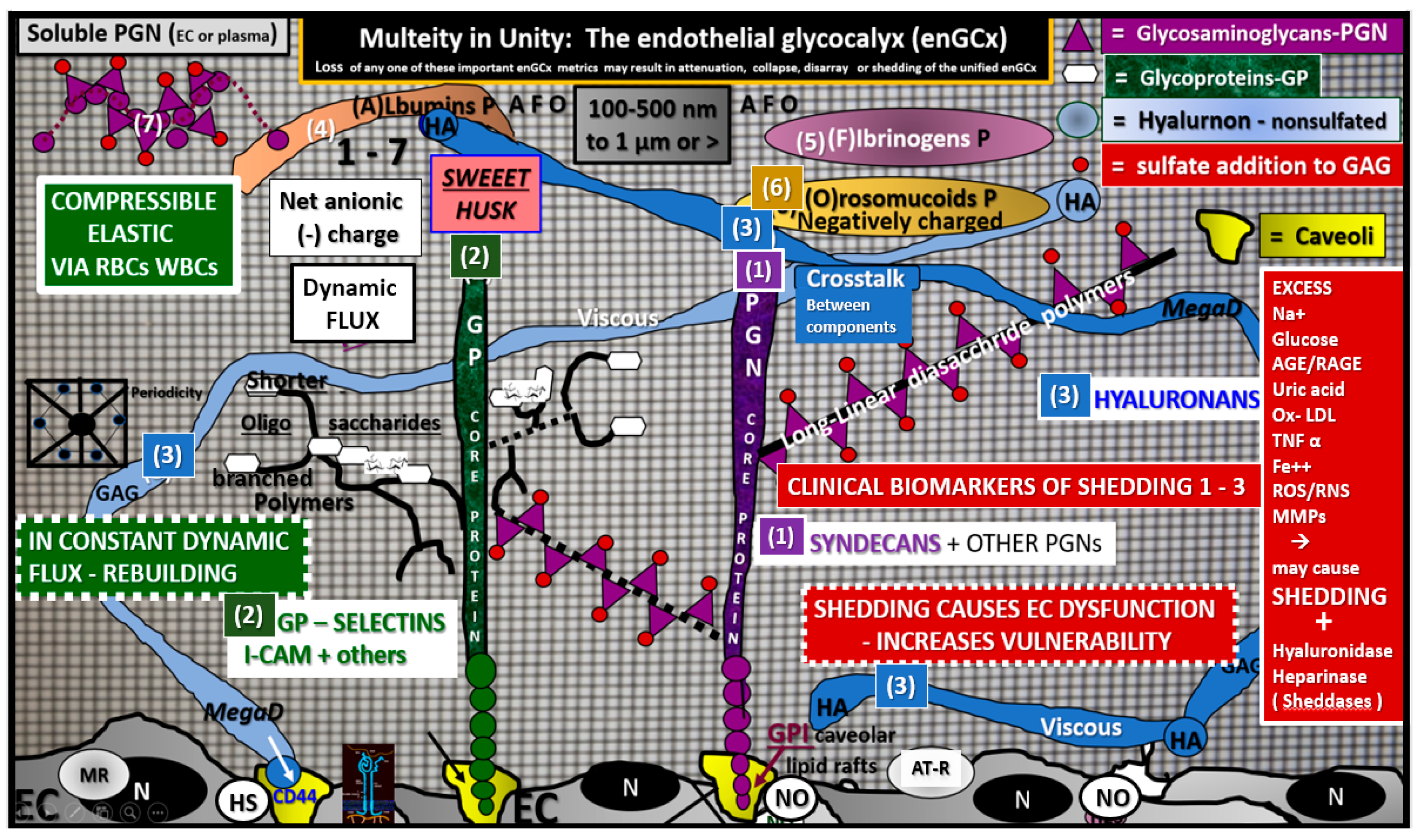
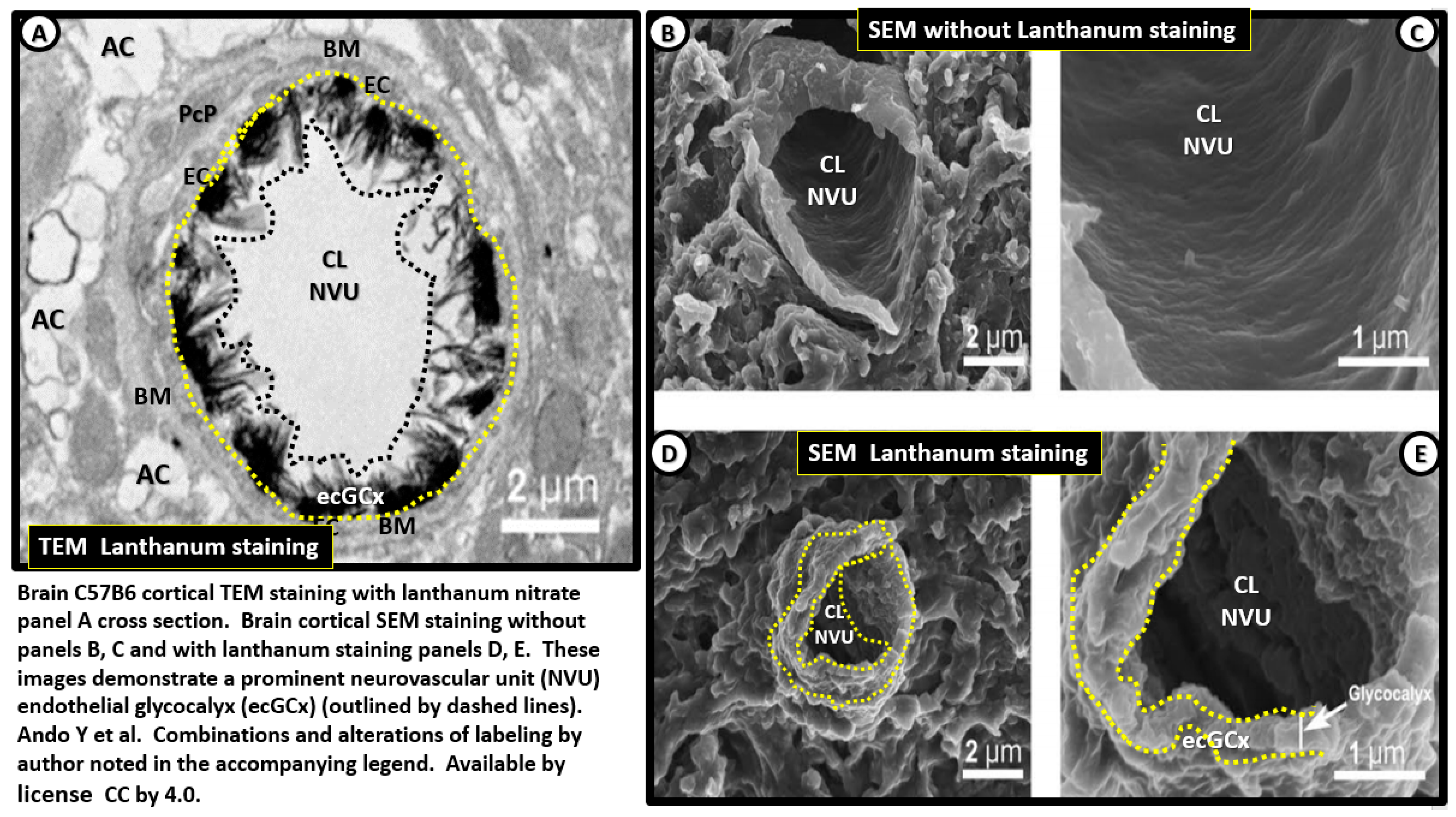
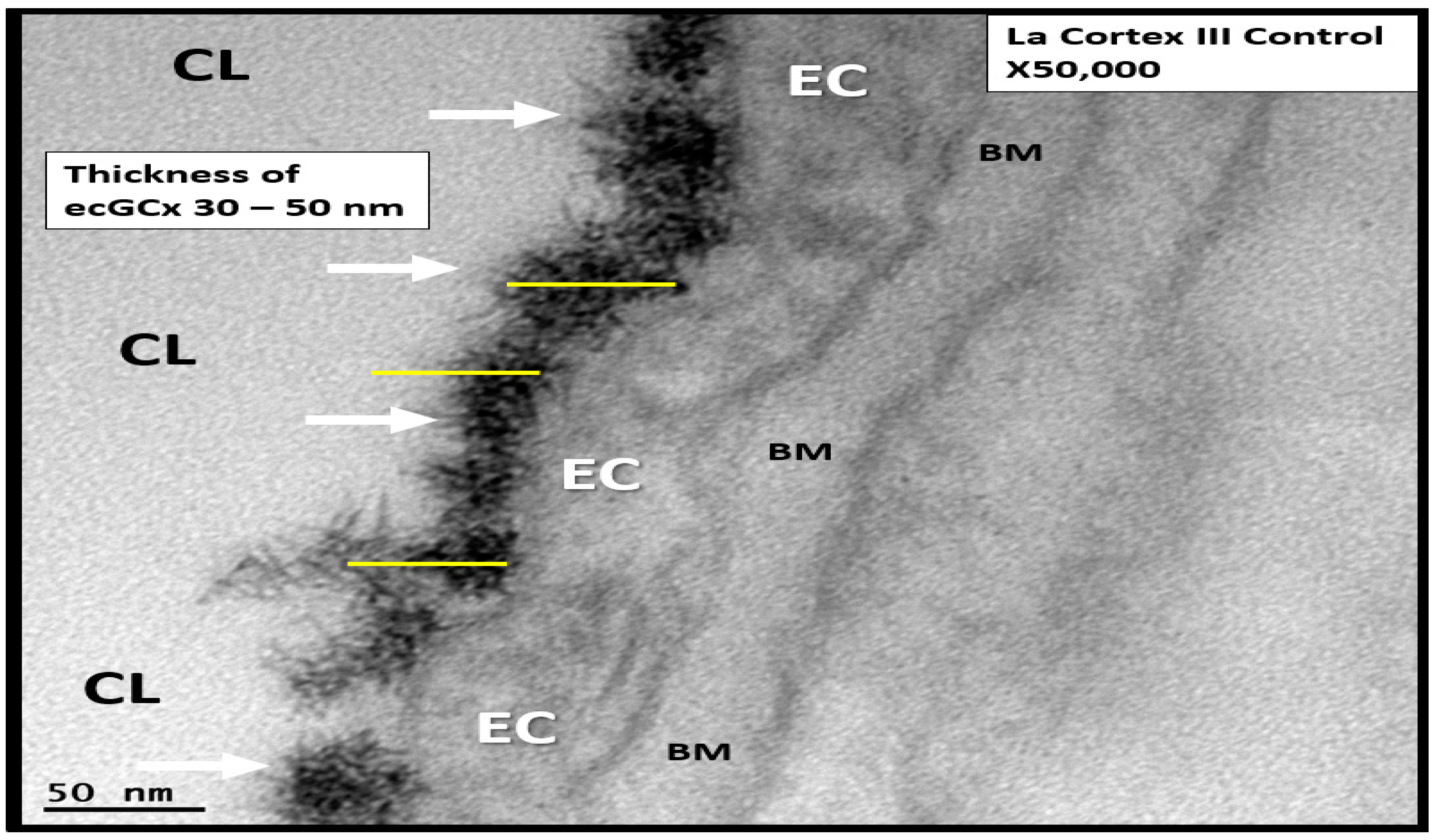
2.2. Endothelial Glycocalyx (ecGCx)
2.3. Endothelial Cell Basement Membrane (BM)
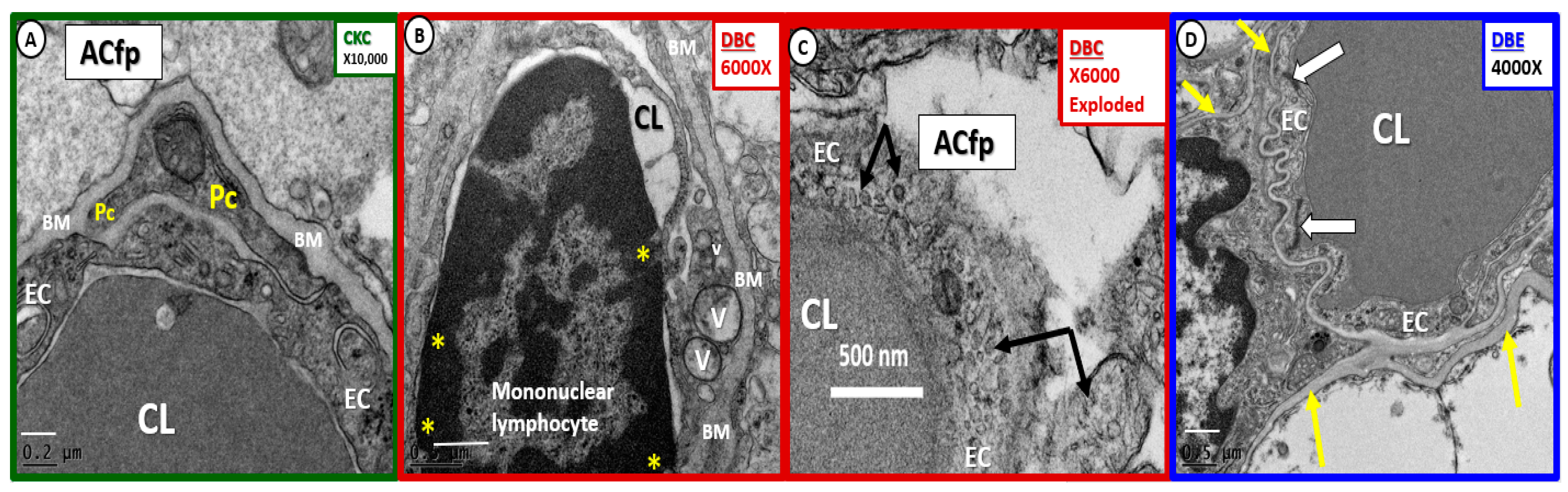
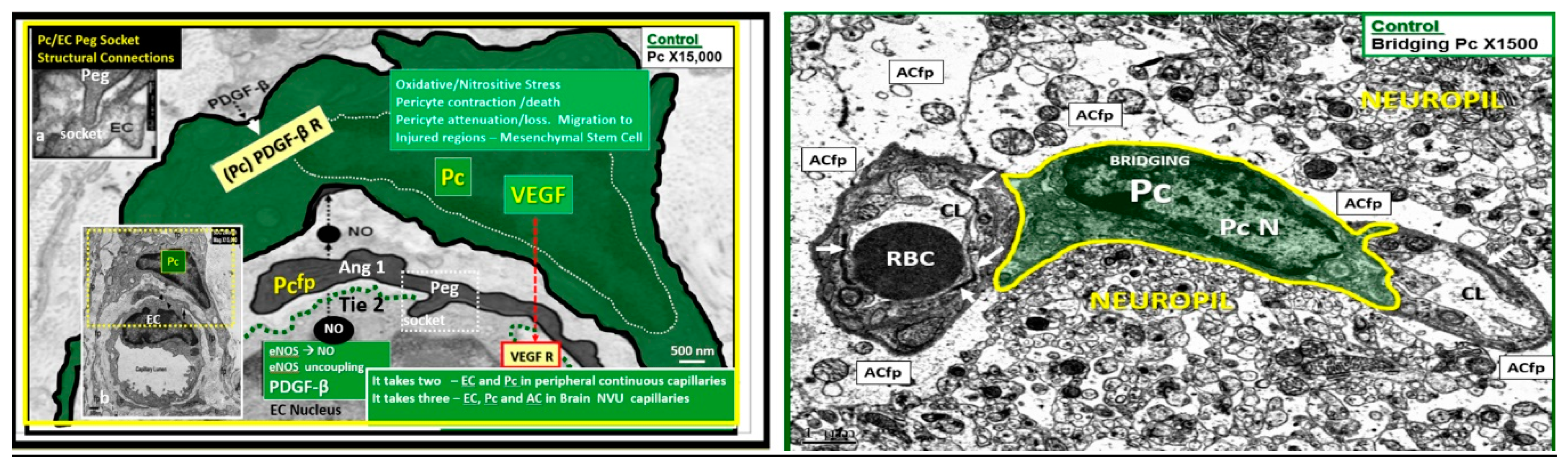
2.4. Neurovascular Unit (NVU) Pericyte(s) (Pc)
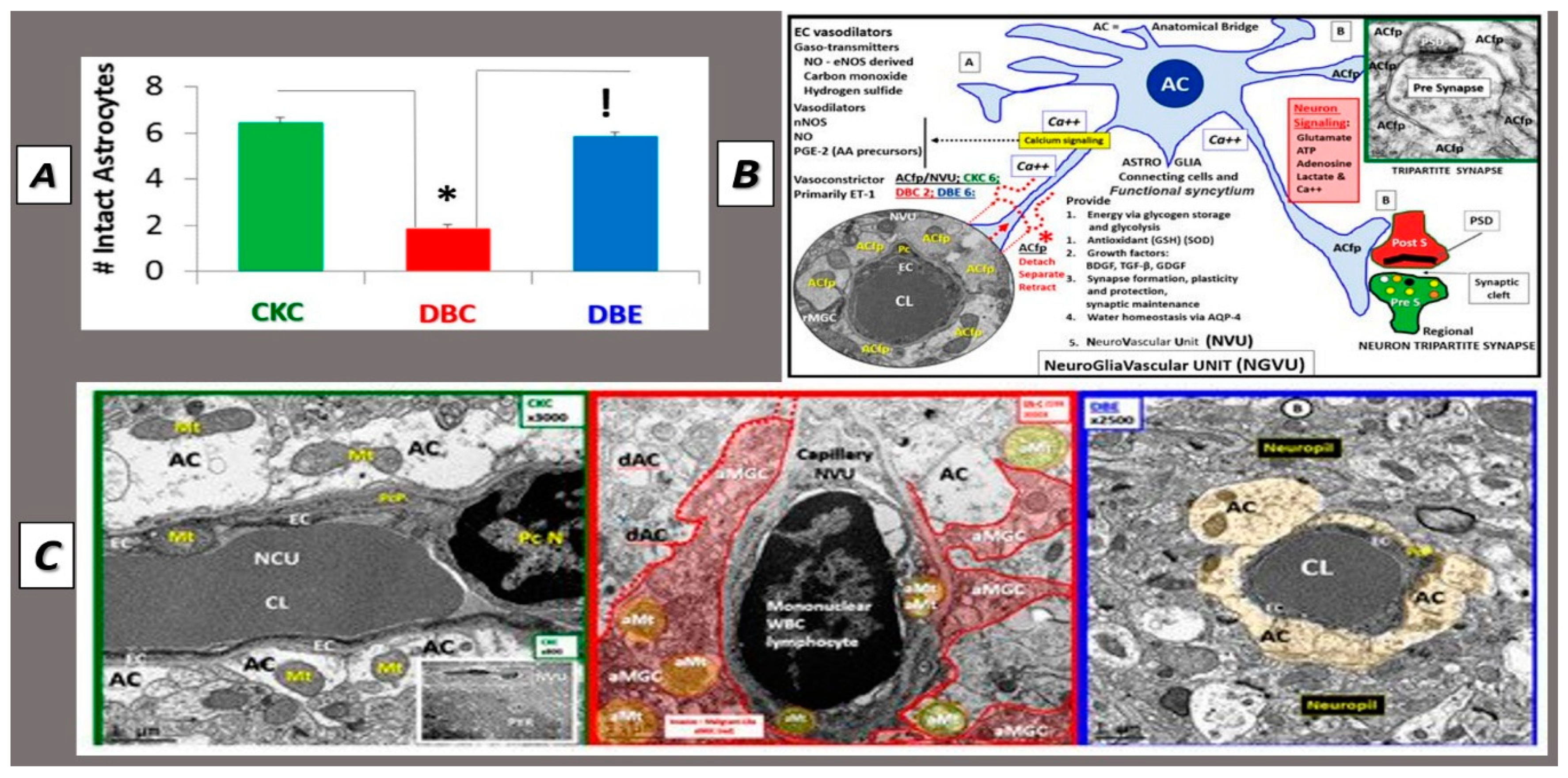
2.5. Astrocyte (AC)–Diabetic Gliopathy
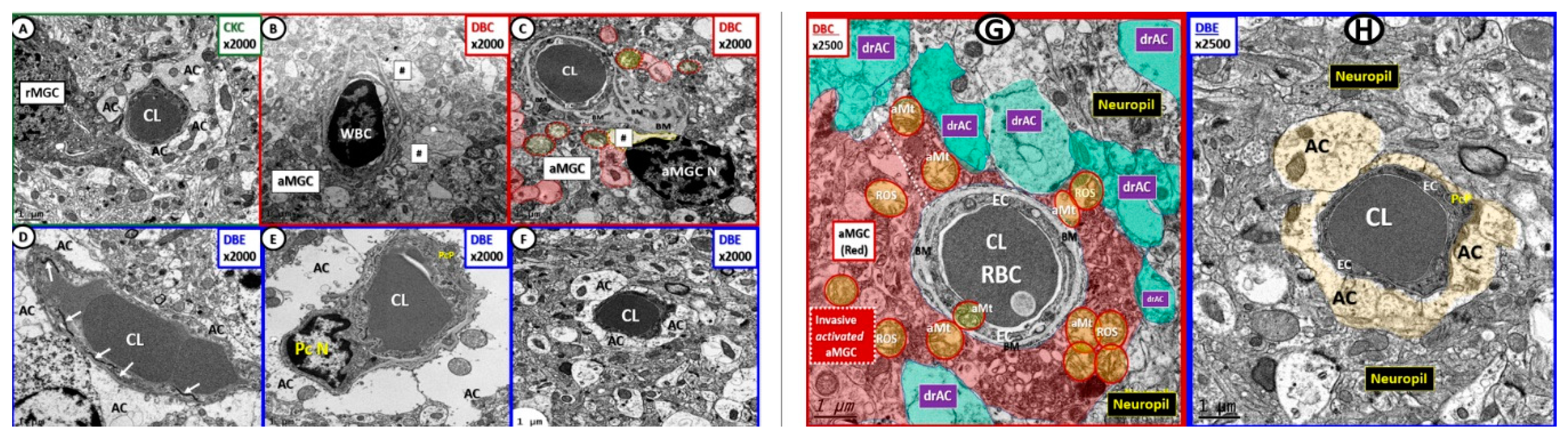
2.6. Microglial Cell(s) (MGC), Neuroinflammation and Diabetic Gliopathy
2.7. Oligodendrocyte and Myelin–Diabetic Gliopathy
2.8. Neurovascular Unit and Barrier Functions
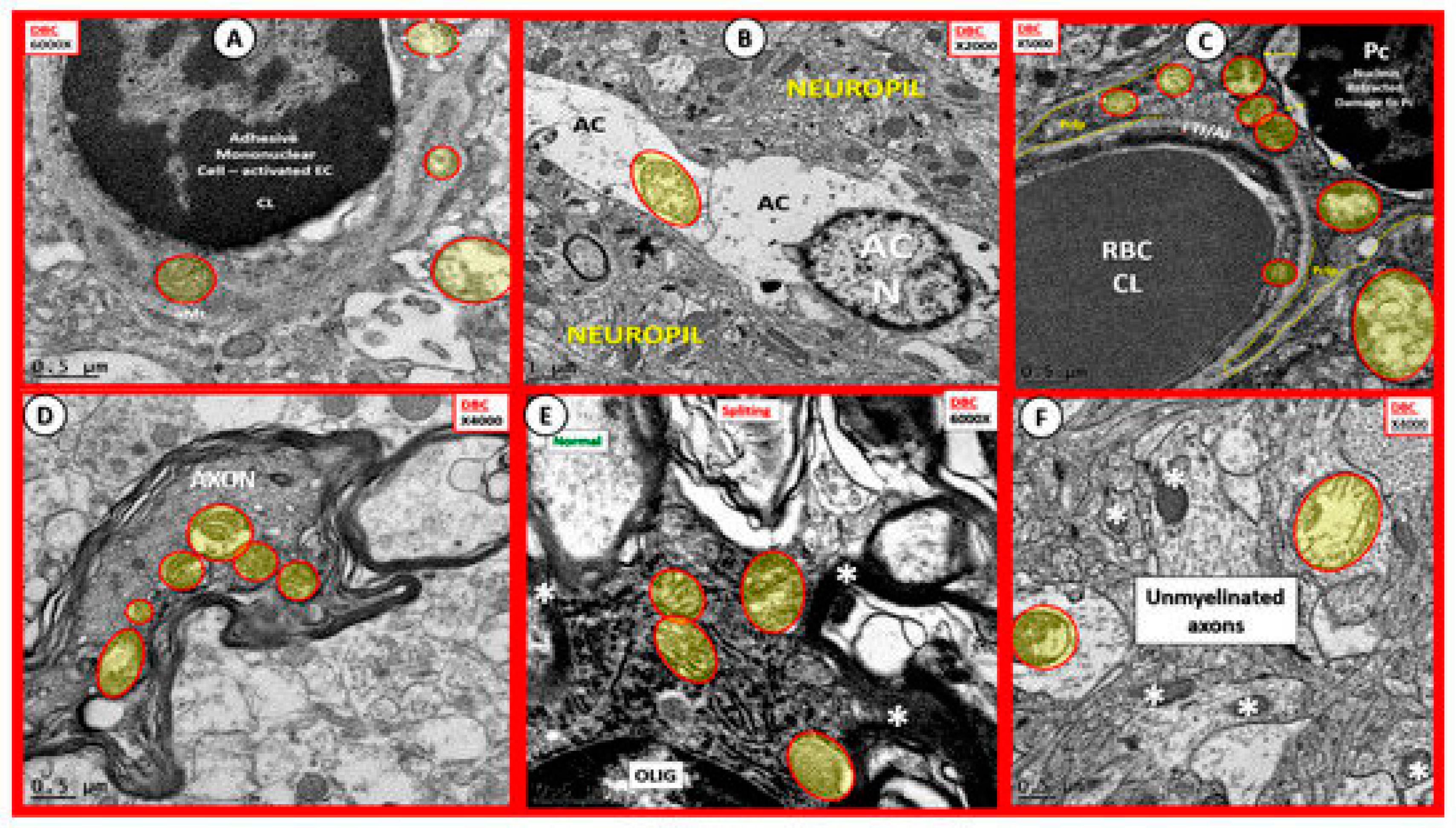
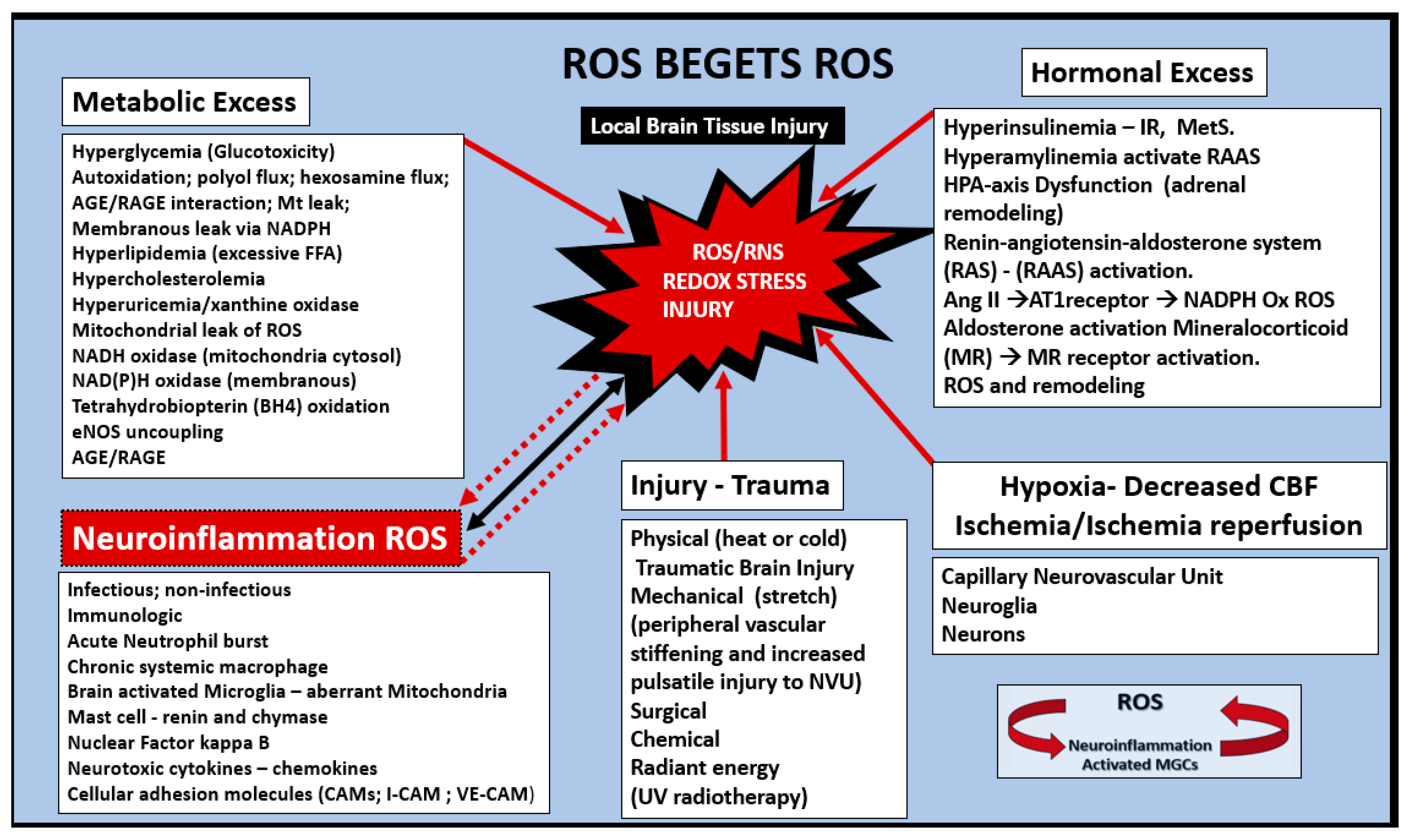
3. Oxidative Stress: Reactive Oxygen/Nitrogen Species (ROS/RNS), Redox Stress and Aberrant Mitochondria (aMt)
Oxidative Nitrosative Stress (ROS/NOS): Aberrant Mitochondrial-Derived ROS/NOS Leakage in T2DM End-Organ Complications
4. Major Existing and Emerging Hypotheses for LOAD
4.1. Endothelial Cell Activation–Dysfunction and Impaired Cerebral Blood Flow (CBF) in Diabetic db/db Models: Hypometabolism (VII) and Vascular (IX) Hypotheses Elaborated
5. Neuropathological Changes in LOAD
6. Discussion and Concluding Remarks
6.1. Metabolic Hypothesis: Hyperglycemia and Hyperinsulinemia
6.2. The Antimicrobial Protection Hypothesis: Antimicrobial Peptide(s) (AMP)
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aα | amyloid alpha |
| AC | astrocyte |
| Aβ | amyloid beta |
| ACfp | astrocyte foot processes |
| AD | Alzheimer’s disease |
| AFib | amyloid fibrinogen/fibrin |
| AGE | advanced glycation end products |
| AMP | antimicrobial peptide(s) |
| aMt | aberrant mitochondria |
| APP | amyloid precursor protein |
| aMGC | activated microglia cell |
| BB | baby boom generation following WWII |
| Boomer | baby boom generation following WWII |
| BBB | blood–brain barrier |
| BIR | brain insulin resistance |
| BM | basement membrane |
| CAA | cerebral amyloid angiopathy |
| CBF | cerebral blood flow |
| CKC | C57B6-J control models |
| CSF | cerebral spinal fluid |
| CVD | cerebro-cardiovascular disease |
| DBC | diabetic db/db models |
| DC | diabetic cognopathy |
| DIO | diet induced obesity |
| EC | endothelial cell |
| ecGCx | endothelial cell glycocalyx |
| eNOS | endothelial nitric oxide synthase |
| FFA | free fatty acids |
| HPA | hypothalamic–pituitary–adrenal (HPA) axis |
| H and E | hyper and excess phenomenon |
| HSV-1 | herpes simplex virus type 1 |
| IAPP | islet amyloid polypeptide |
| IR | insulin resistance |
| LOAD | late-onset Alzheimer’s disease |
| MC | metabolic cognopathy |
| MD | mixed dementia |
| MetS | metabolic syndrome |
| MGC | microglia cell |
| MT | microtubule |
| NO | nitric oxide |
| NFT | neurofibrillary tangles |
| NVU | neurovascular unit |
| OL | oligodendrocyte |
| Pc | pericyte |
| Pcfp | pericyte foot process |
| PHF | paired helical fragments |
| PIR | peripheral insulin resistance |
| RAAS | renin–angiotensin–aldosterone system |
| RAGE | receptor for advanced glycation end products |
| RBC | red blood cell |
| rMGC | ramified microglia cell |
| ROS/RNS | reactive oxygen species/reactive nitrogen species |
| T2DM | type 2 diabetes mellitus |
| Tau | microtubule associated protein |
| TJ/AJ | tight junctions/adherens junctions |
| VaD / VAD | vascular dementia |
| VCID | vascular contributions of impaired cognition and dementia |
| VSMC | vascular smooth muscle cell |
| WBC | white blood cell |
References
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A. Do we have brain to spare? Neurology 2005, 64, 2004–2005. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood–brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in The Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in The Female Diabetic db/db Model—Part II: Microglia and Mitochondria. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in The Female Diabetic db/db Model—Part III: Oligodendrocyte and Myelin. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef]
- Hayden, M.R.; Banks, W.A.; Shah, G.N.; Gu, Z.; Sowers, J.R. Cardiorenal metabolic syndrome and diabetic cognopathy. Cardiorenal Med. 2013, 3, 265–282. [Google Scholar] [CrossRef]
- Salameh, T.S.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood–brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef]
- Wrighten, S.A.; Piroli, G.G.; Grillo, C.A.; Reagan, L.P. A look inside the diabetic brain: Contributors to diabetes induced brain aging. Biochim. Biophys. Acta 2009, 1792, 444–453. [Google Scholar] [CrossRef] [PubMed]
- DeVries, J.H.; Desouza, C.; Bellary, S.; Unger, J.; Hansen, O.K.H.; Zacho, J.; Woo, V. Achieving glycaemic control without weight gain, hypoglycaemia, or gastrointestinal adverse events in type 2 diabetes in the SUSTAIN clinical trial programme. Diabetes Obes. Metab. 2018, 20, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- Reasner, C.A.; Defronzo, R.A. Treatment of type 2 diabetes mellitus: A rational approach based on its pathophysiology. Am. Fam. Physician 2001, 63, 1687. [Google Scholar] [PubMed]
- DeFronzo, R.A.; Riccardd CBonadonna, R.C.; Ferrannini, E. Pathogenesis of NIDDM: A Balanced Overview. Diabetes Care 1992, 15, 318–368. [Google Scholar] [CrossRef]
- Dagogo-Jack, S.; Santiago, J.V. Pathophysiology of type 2 diabetes and modes of action of therapeutic interventions. Arch. Intern. Med. 1997, 157, 1802–1817. [Google Scholar] [CrossRef]
- The Diabetes Prevention Program Research Group; Crandall, J.; Schade, D.; Ma, Y.; Fujimoto, W.Y.; Barrett-Connor, E.; Fowler, S.; Dagogo-Jack, S.; Andres, R. The Influence of Age on the Effects of Lifestyle Modification and Metformin in Prevention of Diabetes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1075–1081, United Nations Department of Economics and Social Affairs Populations Division. Population Ageing and Sustainable Development. No2017/1. June 2017. Available online: https://www.un.org/development/desa/publications/world-population-prospects-the-2017-revision.htmlm (accessed on 25 June 2019).
- United Nations Department of Economics and Social Affairs Populations Division. Population Ageing and Sustainable Development. Available online: https://www.un.org/en/development/desa/population/publications/pdf/popfacts/PopFacts_2017-1.pdf (accessed on 10 July 2019).
- Tobore, T.O. On the Etiopathogenesis and Pathophysiology of Alzheimer’s Disease: A Comprehensive Theoretical Review. J. Alzheimers Dis. 2019. [Google Scholar] [CrossRef]
- International Diabetes Foundation. IDF Diabetes Atlas, 8th ed.; Available online: https://diabetesatlas.org/ (accessed on 1 July 2019).
- Inzucchi, S.E.; Viscoli, C.M.; Young, L.H.; Furie, K.L.; Gorman, M.; Lovejoy, A.M.; Dagogo-Jack, S.; Ismail-Beigi, F.; Korytkowski, M.T.; Pratley, R.E.; et al. IRIS Trial Investigators: Pioglitazone Prevents Diabetes in Patients with Insulin Resistance and Cerebrovascular Disease. Diabetes Care 2016, 39, 1684–1692. [Google Scholar] [CrossRef]
- Trajectory Report. Available online: https://www.alz.org/help-support/resources/publications/trajectory_report (accessed on 10 July 2019).
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome. An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [PubMed]
- Pruzin, J.J.; Nelson, P.T.; Abner, E.L.; Arvanitakis, Z. Review: Relationship of type 2 diabetes to human brain pathology. Neuropathol. Appl. Neurobiol. 2018, 44, 347–362. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Barbagallo, M.; Dominguez, L.J. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Strachan, M.W.; Reynolds, R.M.; Frier, B.M.; Mitchell, R.J.; Price, J.F. The relationship between type 2 diabetes and dementia. Br. Med. Bull. 2008, 88, 131–146. [Google Scholar] [CrossRef]
- Swierczynska, M.M.; Mateska, I.; Peitzsch, M.; Bornstein, S.R.; Chavakis, T.; Eisenhofer, G.; Lamounier-Zepter, V.; Eaton, S. Changes in morphology and function of adrenal cortex in mice fed a high-fat diet. Int. J. Obes. 2015, 39, 321–330. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. The Role of Aldosterone in Obesity-Related Hypertension. Am. J. Hypertens. 2016, 29, 415–423. [Google Scholar] [CrossRef]
- DeMarco, V.G.; Habibi, J.; Jia, G.; Aroor, A.R.; Ramirez-Perez, F.I.; Martinez-Lemus, L.A.; Bender, S.B.; Garro, M.; Hayden, M.R.; Sun, Z.; et al. Low-Dose Mineralocorticoid Receptor Blockade Prevents Western Diet-Induced Arterial Stiffening in Female Mice. Hypertension 2015, 66, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Pike, C.J. Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr. Diabetes Rep. 2014, 14, 476. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef]
- Nguyen, J.C.; Killcross, A.S.; Jenkins, T. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Backman, K.; Joas, E.; Waern, M.; Ostling, S.; Guo, X.; Skoog, I. 37 years of body mass index and dementia: Observations from the prospective population study of women in Gothenburg, Sweden. J. Alzheimers Dis. 2012, 28, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Panza, F.; Colacicco, A.M.; D’Introno, A.; Capurso, C.; Torres, F.; Grigoletto, F.; Maggi, S.; Del Parigi, A.; Reiman, E.M.; et al. Vascular risk factors, incidence of MCI, and rates of progression to dementia. Neurology 2004, 63, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Besser, L.M.; Gill, D.P.; Monsell, S.E.; Brenowitz, W.; Meranus, D.H.; Kukull, W.; Gustafson, D.R. Body mass index, weight change, and clinical progression in mild cognitive impairment and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2014, 28, 36–43. [Google Scholar] [CrossRef]
- Strachan, M.W.; Reynolds, R.M.; Marioni, R.E.; Price, J.F. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat. Rev. Endocrinol. 2011, 7, 108–114. [Google Scholar] [CrossRef]
- Mendez, M.F. Early-onset Alzheimer’s Disease: Nonamnestic Subtypes and Type 2 AD. Arch. Med. Res. 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Abramowitz, W.D.; Keene, C.D.; Hawes, S.E.; Hubbard, R.A.; Longstreth, W.T., Jr.; Woltjer, R.L.; Crane, P.K.; Larson, E.B.; Kukull, W.A. Alzheimer’s disease neuropathologic change, Lewy body disease, and vascular brain injury in clinic- and community-based samples. Neurobiol. Aging 2017, 53, 83–92. [Google Scholar] [CrossRef]
- Rahimi, J.; Kovacs, G.G. Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 2014, 6, 82. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Snyder, H.M.; Corriveau, R.A.; Craft, S.; Faber, J.E.; Greenberg, S.M.; Knopman, D.; Lamb, B.T.; Montine, T.J.; Nedergaard, M.; Schaffer, C.B.; et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 710–717. [Google Scholar] [CrossRef]
- Corriveau, R.A.; Bosetti, F.; Emr, M.; Gladman, J.T.; Koenig, J.I.; Moy, C.S.; Pahigiannis, K.; Waddy, S.P.; Koroshetz, W. The science of vascular contributions to cognitive impairment and dementia (VCID): A framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell. Mol. Neurobiol. 2016, 36, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Corriveau, R.A.; Wilcock, D.M. Vascular contributions to cognitive impairment and dementia (VCID). Biochim. Biophys. Acta 2016, 1862, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Deane, R.; Sagare, A.P.; Bell, R.D.; Winkler, E.A. Low-density lipoprotein receptor-related protein-1: A serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J. Neurochem. 2010, 115, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 1055–1068. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef]
- Xu, W.L.; Atti, A.R.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Midlife overweight and obesity increase late-life dementia risk: A population-based twin study. Neurology 2011, 76, 1568–1574. [Google Scholar] [CrossRef]
- Lizama, C.O.; Zovein, A.C. Polarizing pathways: Balancing endothelial polarity, permeability, and lumen formation. Exp. Cell Res. 2013, 319, 1247–1254. [Google Scholar] [CrossRef]
- Bors, L.; Toth, K.; Toth, E.Z.; Bajza, A.; Csorba, A.; Sziegeti, K.; Mathe, D.; Perlaki, G.; Orsi, G.; Toth, G.K. Age-dependent changes at the blood–brain barrier. A comparative structural and functional study in young adult and middle-aged rats. Brain Res. Bull. 2018, 139, 269–277. [Google Scholar] [CrossRef]
- Ando, Y.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Tomita, H.; Zaikokuji, R.; Hotta, Y.; Miyazaki, N.; Yano, H.; et al. Brain-Specific Ultrastructure of Capillary Endothelial Glycocalyx and Its Possible Contribution for Blood Brain Barrier. Sci. Rep. 2018, 8, 17523. [Google Scholar] [CrossRef]
- Yoon, J.H.; Jeong, Y. In vivo imaging for neurovascular disease research. Arch. Pharm. Res. 2019, 42, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflügers Archiv-Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Haeren, R.H.L.; Rijkers, K.; Schijns, O.E.M.G.; Dings, J.; Hoogland, G.; van Zandvoort, M.A.M.J.; Vink, H.; van Overbeeke, J.J. In vivo assessment of the human cerebral microcirculation and its glycocalyx: A technical report. J. Neurosci. Methods 2018, 303, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Machin, D.R.; Bloom SLMachin, D.R.; Bloom, S.I.; Campbell, R.A.; Phuong, T.T.T.; Gates, P.E.; Lesniewski, L.A.; Rondina, M.T.; Donato, A.J. Advanced age results in a diminished endothelial glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H531–H539. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood–brain barrier in systemic inflammation. Brain Behav. Immunity 2017, 60, 1–12. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R.; Tyagi, S.C. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: The matrix preloaded. Cardiovasc. Diabetol. 2005, 4, 9. [Google Scholar] [CrossRef]
- Hurtado-Alvarado, G.; Cabañas-Morales, A.M.; Gómez-Gónzalez, B. Pericytes: Brain-immune interface modulators. Front. Integr. Neurosci. 2014, 7, 80. [Google Scholar] [CrossRef]
- Hayden, M.R.; Yang, Y.; Habibi, J.; Bagree, S.V.; Sowers, J.R. Pericytopathy: Oxidative stress and impaired cellular longevity in the pancreas and skeletal muscle in metabolic syndrome and type 2 diabetes. Oxid. Med. Cell. Longev. 2010, 3, 290–303. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood–brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D.; Hall, C.N. Pericyte-mediated regulation of capillary diameter: A component of neurovascular coupling in health and disease. Front. Neuroenerget. 2010, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C.; Kolb, L.; Sowers, J.R.; Khanna, R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: The emerging role of sodium thiosulfate. Cardiovasc. Diabetol. 2005, 4, 4. [Google Scholar] [CrossRef]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Lastra, G.; Patel, K.; Wasekar, C.; Manrique, C.M.; Ozerdem, U.; Stas, S.; Sowers, J.R. Ultrastructure of islet microcirculation, pericytes and the islet exocrine interface in the HIP rat model of diabetes. Exp. Biol. Med. 2008, 233, 1109–1123. [Google Scholar] [CrossRef]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Wasekar, C.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J.R. Ultrastructural islet study of early fibrosis in the Ren2 rat model of hypertension. Emerging role of the islet pancreatic pericyte-stellate cell. JOP 2007, 8, 725–738. [Google Scholar]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J. Astrocyte–endothelial interactions and blood–brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770. [Google Scholar] [CrossRef]
- Petzold, G.C.; Murthy, V.N. Role of astrocytes in neurovascular coupling. Neuron 2011, 71, 782–797. [Google Scholar] [CrossRef]
- Huang, L.; Nakamura, Y.; Lo, E.H.; Hayakawa, K. Astrocyte signaling in the neurovascular unit after central nervous system injury. Int. J. Mol. Sci. 2019, 20, 282. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef]
- Hayden, M.R. Hypothesis: Astrocyte Foot Processes Detachment from the Neurovascular Unit in Female Diabetic Mice May Impair Modulation of Information Processing-Six Degrees of Separation. Brain Sci. 2019, 9, 83. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef]
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The indispensable roles of microglia and astrocytes during brain development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics During Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef] [PubMed]
- Tambuyzer, B.R.; Ponsaerts, P.; Nouwen, E.J. Microglia: Gatekeepers of the central nervous system immunology. J. Leukoc. Biol. 2009, 85, 352–370. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, B.; Cserép, C.; Orsolits, B.; Dénes, Á. New Insights into Microglia-Neuron Interactions: A Neuron’s Perspective. Neuroscience 2019, 405, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Zhang, Z.; Ye, Y.; Zhou, J. Role of microglia-neuron interactions in diabetic encephalopathy. Ageing Res. Rev. 2018, 42, 28–39. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–999. [Google Scholar] [CrossRef]
- Wang, M.; Roussos, P.; McKenzie, A.; Zhou, X.; Kajiwara, Y.; Brennand, K.J.; De Luca, G.C.; Crary, J.F.; Casaccia, P.; Buxbaum, J.D.; et al. Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 2016, 8, 104. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Del Rio-Hortega, P. Microglia. In Cytology and Cellular Pathology of the Nervous System; Penfield, W., Ed.; Hoeber: New York, NY, USA, 1932; pp. 482–534. [Google Scholar]
- Michalski, J.P.; Kothary, R. Oligodendrocytes in a nutshell. Front. Cell. Neurosci. 2015, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Palay, S.L.; Webster, H. The Fine Structure of the Nervous System—Neurons and Their Supportive Cells—The Cellular Sheaths of Neurons, 3rd ed.; Oxford University Press: New York, NY, USA, 1991; pp. 212–261. ISBN 0-19-506571-9. [Google Scholar]
- Pukos, N.; Yoseph, R.; McTigue, D.M. To Be or Not to Be: Environmental Factors that Drive Myelin Formation during Development and after CNS Trauma. Neuroglia 2018, 1, 63–90. [Google Scholar] [CrossRef]
- Reijmer, Y.D.; Brundel, M.; de Bresser, J.; Kappelle, L.J.; Leemans, A.; Biessels, G.J. Utrecht Vascular Cognitive Impairment Study Group. Microstructural white matter abnormalities and cognitive functioning in type 2 diabetes: A diffusion tensor imaging study. Diabetes Care 2013, 36, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Van Agtmaal, M.J.M.; Houben, A.J.H.M.; de Wit, V.; Henry, R.M.A.; Schaper, N.C.; Dagnelie, P.C.; van der Kallen, C.J.; Koster, A.; Sep, S.J.; Kroon, A.A.; et al. Prediabetes Is Associated with Structural Brain Abnormalities: The Maastricht Study. Diabetes Care 2018, 41, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Den Heijer, T.; Vermeer, S.E.; van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef]
- Brundel, M.; van den Heuvel, M.; de Bresser, J.; Kappelle, L.J.; Biessels, G.J. Utrecht Diabetic Encephalopathy Study Group: Cerebral cortical thickness in patients with type 2 diabetes. J. Neurol Sci. 2010, 299, 126–130. [Google Scholar] [CrossRef]
- Kutuzov, N.; Flyvbjerg, H.; Lauritzen, M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2018, 115, E9429–E9438. [Google Scholar] [CrossRef]
- Frederick, R.L.; Shaw, J.M. Moving Mitochondria: Establishing Distribution of an Essential Organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F. The Association of Tau With Mitochondrial Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Sowers, K.M.; Hayden, M.R. Calcific uremic arteriolopathy: Pathophysiology, reactive oxygen species and therapeutic approaches. Oxid. Med. Cell. Longev. 2010, 3, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Whaley-Connell, A.; Sowers, J.R. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: Paying homage to the podocyte. Am. J. Nephrol. 2005, 25, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hayden, M.R.; Sowers, S.; Bagree, S.V.; Sowers, J.R. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxid. Med. Cell. Longev. 2010, 3, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Sowers, K.M.; Pulakat, L.; Joginpally, T.; Krueger, B.; Whaley-Connell, A.; Sowers, J.R. Possible Mechanisms of Local Tissue Renin-Angiotensin System Activation in the Cardiorenal Metabolic Syndrome and Type 2 Diabetes Mellitus. Cardiorenal Med. 2011, 1, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 diabetes mellitus: Implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid. Redox Signal. 2007, 9, 891–910. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Redox imbalance in diabetes. Antioxid. Redox Signal. 2007, 9, 865–867. [Google Scholar] [CrossRef]
- Whaley-Connell, A.T.; Chowdhury, N.A.; Hayden, M.R.; Stump, C.S.; Habibi, J.; Wiedmeyer, C.E.; Gallagher, P.E.; Tallant, E.A.; Cooper, S.A.; Link, C.D.; et al. Oxidative stress and glomerular filtration barrier injury: Role of the renin-angiotensin system in the Ren2 transgenic rat. Am. J. Physiol. Renal Physiol. 2006, 291, F1308–F1314. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Neural redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic neuropathy. Med. Sci. Monit. 2004, 10, RA291–RA307. [Google Scholar]
- Hayden, M.R.; Tyagi, S.C. Myocardial redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and congestive heart failure. Med. Sci. Monit. 2003, 9, SR35–SR52. [Google Scholar]
- Hunt, M.J.; Aru, G.M.; Hayden, M.R.; Moore, C.K.; Hoit, B.D.; Tyagi, S.C. Induction of oxidative stress and disintegrin metalloproteinase in human heart end-stage failure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L239–L245. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Islet redox stress: The manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP 2002, 3, 86–108. [Google Scholar] [PubMed]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S265–S279. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Soiza, R. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197–226. [Google Scholar]
- Ernst, E.; Matrai, A. Altered red and white blood cell rheology in type II diabetes. Diabetes 1986, 35, 1412–1415. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Misiti, F. Erythrocytes as Potential Link between Diabetes and Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 276. [Google Scholar] [CrossRef]
- Cruz Hernández, J.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, M.; Berg, M. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef]
- Roher, A.E.; Debbins, J.P.; Malek-Ahmadi, M.; Chen, K.; Pipe, J.G.; Maze, S.; Belden, C.; Maarouf, C.L.; Thiyyagura, P.; Mo, H.; et al. Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 2012, 8, 599–611. [Google Scholar] [CrossRef]
- Mistur, R.; Mosconi, L.; Santi, S.D.; Guzman, M.; Li, Y.; Tsui, W.; de Leon, M.J. Current challenges for the early detection of Alzheimer’s disease: Brain imaging and CSF studies. J. Clin. Neurol. 2009, 5, 153–166. [Google Scholar] [CrossRef]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Greenberg, S.M. Beta-amyloid, blood vessels and brain function. Stroke 2009, 40, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Insulin Signaling Impairment in the Brain as a Risk Factor in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 88. [Google Scholar] [CrossRef]
- Craft, S. The Role of Metabolic Disorders in Alzheimer’s Disease and Vascular Dementia: Two Roads Converged? Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Wadman, M. US government sets out Alzheimer’s plan. Nature 2012, 485, 426–427. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Kalaitzidis, G.; Malli, A.; Kalaitzoglou, D.; Myserlis, P.G.; Lioutas, V.A. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or earlystage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., 2nd; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition modulates beta-amyloid in early, A.D. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. 2017, 59, 421–432. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. “A” is for amylin and amyloid in type 2 diabetes mellitus. JOP 2001, 2, 124–139. [Google Scholar] [PubMed]
- Hayden, M.R.; Tyagi, S.C.; Kerklo, M.M.; Nicolls, M.R. Type 2 diabetes mellitus as a conformational disease. JOP 2005, 6, 287–302. [Google Scholar] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Despa, F.; Decarli, C. Amylin: What might be its role in Alzheimer’s disease and how could this affect therapy? Expert Rev. Proteom. 2013, 10, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.; Despa, F. Hyperamylinemia as a risk factor for accelerated cognitive decline in diabetes. Expert Rev. Proteom. 2015, 12, 575–577. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef]
- Ly, H.; Verma, N.; Wu, F.; Liu, M.; Saatman, K.E.; Nelson, P.T.; Slevin, J.T.; Goldstein, L.B.; Biessels, G.J.; Despa, F. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann. Neurol. 2017, 82, 208–222. [Google Scholar] [CrossRef]
- Fawver, J.N.; Ghiwot, Y.; Koola, C.; Carrera, W.; Rodriguez-Rivera, J.; Hernandez, C.; Dineley, K.T.; Kong, Y.; Li, J.; Jhamandas, J.; et al. Islet amyloid polypeptide (IAPP): A second amyloid in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 928–940. [Google Scholar] [CrossRef]
- Grizzanti, J.; Corrigan, R.; Casadesus, G. Neuroprotective Effects of Amylin Analogues on Alzheimer’s Disease Pathogenesis and Cognition. J. Alzheimers Dis. 2018, 66, 11–23. [Google Scholar] [CrossRef]
- Marrero, D.G.; Crean, J.; Zhang, B.; Kellmeyer, T.; Gloster, M.; Herrmann, K.; Rubin, R.; Fineberg, N.; Kolterman, O. Effect of adjunctive pramlintide treatment on treatment satisfaction in patients with type 1 diabetes. Diabetes Care 2007, 30, 210–216. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Zamolodchikov, D.; Ahn, H.J.; Strickland, S.; Norris, E.H. Fibrinogen and altered hemostasis in Alzheimer’s disease. J. Alzheimers Dis. 2012, 32, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-B is an antimicrobial peptide: A review of the evidence. J. Alzheimers Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Appleby, B.S.; Nacopoulos, D.; Milano, N.; Zhong, K.; Cummings, J.L. A Review: Treatment of Alzheimer’s disease discovered in repurposed agents. Dement. Geriatr. Cogn. Disord. 2013, 35, 1–22. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, H.S.; Shin, K.Y.; Kim, E.M.; Kim, M.; Kim, H.S.; Park, C.H.; Jeong, Y.H.; Yoo, J.; Lee, J.P.; et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology 2007, 32, 2393–2404. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. The amyloid hypothesis: Let sleeping dogmas lie? Neurobiol. Aging 2002, 23, 1101–1105. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Bourgade, K.; Khalil, A.; Zerif, E.; Larbi, A.; Hirokawa, K.; Pawelec, G.; Bocti, C.; Lacombe, G.; et al. Can an Infection Hypothesis Explain the Beta Amyloid Hypothesis of Alzheimer’s Disease? Front. Aging Neurosci. 2018, 10, 224. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Banks, W.A. The blood–brain barrier as an endocrine tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455. [Google Scholar] [CrossRef]


























© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. https://doi.org/10.3390/brainsci9100262
Hayden MR. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sciences. 2019; 9(10):262. https://doi.org/10.3390/brainsci9100262
Chicago/Turabian StyleHayden, Melvin R. 2019. "Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy" Brain Sciences 9, no. 10: 262. https://doi.org/10.3390/brainsci9100262
APA StyleHayden, M. R. (2019). Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sciences, 9(10), 262. https://doi.org/10.3390/brainsci9100262