Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches

Abstract
1. Introduction
2. Causes and Consequences of Neurodegeneration
3. Intra-Cellular Signaling Mechanisms
4. Current Treatment Paradigm
5. Future Strategies
5.1. Inhibiting and Disaggregating Protein Aggregates
5.1.1. Induction of Endogenous Hsp70 Production
5.1.2. Application of Exogenous Hsp70
5.1.3. Constitutive Expression of Hsp70
5.1.4. Inhibition of Hsp70 ATPase
5.2. Immuno-Modulation
5.3. Stimulating Autophagy
5.4. Clearance by Glymphatic System
5.5. Neurogenesis and Neurotrophic Factors
5.6. Insulin and Neurodegeneration
5.7. Cholinergic System in AD
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Dickinson-Anson, H.; Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 2027–2033. [Google Scholar] [CrossRef]
- Altman, J. Are new neurons formed in the brains of adult mammals? Science (New York) 1962, 135, 1127–1128. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- David, M.A.; Tayebi, M. Detection of protein aggregates in brain and cerebrospinal fluid derived from multiple sclerosis patients. Front. Neurol. 2014, 5, 251. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking traumatic brain injury to chronic traumatic encephalopathy: Identification of potential mechanisms leading to neurofibrillary tangle development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M. Protein misfolding inside cells: The case of huntingtin and Huntington’s disease. IUBMB Life 2008, 60, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.B. Molecular basis of the neurodegenerative disorders. N. Engl. J. Med. 1999, 340, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Estrada, L.D. Protein misfolding and neurodegeneration. Arch. Neurol. 2008, 65, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Tuite, M.F.; Melki, R. Protein Misfolding and Aggregation in Ageing and Disease: Molecular Processes and Therapeutic Perspectives; Taylor & Francis: Abingdon, UK, 2007. [Google Scholar]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.R.; Kim, S.H. Synapses in neurodegenerative diseases. BMB Rep. 2017, 50, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New hopes and challenges for treatment of neurodegenerative disorders: Great opportunities for young neuroscientists. Basic Clin. Neurosci. 2013, 4, 3–4. [Google Scholar] [PubMed]
- Duraes, F.; Pinto, M.; Sousa, E. Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals (Basel, Switzerland) 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current therapeutic molecules and targets in neurodegenerative diseases based on in silico drug design. Curr. Neuropharmacol. 2018, 16, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Coppede, F. The potential of epigenetic therapies in neurodegenerative diseases. Front. Genet. 2014, 5, 220. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Ghoumari, A.M.; Bielecki, B.; Steibel, J.; Boehm, N.; Liere, P.; Macklin, W.B.; Kumar, N.; Habert, R.; Mhaouty-Kodja, S.; et al. The neural androgen receptor: A therapeutic target for myelin repair in chronic demyelination. Brain J. Neurol. 2013, 136, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Oestrogen as a neuroprotective hormone. Nat. Rev. Neurosci. 2002, 3, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: Comparison with insulin-like growth factors I and II. Neuroscience 1992, 46, 909–923. [Google Scholar] [CrossRef]
- Woolley, J.D.; Khan, B.K.; Murthy, N.K.; Miller, B.L.; Rankin, K.P. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: Rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J. Clin. Psychiatry 2011, 72, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Montero-Odasso, M.; Pieruccini-Faria, F.; Bartha, R.; Black, S.E.; Finger, E.; Freedman, M.; Greenberg, B.; Grimes, D.A.; Hegele, R.A.; Hudson, C.; et al. Motor phenotype in neurodegenerative disorders: Gait and balance platform study design protocol for the ontario neurodegenerative research initiative (ONDRI). J. Alzheimer’s Dis. 2017, 59, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Vadakkan, K.I. Neurodegenerative disorders share common features of “loss of function” states of a proposed mechanism of nervous system functions. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 83, 412–430. [Google Scholar] [CrossRef] [PubMed]
- Hervas, R.; Oroz, J.; Galera-Prat, A.; Goni, O.; Valbuena, A.; Vera, A.M.; Gomez-Sicilia, A.; Losada-Urzaiz, F.; Uversky, V.N.; Menendez, M.; et al. Common features at the start of the neurodegeneration cascade. PLoS Boil. 2012, 10, e1001335. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.M. Neurodegeneration: A question of balance. Nature 2008, 452, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 131–168. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar] [PubMed]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s disease therapy and prevention strategies. Annu. Rev Med 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Sanabria-Castro, A.; Alvarado-Echeverria, I.; Monge-Bonilla, C. Molecular pathogenesis of Alzheimer’s disease: An update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L. Alzheimer’s disease and other dementias: Update on research. Lancet Neurol. 2017, 16, 4–5. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Cali, T.; Szabo, I.; Brini, M. Alpha-synuclein at the intracellular and the extracellular side: Functional and dysfunctional implications. Boil. Chem. 2017, 398, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Alkhuja, S. Parkinson disease: Research update and clinical management. South Med. J. 2013, 106, 334. [Google Scholar] [CrossRef] [PubMed]
- Frucht, S.J. Parkinson disease: An update. Neurologist 2004, 10, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for Lewy-body formation in Parkinson disease. Nat. Med. 2001, 7, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, R.; Fujishiro, H.; Maraganore, D.M.; Klos, K.J.; DelleDonne, A.; Heckman, M.G.; Crook, J.E.; Josephs, K.A.; Parisi, J.E.; Boeve, B.F.; et al. Comparison of risk factor profiles in incidental Lewy body disease and Parkinson disease. Arch. Neurol. 2009, 66, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Wood, H. Parkinson disease: Antibodies reveal age of Lewy pathology in PD. Nat. Rev. Neurol. 2017, 13, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Sendek, S.; Yang, C. Relationship between motor symptoms, cognition, and demographic characteristics in treated mild/moderate Parkinson’s disease. PLoS ONE 2015, 10, e0123231. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Bernhard, F.P.; Roeben, B.; Nussbaum, S.; Heger, T.; Martus, P.; Hobert, M.A.; Maetzler, W.; Berg, D. Progression markers of motor deficits in Parkinson’s disease: A biannual 4-year prospective study. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Kaplan, A.; Stockwell, B.R. Therapeutic approaches to preventing cell death in Huntington disease. Prog. Neurobiol. 2012, 99, 262–280. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.; McLear, J. The therapeutic potential of intrabodies in neurologic disorders: Focus on Huntington and Parkinson diseases. BioDrugs 2006, 20, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, G.; Cicchetti, F. Mutant huntingtin protein expression and blood-spinal cord barrier dysfunction in Huntington disease. Ann. Neurol. 2017, 82, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Hong, Y.; Li, X.J.; Li, S.H. Subcellular clearance and accumulation of Huntington disease protein: A mini-review. Front. Mol. Neurosci. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Diamond, M.I. Huntington disease and the huntingtin protein. Prog. Mol. Biol. Transl. Sci. 2012, 107, 189–214. [Google Scholar] [PubMed]
- Frank, S. Treatment of Huntington’s disease. Neurotherapeutics 2014, 11, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Koutsis, G.; Panas, M.; Paraskevas, G.P.; Bougea, A.M.; Kladi, A.; Karadima, G.; Kapaki, E. From mild ataxia to Huntington disease phenocopy: The multiple faces of spinocerebellar ataxia 17. Case Rep. Neurol. Med. 2014, 2014, 643289. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, R.H.; Smith, C.O.; Lim, J.G.; Bachman, D.F.; Byrd, E.; Bird, T.D. The unique co-occurrence of spinocerebellar ataxia type 10 (SCA10) and Huntington disease. J. Neurol. Sci. 2013, 324, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Van Hoesen, G.W. Neuron numbers in Alzheimer’s disease: Cell-specific pathology. Neurobiol. Aging 1987, 8, 555–556. [Google Scholar] [CrossRef]
- Bendfeldt, K.; Kappos, L.; Radue, E.W.; Borgwardt, S.J. Progression of gray matter atrophy and its association with white matter lesions in relapsing-remitting multiple sclerosis. J. Neurol. Sci. 2009, 285, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Colasanti, A.; Rabiner, E.A.; Gunn, R.N.; Malik, O.; Ciccarelli, O.; Nicholas, R.; Van Vlierberghe, E.; Van Hecke, W.; Searle, G.; et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain J. Neurol. 2017, 140, 2927–2938. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, Y.; Ciccarelli, O.; Hemingway, C. Abnormal white matter development in children with multiple sclerosis and monophasic acquired demyelination. Brain J. Neurol. 2017, 140, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Mechanisms of white matter damage in multiple sclerosis. Glia 2014, 62, 1816–1830. [Google Scholar] [CrossRef] [PubMed]
- Longoni, G.; Brown, R.A.; MomayyezSiahkal, P.; Elliott, C.; Narayanan, S.; Bar-Or, A.; Marrie, R.A.; Yeh, E.A.; Filippi, M.; Banwell, B.; et al. White matter changes in paediatric multiple sclerosis and monophasic demyelinating disorders. Brain J. Neurol. 2017, 140, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Josephy-Hernandez, S.; Jmaeff, S.; Pirvulescu, I.; Aboulkassim, T.; Saragovi, H.U. Neurotrophin receptor agonists and antagonists as therapeutic agents: An evolving paradigm. Neurobiol. Dis. 2017, 97, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Quarantelli, M.; Lanzillo, R.; Cocozza, S.; Carotenuto, A.; Carotenuto, B.; Alfano, B.; Prinster, A.; Triassi, M.; Nardone, A.; et al. Grey:White matter ratio at diagnosis and the risk of 10-year multiple sclerosis progression. Eur. J. Neurol. 2017, 24, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kamoshita, S.; Eto, Y.; Tourtellotte, W.W.; Gonatas, J.O. Myelin in multiple sclerosis. Composition of myelin from normal-appearing white matter. Arch. Neurol. 1973, 28, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Chalah, M.A.; Ayache, S.S. Psychiatric event in multiple sclerosis: Could it be the tip of the iceberg? Rev. Bras. Psiquiatr. 2017, 39, 365–368. [Google Scholar] [CrossRef] [PubMed]
- de Cerqueira, A.C.; Semionato de Andrade, P.; Godoy Barreiros, J.M.; Teixeira, A.L.; Nardi, A.E. Psychiatric disorders in patients with multiple sclerosis. Compr. Psychiatry 2015, 63, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Honer, W.G.; Hurwitz, T.; Li, D.K.; Palmer, M.; Paty, D.W. Temporal lobe involvement in multiple sclerosis patients with psychiatric disorders. Arch. Neurol. 1987, 44, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Zambon, A.A.; Cecchetti, G.; Caso, F.; Santangelo, R.; Baldoli, C.; Natali Sora, M.G.; Comi, G.; Magnani, G.; Martinelli, V. Primary progressive multiple sclerosis presenting with severe predominant cognitive impairment and psychiatric symptoms: A challenging case. Mult. Scler. 2017, 23, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Desikan, R.S.; Fan, C.C.; Wang, Y.; Schork, A.J.; Cabral, H.J.; Cupples, L.A.; Thompson, W.K.; Besser, L.; Kukull, W.A.; Holland, D.; et al. Genetic assessment of age-associated Alzheimer disease risk: Development and validation of a polygenic hazard score. PLoS Med. 2017, 14, e1002258. [Google Scholar] [CrossRef] [PubMed]
- Gaiteri, C.; Mostafavi, S.; Honey, C.J.; De Jager, P.L.; Bennett, D.A. Genetic variants in Alzheimer disease-molecular and brain network approaches. Nat. Rev. Neurol. 2016, 12, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hollingworth, P.; Moore, P.; Foy, C.; Archer, N.; Powell, J.; Nowotny, P.; Holmans, P.; O’Donovan, M.; Tacey, K.; et al. Genetic association of the app binding protein 2 gene (APBB2) with late onset Alzheimer disease. Hum. Mutat. 2005, 25, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.N.; Richter, R.W.; Risser, R.C.; Taubman, K.; Prado-Farmer, I.; Ebalo, E.; Posey, J.; Kingfisher, D.; Dean, D.; Weiner, M.F.; et al. Genetic factors for the development of Alzheimer disease in the Cherokee Indian. Arch. Neurol. 1996, 53, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.X.; Rosado, L.; Ross, B.M.; Verbitsky, M.; Kisselev, S.; Louis, E.D.; Comella, C.; et al. Frequency of known mutations in early-onset Parkinson disease: Implication for genetic counseling: The consortium on risk for early onset Parkinson disease study. Arch. Neurol. 2010, 67, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.K.; Staijich, J.M.; Yamaoka, L.H.; Speer, M.C.; Vance, J.M.; Roses, A.D.; Pericak-Vance, M.A. Genetic complexity and Parkinson’s disease. Deane laboratory Parkinson disease research group. Science (New York) 1997, 277, 387–388. [Google Scholar] [CrossRef]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P. History of genetic disease: The molecular genetics of Huntington disease—A history. Nat. Rev. Genet. 2005, 6, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Pericak-Vance, M.A.; Conneally, P.M.; Merritt, A.D.; Roos, R.; Norton, J.A., Jr.; Vance, J.M. Genetic linkage studies in Huntington disease. Cytogenet. Cell Genet. 1978, 22, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M. Neurodegenerative disease. Genetic discrimination in Huntington disease. Nat. Rev. Neurol. 2009, 5, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Barral, S.; Lee, J.H.; Leurgans, S.E.; Foroud, T.M.; Sweet, R.A.; Graff-Radford, N.; Bird, T.D.; Mayeux, R.; Bennett, D.A. Heritability of different forms of memory in the late onset Alzheimer’s disease family study. J. Alzheimer’s Dis. 2011, 23, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Ebers, G.C.; Sadovnick, A.D. Genetics of multiple sclerosis. Lancet Neurol. 2004, 3, 104–110. [Google Scholar] [CrossRef]
- Axisa, P.P.; Hafler, D.A. Multiple sclerosis: Genetics, biomarkers, treatments. Curr. Opin. Neurol. 2016, 29, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Oksenberg, J.R. The genetics of multiple sclerosis: From 0 to 200 in 50 years. Trends Genet. 2017, 33, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, R.; Fumagalli, M.; Guerini, F.R.; Riva, S.; Galimberti, D.; Comi, G.P.; Agliardi, C.; Scarpini, E.; Pozzoli, U.; Forni, D.; et al. Identification of a new susceptibility variant for multiple sclerosis in OAS1 by population genetics analysis. Hum. Genet. 2012, 131, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tizaoui, K. Multiple sclerosis genetics: Results from meta-analyses of candidate-gene association studies. Cytokine 2017. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Fransquet, P.; Wrigglesworth, J.; Lacaze, P. Phenotypic heterogeneity in dementia: A challenge for epidemiology and biomarker studies. Front. Public Health 2018, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Head, E.; Schmitt, F.A.; Davis, P.R.; Neltner, J.H.; Jicha, G.A.; Abner, E.L.; Smith, C.D.; Van Eldik, L.J.; Kryscio, R.J. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011, 121, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Petrovitch, H.; White, L.; Ross, G.; Steinhorn, S.; Li, C.; Masaki, K.; Davis, D.; Nelson, J.; Hardman, J.; Curb, J. Accuracy of clinical criteria for ad in the Honolulu–Asia aging study, a population-based study. Neurology 2001, 57, 226–234. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.; Lowe, J.; Emre, M.; O’brien, J.; Feldman, H.; Cummings, J.; Duda, J.; Lippa, C.; Perry, E. Diagnosis and management of dementia with Lewy bodies third report of the DLB consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Morales, R.; Esteller, M. Opening up the DNA methylome of dementia. Mol. Psychiatry 2017, 22, 485. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Karlawish, J.; Johnson, K.A. Preclinical Alzheimer disease—The challenges ahead. Nat. Rev. Neurol. 2013, 9, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Harris, C.R.; Puzio-Kuter, A.M. The interfaces between signal transduction pathways: IGF-1/mTor, p53 and the Parkinson disease pathway. Oncotarget 2012, 3, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. 2012, 4, 941–952. [Google Scholar] [CrossRef]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR signaling in Parkinson’s disease. Neuromol. Med. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mtor signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Valjent, E.; Fisone, G. Mtorc1 signaling in Parkinson’s disease and L-DOPA-induced dyskinesia: A sensitized matter. Cell Cycle 2010, 9, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chen, G.; He, W.; Xiao, M.; Yan, L.J. Activation of mTOR: A culprit of Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2015, 11, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, J.T.; Miao, D.; Wu, Z.C.; Tan, M.S.; Tan, L. Targeting the mTOR signaling network for Alzheimer’s disease therapy. Mol. Neurobiol. 2014, 49, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR inhibitor rapamycin mitigates perforant pathway neurodegeneration and synapse loss in a mouse model of early-stage Alzheimer-type tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORc1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hohlfeld, R.; Wekerle, H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: From pipe dreams to (therapeutic) pipelines. Proc. Natl. Acad. Sci. USA 2004, 101 (Suppl. 2), 14599–14606. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain J. Neurol. 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Mtor activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 2015, 1346, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; De Rosa, V.; Carrieri, P.B.; Montella, S.; Bruzzese, D.; Porcellini, A.; Procaccini, C.; La Cava, A.; Matarese, G. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat. Med. 2014, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. Mtorc1 and mTORc2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORc1 and mTORc2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Tyler, W.A.; Gangoli, N.; Gokina, P.; Kim, H.A.; Covey, M.; Levison, S.W.; Wood, T.L. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6367–6378. [Google Scholar] [CrossRef] [PubMed]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and cns myelination. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [PubMed]
- Lebrun-Julien, F.; Bachmann, L.; Norrmen, C.; Trotzmuller, M.; Kofeler, H.; Ruegg, M.A.; Hall, M.N.; Suter, U. Balanced mTORc1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8432–8448. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Lengfeld, J.E.; Lutz, S.E.; Smith, J.R.; Diaconu, C.; Scott, C.; Kofman, S.B.; Choi, C.; Walsh, C.M.; Raine, C.S.; Agalliu, I.; et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1168–E1177. [Google Scholar] [CrossRef] [PubMed]
- Bayat, V.; Jaiswal, M.; Bellen, H.J. The bmp signaling pathway at the drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr. Opin. Neurobiol. 2011, 21, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Holbert, S.; Dedeoglu, A.; Humbert, S.; Saudou, F.; Ferrante, R.J.; Neri, C. Cdc42-interacting protein 4 binds to huntingtin: Neuropathologic and biological evidence for a role in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2712–2717. [Google Scholar] [CrossRef] [PubMed]
- Lione, L.A.; Carter, R.J.; Hunt, M.J.; Bates, G.P.; Morton, A.J.; Dunnett, S.B. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 10428–10437. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Scheff, M.A.; Mufson, E.J. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2011, 24, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Meyermann, R.; Schluesener, H. Detection of two transforming growth factor-beta-related morphogens, bone morphogenetic proteins-4 and -5, in RNA of multiple sclerosis and Creutzfeldt-Jakob disease lesions. Acta Neuropathol. 1995, 90, 76–79. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Avila, M.E.; Medina, M.A.; Perez-Palma, E.; Bustos, B.I.; Alarcon, M.A. Wnt/beta-catenin signaling in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 745–754. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Serapide, M.F.; Pluchino, S.; Marchetti, B. Wnt/B-catenin signaling is required to rescue midbrain dopaminergic progenitors and promote neurorepair in ageing mouse model of Parkinson’s disease. Stem Cells (Dayton, Ohio) 2014, 32, 2147–2163. [Google Scholar] [CrossRef] [PubMed]
- Finkel, S.I. Effects of rivastigmine on behavioral and psychological symptoms of dementia in Alzheimer’s disease. Clin. Ther. 2004, 26, 980–990. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeong, S.K.; Kim, B.; Park, K.; Dash, A. Donepezil across the spectrum of Alzheimer’s disease: Dose optimization and clinical relevance. Acta Neurol. Scand. 2015, 131, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Quinn, N. Drug treatment of Parkinson’s disease. BMJ Br. Med J. 1995, 310, 575. [Google Scholar] [CrossRef]
- Bonuccelli, U.; Colzi, A.; Del Dotto, P. Pergolide in the treatment of patients with early and advanced Parkinson’s disease. Clin. Neuropharmacol. 2002, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, W.-D.; Jankovic, J. Are dopamine receptor agonists neuroprotective in Parkinson’s disease? Drugs Aging 2001, 18, 389–396. [Google Scholar] [CrossRef] [PubMed]
- McMurray, C.T. Huntington’s disease: New hope for therapeutics. Trends Neurosci. 2001, 24, S32–S38. [Google Scholar] [CrossRef]
- Faissner, S.; Gold, R. Oral therapies for multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stangel, M.; Kuhlmann, T.; Matthews, P.M.; Kilpatrick, T.J. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Zeydan, B.; Rodriguez, M.; Kantarci, O.H. Timing of future remyelination therapies and their potential to stop multiple sclerosis progression. Adv. Exp. Med. Biol. 2017, 958, 161–170. [Google Scholar] [PubMed]
- Plemel, J.R.; Liu, W.Q.; Yong, V.W. Remyelination therapies: A new direction and challenge in multiple sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Herczenik, E.; Gebbink, M.F. Molecular and cellular aspects of protein misfolding and disease. FASEB J. 2008, 22, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Smith, M.A.; Sayre, L.M.; Monnier, V.M.; Perry, G. Radical ageing in Alzheimer’s disease. Trends Neurosci. 1995, 18, 172–176. [Google Scholar] [CrossRef]
- Greenbaum, E.A.; Graves, C.L.; Mishizen-Eberz, A.J.; Lupoli, M.A.; Lynch, D.R.; Englander, S.W.; Axelsen, P.H.; Giasson, B.I. The E46K mutation in α-synuclein increases amyloid fibril formation. J. Boil. Chem. 2005, 280, 7800–7807. [Google Scholar] [CrossRef] [PubMed]
- Azuaga, A.I.; Dobson, C.M.; Mateo, P.L.; Conejero-Lara, F. Unfolding and aggregation during the thermal denaturation of streptokinase. FEBS J. 2002, 269, 4121–4133. [Google Scholar]
- Iametti, S.; Gregori, B.; Vecchio, G.; Bonomi, F. Modifications occur at different structural levels during the heat denaturation of β-lactoglobulin. FEBS J. 1996, 237, 106–112. [Google Scholar] [CrossRef]
- Giri, K.; Bhattacharyya, N.P.; Basak, S. Ph-dependent self-assembly of polyalanine peptides. Biophys. J. 2007, 92, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.K.; Alam, P.; Khan, J.M.; Siddiqui, M.K.; Kalaiarasan, P.; Subbarao, N.; Ahmad, Z.; Khan, R.H. Biophysical insight into the anti-amyloidogenic behavior of taurine. Int. J. Boil. Macromol. 2015, 80, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science (New York) 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Chen, S.; Brown, I.R. Neuronal expression of constitutive heat shock proteins: Implications for neurodegenerative diseases. Cell Stress Chaperones 2007, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; Shorter, J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 2015, 9, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.; Burke, R.; Doig, A. Inhibition of toxicity in the Alzheimer’s disease peptide fragment β (25–35) using N-methylated derivatives. Biochem. Soc. Trans. 2000, 28, A72. [Google Scholar] [CrossRef]
- Madine, J.; Doig, A.J.; Middleton, D.A. Design of an N-methylated peptide inhibitor of α-synuclein aggregation guided by solid-state NMR. J. Am. Chem. Soc. 2008, 130, 7873–7881. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.-C.; Tan, M.-S.; Wang, H.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat shock protein 70 in Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 435203. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 2011, 31, 5225–5234. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, T.; Rokutan, K.; Nikawa, T.; Kishi, K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 1996, 111, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.M.; Tang, D.W.; Hanif, A.; Brown, I.R. Induction of heat shock proteins in cerebral cortical cultures by celastrol. Cell Stress Chaperones 2013, 18, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Bobkova, N.V.; Garbuz, D.G.; Nesterova, I.; Medvinskaya, N.; Samokhin, A.; Alexandrova, I.; Yashin, V.; Karpov, V.; Kukharsky, M.S.; Ninkina, N.N. Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Vinokurov, M.; Ostrov, V.; Yurinskaya, M.; Garbuz, D.; Murashev, A.; Antonova, O.; Evgen’ev, M. Recombinant human hsp70 protects against lipoteichoic acid-induced inflammation manifestations at the cellular and organismal levels. Cell Stress Chaperones 2012, 17, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, S.B.; Kashlan, O.B.; Watkins, J.N.; Suaud, L.; Yan, W.; Kleyman, T.R.; Rubenstein, R.C. Differential effects of hsc70 and hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc. Natl. Acad. Sci. USA 2006, 103, 5817–5822. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary III, J.C.; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M. Imbalance of hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Dynamic remodeling of transcription complexes by molecular chaperones. Cell 2002, 110, 281–284. [Google Scholar] [CrossRef]
- Kim, S.A.; Yoon, J.H.; Lee, S.H.; Ahn, S.G. Polo-like kinase 1 phosphorylates heat shock transcription factor 1 and mediates its nuclear translocation during heat stress. J. Boil. Chem. 2005, 280, 12653–12657. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mivechi, N.F. Hsf-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J. Boil. Chem. 2003, 278, 17299–17306. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guettouche, T.; Fenna, M.; Boellmann, F.; Pratt, W.B.; Toft, D.O.; Smith, D.F.; Voellmy, R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Boil. Chem. 2001, 276, 45791–45799. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, I.; Shorter, J.; Wiseman, R.L.; Chiti, F.; Dickey, C.A.; McLean, P.J. Chaperones in neurodegeneration. J. Neurosci. 2015, 35, 13853–13859. [Google Scholar] [CrossRef] [PubMed]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Shorter, J. Engineering therapeutic protein disaggregases. Mol. Boil. Cell 2016, 27, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. Hsp104: A weapon to combat diverse neurodegenerative disorders. Neurosignals 2008, 16, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; Shorter, J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Model Mech. 2014, 7, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lim, H.S.; Masliah, E.; Lee, H.J. Protein aggregate spreading in neurodegenerative diseases: Problems and perspectives. Neurosci. Res. 2011, 70, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta (1-42) aggregation in vitro. J. Boil. Chem. 2006. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of huntingtin in health and disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, M.M.; Christodoulou, J.; Wilson, M.R.; Dobson, C.M. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J. Boil. Chem. 2005, 280, 14733–14740. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Molecular chaperones in Parkinson’s disease—Present and future. J. Park. Dis. 2011, 1, 299–320. [Google Scholar]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 1998, 42, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Y.; He, J.C.; Wang, Y.; Huang, Q.Y.; Chen, J.F. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J. Boil. Chem. 2005, 280, 39962–39969. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein quality control by molecular chaperones in neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; O’Reilly, E.J.; Schwarzschild, M.A.; Ascherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.-C.; Arlicot, N. Molecular targets for pet imaging of activated microglia: The current situation and future expectations. Int. J. Mol. Sci. 2017, 18, 802. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain J. Neurol. 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.P.; Ribeiro, F.M.; Furr-Stimming, E.; Teixeira, A.L. Neuroimmunology of Huntington’s disease: Revisiting evidence from human studies. Mediat. Inflamm. 2016, 2016, 8653132. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-M.; Yang, S.; Huang, S.-S.; Tang, B.-S.; Guo, J.-F. Microglial activation in the pathogenesis of Huntington’s disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Wu, Y.R.; Chen, Y.C.; Chen, C.M. Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Lahiri, N.; Niccolini, F.; Su, P.; Wu, K.; Giannetti, P.; Scahill, R.I.; Turkheimer, F.E.; Tabrizi, S.J.; Piccini, P. Increased central microglial activation associated with peripheral cytokine levels in premanifest Huntington’s disease gene carriers. Neurobiol. Dis. 2015, 83, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Träger, U.; Andre, R.; Lahiri, N.; Magnusson-Lind, A.; Weiss, A.; Grueninger, S.; McKinnon, C.; Sirinathsinghji, E.; Kahlon, S.; Pfister, E.L. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain J. Neurol. 2014, 137, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: Evidence from observational studies. Cochrane Database Syst. Rev. 2011, Cd008454. [Google Scholar]
- Du, Y.; Ma, Z.; Lin, S.; Dodel, R.C.; Gao, F.; Bales, K.R.; Triarhou, L.C.; Chernet, E.; Perry, K.W.; Nelson, D.L.; et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 14669–14674. [Google Scholar] [CrossRef] [PubMed]
- Lofrumento, D.D.; Nicolardi, G.; Cianciulli, A.; Nuccio, F.D.; Pesa, V.L.; Carofiglio, V.; Dragone, T.; Calvello, R.; Panaro, M.A. Neuroprotective effects of resveratrol in an MPTP mouse model of Parkinson’s-like disease: Possible role of socs-1 in reducing pro-inflammatory responses. Innate Immun. 2014, 20, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shi, J.-S.; Zhou, H.; Wilson, B.C.; Hong, J.-S.; Gao, H.-M. Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol. Pharmacol. 2010, 94. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Zhang, Y.-X.; Zhou, H.-X.; Sun, F.-W.; Zhang, Z.-F.; Wei, Z.-F.; Zhang, C.-Y.; Si, D.-W. Tanshinone IIA prevents the loss of nigrostriatal dopaminergic neurons by inhibiting NADPH oxidase and iNOS in the MPTP model of Parkinson’s disease. J. Neurol. Sci. 2015, 348, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jing, H.; Yang, H.; Liu, Z.; Guo, H.; Chai, L.; Hu, L. Tanshinone I selectively suppresses pro-inflammatory genes expression in activated microglia and prevents nigrostriatal dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. J. Ethnopharmacol. 2015, 164, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Borah, A.; Paul, R.; Choudhury, S.; Choudhury, A.; Bhuyan, B.; Das Talukdar, A.; Dutta Choudhury, M.; Mohanakumar, K.P. Neuroprotective potential of silymarin against CNS disorders: Insight into the pathways and molecular mechanisms of action. CNS Neurosci. Ther. 2013, 19, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Zhang, Y.; Santamaria, P.; Olson, K.E.; Schutt, C.R.; Bhatti, D.; Shetty, B.L.D.; Lu, Y.; Estes, K.A.; Standaert, D.G. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Park. Dis. 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Dobson, L.; Träger, U.; Farmer, R.; Hayardeny, L.; Loupe, P.; Hayden, M.R.; Tabrizi, S.J. Laquinimod dampens hyperactive cytokine production in Huntington’s disease patient myeloid cells. J. Neurochem. 2016, 137, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Na, S.Y.; Mracsko, E.; Liesz, A.; Hunig, T.; Veltkamp, R. Amplification of regulatory t cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke 2015, 46, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Lee, G.; Lee, C.; Ye, M.; Chung, H.S.; Kim, H.; Bae, S.J.; Hwang, D.S.; Bae, H. Bee venom phospholipase A2, a novel Foxp3+ regulatory t cell inducer, protects dopaminergic neurons by modulating neuroinflammatory responses in a mouse model of Parkinson’s disease. J. Immunol. 2015, 195, 4853–4860. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h [A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Williams, A.D.; McWilliams-Koeppen, H.P.; Acero, L.; Weber, A.; Ehrlich, H.; Schwarz, H.P.; Solomon, A. Anti-amyloidogenic activity of IgGs contained in normal plasma. J. Clin. Immunol. 2010, 30, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay, R.; Pensalfini, A.; Yeung, S.; Head, E. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 2010, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.H.; Shaw, P. Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 2007, 100, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.-L.; Klionsky, D.J. How to live long and prosper: Autophagy, mitochondria, and aging. Physiology 2008, 23, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective pharmacogenetic inhibition of mammalian target of rapamycin complex I (mTORc1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Pupyshev, A.B.; Korolenko, T.A.; Tikhonova, M.A. A therapeutic target for inhibition of neurodegeneration: Autophagy. Neurosci. Behav. Physiol. 2017, 47, 1109–1127. [Google Scholar] [CrossRef]
- Nah, J.; Yuan, J.; Jung, Y.-K. Autophagy in neurodegenerative diseases: From mechanism to therapeutic approach. Mol. Cells 2015, 38, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Wang, C.; Peng, X.; Gan, B.; Guan, J.-L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Boil. Chem. 2010, 285, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Panhelainen, A.; Schmitz, Y.; Larsen, K.E.; Kanter, E.; Wu, M.; Sulzer, D.; Mosharov, E.V. Changes in neuronal dopamine homeostasis following 1-methyl-4-phenylpyridinium (MPP+) exposure. J. Boil. Chem. 2015, 290, 6799–6809. [Google Scholar] [CrossRef] [PubMed]
- Javitch, J.A.; D’Amato, R.J.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Gandy, S. Latrepirdine (Dimebon(R)), a potential alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy 2013, 9, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Chau, S.; Herrmann, N.; Ruthirakuhan, M.T.; Chen, J.J.; Lanctot, K.L. Latrepirdine for Alzheimer’s Disease; The Cochrane Library: London, UK, 2015. [Google Scholar]
- Forlenza, O.V.; de Paula, V.J.; Machado-Vieira, R.; Diniz, B.S.; Gattaz, W.F. Does lithium prevent Alzheimer’s disease? Drugs Aging 2012, 29, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’kane, C.J. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2005, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lao, U.; Edgar, B.A. Tor-mediated autophagy regulates cell death in drosophila neurodegenerative disease. J. Cell Boil. 2009, 186, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.-H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.-J.; Kim, J.S. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau: Effects on cognitive impairments. J. Boil. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wei, L.-L.; Zhang, M.; Li, T.-X.; Yang, C.; Deng, S.-P.; Zeng, Q.-C. Rapamycin inhibits activation of ampk-mTOR signaling pathway-induced Alzheimer’s disease lesion in hippocampus of rats with type 2 diabetes mellitus. Int. J. Neurosci. 2018, 2018, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. Tor signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Buescher, J.L.; Oatis, B.; Funk, J.A.; Nash, A.J.; Carrier, R.L.; Hoyt, K.R. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci. Lett. 2007, 411, 98–103. [Google Scholar]
- Goedert, M.; Jakes, R.; Qi, Z.; Wang, J.H.; Cohen, P. Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic amp-dependent protein kinase. J Neurochem. 1995, 65, 2804–2807. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Olopade, P.; Selby, S.T.; Pagan, F.; Moussa, C.E. Tyrosine kinase inhibition regulates early systemic immune changes and modulates the neuroimmune response in α-synucleinopathy. J. Clin. Cell. Immunol. 2014, 5, 259. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.; Selby, S.; Turner, R.; Moussa, C.-H. Nilotinib and Bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer’s disease models. Neuroscience 2015, 304, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science (New York) 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M. Neuroscience. Garbage truck of the brain. Science (New York) 2013, 340, 1529–1530. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Goldman, S.A. Brain drain. Sci. Am. 2016, 314, 44–49. [Google Scholar] [CrossRef] [PubMed]
- de Leon, M.J.; Li, Y.; Okamura, N.; Tsui, W.H.; Saint Louis, L.A.; Glodzik, L.; Osorio, R.S.; Fortea, J.; Butler, T.; Pirraglia, E.; et al. CSF clearance in Alzheimer disease measured with dynamic pet. J. Nucl. Med. 2017, 58, 1471. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Madison, C.; Baker, S.; Rabinovici, G.; Jagust, W. Dynamic relationships between age, amyloid-beta deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain J. Neurol. 2016, 139, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Neurogenesis in the adult brain. J. Neurosci. 2002, 22, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York) 1992, 255, 1707–1710. [Google Scholar] [CrossRef]
- Shihabuddin, L.S.; Horner, P.J.; Ray, J.; Gage, F.H. Adult spinal cord stem cells generate neurons after transplantation in the adult dentate gyrus. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 8727–8735. [Google Scholar] [CrossRef]
- Pencea, V.; Bingaman, K.D.; Freedman, L.J.; Luskin, M.B. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp. Neurol. 2001, 172, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The neurotrophins and their role in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M.; Altar, C.A.; Cedarbaum, J.M.; Hyman, C.; Wiegand, S.J. The therapeutic potential of neurotrophic factors in the treatment of Parkinson’s disease. Exp. Neurol. 1993, 124, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Grondin, R.; Gash, D.M. Glial cell line-derived neurotrophic factor (GDNF): A drug candidate for the treatment of Parkinson’s disease. J. Neurol. 1998, 245, P35–P42. [Google Scholar] [CrossRef] [PubMed]
- Bowenkamp, K.E.; Lapchak, P.A.; Hoffer, B.J.; Miller, P.J.; Bickford, P.C. Intracerebroventricular glial cell line-derived neurotrophic factor improves motor function and supports nigrostriatal dopamine neurons in bilaterally 6-hydroxydopamine lesioned rats. Exp. Neurol. 1997, 145, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: Stress and depression. Dialogues Clin. Neurosci. 2009, 11, 239–255. [Google Scholar] [PubMed]
- Tsiperson, V.; Huang, Y.; Bagayogo, I.; Song, Y.; VonDran, M.W.; DiCicco-Bloom, E.; Dreyfus, C.F. Brain-derived neurotrophic factor deficiency restricts proliferation of oligodendrocyte progenitors following cuprizone-induced demyelination. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef] [PubMed]
- Gold, S.M.; Chalifoux, S.; Giesser, B.S.; Voskuhl, R.R. Immune modulation and increased neurotrophic factor production in multiple sclerosis patients treated with testosterone. J. Neuroinflamm. 2008, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.E. Role of neurotrophic factors in neuronal development. Curr. Opin. Neurobiol. 1996, 6, 64–70. [Google Scholar] [CrossRef]
- Poo, M.M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.X.; Kim, C.K.; Cho, J.-H.; Lee, K.-H.; Ahn, J.-Y. Neuroprotection signaling pathway of nerve growth factor and brain-derived neurotrophic factor against staurosporine induced apoptosis in hippocampal H19-7 cells. Exp. Mol. Med. 2010, 42, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Middlemas, D.S.; Kihl, B.K.; Zhou, J.; Zhu, X. Brain-derived neurotrophic factor promotes survival and chemoprotection of human neuroblastoma cells. J. Boil. Chem. 1999, 274, 16451–16460. [Google Scholar] [CrossRef]
- Weissmiller, A.M.; Wu, C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl. Neurodegener. 2012, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Delcroix, J.D.; Swaab, D.F. Alzheimer’s disease and NGF signaling. J. Neural Transm. (Vienna, Austria 1996) 2004, 111, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Mufson, E.J. Ngf receptor gene expression is decreased in the nucleus basalis in Alzheimer’s disease. Exp. Neurol. 1989, 106, 222–236. [Google Scholar] [CrossRef]
- Costa, A.; Peppe, A.; Carlesimo, G.A.; Zabberoni, S.; Scalici, F.; Caltagirone, C.; Angelucci, F. Brain-derived neurotrophic factor serum levels correlate with cognitive performance in Parkinson’s disease patients with mild cognitive impairment. Front. Behav. Neurosci. 2015, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced bdnf mrna expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Kastin, A.J. Permeability of the blood–brain barrier to neurotrophins. Brain Res. 1998, 788, 87–94. [Google Scholar] [CrossRef]
- Pollack, S.J.; Harper, S.J. Small molecule Trk receptor agonists and other neurotrophic factor mimetics. Curr. Drug Targets CNS Neurol. Disord. 2002, 1, 59–80. [Google Scholar] [CrossRef] [PubMed]
- Baulieu, E.E.; Robel, P. Neurosteroids: A new brain function? J. Steroid Biochem. Mol. Boil. 1990, 37, 395–403. [Google Scholar] [CrossRef]
- Lv, W.; Du, N.; Liu, Y.; Fan, X.; Wang, Y.; Jia, X.; Hou, X.; Wang, B. Low testosterone level and risk of Alzheimer’s disease in the elderly men: A systematic review and meta-analysis. Mol. Neurobiol. 2016, 53, 2679–2684. [Google Scholar] [CrossRef] [PubMed]
- Paganini-Hill, A.; Henderson, V.W. Estrogen deficiency and risk of Alzheimer’s disease in women. Am. J. Epidemiol. 1994, 140, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.C.; Rosario, E.R.; Villamagna, A.; Pike, C.J. Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer-like pathology in female 3xtransgenic-Alzheimer’s disease mice. Endocrinology 2010, 151, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Pike, C.J. Age-related testosterone depletion and the development of Alzheimer disease. JAMA 2004, 292, 1431–1432. [Google Scholar] [CrossRef] [PubMed]
- Seidl, J.N.; Massman, P.J. Relationships between testosterone levels and cognition in patients with Alzheimer disease and nondemented elderly men. J. Geriatr. Psychiatry Neurol. 2015, 28, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; DeLong, M.R.; Hanfelt, J.; Gearing, M.; Levey, A. Plasma testosterone levels in Alzheimer and Parkinson diseases. Neurology 2004, 62, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; McDonald, W.M.; DeLong, M.R. Refractory nonmotor symptoms in male patients with Parkinson disease due to testosterone deficiency: A common unrecognized comorbidity. Arch. Neurol. 2002, 59, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Markianos, M.; Panas, M.; Kalfakis, N.; Vassilopoulos, D. Plasma testosterone in male patients with Huntington’s disease: Relations to severity of illness and dementia. Ann. Neurol. 2005, 57, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Bove, R.; Musallam, A.; Healy, B.C.; Raghavan, K.; Glanz, B.I.; Bakshi, R.; Weiner, H.; De Jager, P.L.; Miller, K.K.; Chitnis, T. Low testosterone is associated with disability in men with multiple sclerosis. Mult. Scler. 2014, 20, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Trenova, A.G.; Slavov, G.S.; Manova, M.G.; Kostadinova, II; Vasileva, T.V. Female sex hormones and cytokine secretion in women with multiple sclerosis. Neurol. Res. 2013, 35, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska-Pniewska, B.; Golebiowski, M.; Zajda, M.; Szeszkowski, W.; Podlecka-Pietowska, A.; Nojszewska, M. Sex hormone patterns in women with multiple sclerosis as related to disease activity—A pilot study. Neurol. I Neurochir. Polska 2011, 45, 536–542. [Google Scholar] [CrossRef]
- Baker, L.D.; Sambamurti, K.; Craft, S.; Cherrier, M.; Raskind, M.A.; Stanczyk, F.Z.; Plymate, S.R.; Asthana, S. 17beta-estradiol reduces plasma Abeta40 for HRT-naive postmenopausal women with Alzheimer disease: A preliminary study. Am. J. Geriatr. Psychiatry 2003, 11, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Valen-Sendstad, A.; Engedal, K.; Stray-Pedersen, B.; Group, A.S.; Strobel, C.; Barnett, L.; Meyer, N.; Nurminemi, M. Effects of hormone therapy on depressive symptoms and cognitive functions in women with Alzheimer disease: A 12 month randomized, double-blind, placebo-controlled study of low-dose estradiol and norethisterone. Am. J. Geriatr. Psychiatry 2010, 18, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Cherrier, M.M.; Matsumoto, A.M.; Amory, J.K.; Asthana, S.; Bremner, W.; Peskind, E.R.; Raskind, M.A.; Craft, S. Testosterone improves spatial memory in men with Alzheimer disease and mild cognitive impairment. Neurology 2005, 64, 2063–2068. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.H.; Masterman, D.A.; Mulnard, R.; Cotman, C.; Miller, B.; Yaffe, K.; Reback, E.; Porter, V.; Swerdloff, R.; Cummings, J.L. Effects of testosterone on cognition and mood in male patients with mild Alzheimer disease and healthy elderly men. Arch. Neurol. 2006, 63, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Moffat, S.D.; Zonderman, A.B.; Metter, E.J.; Kawas, C.; Blackman, M.R.; Harman, S.M.; Resnick, S.M. Free testosterone and risk for Alzheimer disease in older men. Neurology 2004, 62, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Carruthers, M. Testosterone as the missing link between pesticides, Alzheimer disease, and Parkinson disease. JAMA Neurol. 2014, 71, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; Fernandez, H.H.; Rodriguez, R.L.; Romrell, J.; Suelter, M.; Munson, S.; Louis, E.D.; Mulligan, T.; Foster, P.S.; Shenal, B.V.; et al. Testosterone therapy in men with Parkinson disease: Results of the test-PD study. Arch. Neurol. 2006, 63, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; Walter, B.L.; McDonald, W.M.; Tenover, J.L.; Green, J.; Juncos, J.L.; DeLong, M.R. Beneficial effects of testosterone replacement for the nonmotor symptoms of Parkinson disease. Arch. Neurol. 2002, 59, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- Gold, S.M.; Voskuhl, R.R. Estrogen and testosterone therapies in multiple sclerosis. Prog. Brain Res. 2009, 175, 239–251. [Google Scholar] [PubMed]
- Kurth, F.; Luders, E.; Sicotte, N.L.; Gaser, C.; Giesser, B.S.; Swerdloff, R.S.; Montag, M.J.; Voskuhl, R.R.; Mackenzie-Graham, A. Neuroprotective effects of testosterone treatment in men with multiple sclerosis. Neuroimage Clin. 2014, 4, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Sicotte, N.L.; Giesser, B.S.; Tandon, V.; Klutch, R.; Steiner, B.; Drain, A.E.; Shattuck, D.W.; Hull, L.; Wang, H.J.; Elashoff, R.M.; et al. Testosterone treatment in multiple sclerosis: A pilot study. Arch. Neurol. 2007, 64, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Vukusic, S.; Ionescu, I.; El-Etr, M.; Schumacher, M.; Baulieu, E.E.; Cornu, C.; Confavreux, C.; Prevention of Post-Partum Relapses with Progestin; Estradiol in Multiple Sclerosis Study Group. The prevention of post-partum relapses with progestin and estradiol in multiple sclerosis (POPART’MUS) trial: Rationale, objectives and state of advancement. J. Neurol. Sci. 2009, 286, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Palaszynski, K.M.; Loo, K.K.; Ashouri, J.F.; Liu, H.B.; Voskuhl, R.R. Androgens are protective in experimental autoimmune encephalomyelitis: Implications for multiple sclerosis. J. Neuroimmunol. 2004, 146, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Page, S.T.; Plymate, S.R.; Bremner, W.J.; Matsumoto, A.M.; Hess, D.L.; Lin, D.W.; Amory, J.K.; Nelson, P.S.; Wu, J.D. Effect of medical castration on CD4+ CD25+ T cells, CD8+ T cell IFN-gamma expression, and NK cells: A physiological role for testosterone and/or its metabolites. Am. J. Physiology. Endocrinol. Metab. 2006, 290, E856–E863. [Google Scholar] [CrossRef] [PubMed]
- Malkin, C.J.; Pugh, P.J.; Jones, R.D.; Kapoor, D.; Channer, K.S.; Jones, T.H. The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J. Clin. Endocrinol. Metab. 2004, 89, 3313–3318. [Google Scholar] [CrossRef] [PubMed]
- Liva, S.M.; Voskuhl, R.R. Testosterone acts directly on CD4+ T lymphocytes to increase IL-10 production. J. Immunol. (Baltimore, Md. 1950) 2001, 167, 2060–2067. [Google Scholar] [CrossRef]
- Simpkins, J.W.; Singh, M.; Brock, C.; Etgen, A.M. Neuroprotection and estrogen receptors. Neuroendocrinology 2012, 96, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.N.; Dorsa, D.M. Roles of estrogen receptors alpha and beta in sexually dimorphic neuroprotection against glutamate toxicity. Neuroscience 2010, 170, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Deecher, D.C.; Daoud, P.; Bhat, R.A.; O’Connor, L.T. Endogenously expressed estrogen receptors mediate neuroprotection in hippocampal cells (HT22). J. Cell. Biochem. 2005, 95, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Labombarda, F.; Ghoumari, A.M.; Liere, P.; De Nicola, A.F.; Schumacher, M.; Guennoun, R. Neuroprotection by steroids after neurotrauma in organotypic spinal cord cultures: A key role for progesterone receptors and steroidal modulators of Gaba(A) receptors. Neuropharmacology 2013, 71, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Margaill, I.; Zhang, S.; Labombarda, F.; Coqueran, B.; Delespierre, B.; Liere, P.; Marchand-Leroux, C.; O’Malley, B.W.; Lydon, J.P.; et al. Progesterone receptors: A key for neuroprotection in experimental stroke. Endocrinology 2012, 153, 3747–3757. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.B.; Wise, P.M. Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology 2001, 142, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, M.T.; Marino, M. Estrogen/huntingtin: A novel pathway involved in neuroprotection. Neural Regen. Res. 2016, 11, 402–403. [Google Scholar] [PubMed]
- Shen, D.; Tian, X.; Zhang, B.; Song, R. Mechanistic evaluation of neuroprotective effect of estradiol on rotenone and 6-ohda induced Parkinson’s disease. Pharmacol. Rep. PR 2017, 69, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Dahodwala, N. Neuroprotection by sex steroid hormones in Parkinson’s disease (P3.068). Neurology 2014, 82. [Google Scholar]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of estrogen and other sex hormones in brain aging. Neuroprotection and DNA repair. Front. Aging Neurosci. 2017, 9, 430. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.M.; Galea, L.A. Repeated estradiol administration alters different aspects of neurogenesis and cell death in the hippocampus of female, but not male, rats. Neuroscience 2008, 152, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.L.; Waters, E.M.; Romeo, R.D.; Wood, G.E.; Milner, T.A.; McEwen, B.S. Uncovering the mechanisms of estrogen effects on hippocampal function. Front. Neuroendocr. 2008, 29, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J.G.; Wolfe, C.A.; Tobet, S.A. Estrogen modulates neuronal movements within the developing preoptic area/anterior hypothalamus. Eur. J. Neurosci. 2007, 26, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Tiwari-Woodruff, S.; Morales, L.B.J.; Lee, R.; Voskuhl, R.R. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)α and ERβ ligand treatment. Proc. Natl. Acad. Sci. USA 2007, 104, 14813–14818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-G.; Wang, R.; Tang, H.; Dong, Y.; Chan, A.; Sareddy, G.R.; Vadlamudi, R.K.; Brann, D.W. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol. Cell. Endocrinol. 2014, 389, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Shivers, K.-Y.; Amador, N.; Abrams, L.; Hunter, D.; Jenab, S.; Quiñones-Jenab, V. Estrogen alters baseline and inflammatory-induced cytokine levels independent from hypothalamic–pituitary–adrenal axis activity. Cytokine 2015, 72, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Mulcahey, T.A.; Filipek, N.C.; Wise, P.M. Production of proinflammatory cytokines and chemokines during neuroinflammation: Novel roles for estrogen receptors α and β. Endocrinology 2010, 151, 4916–4925. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-Y. Neuroprotection signaling of nuclear Akt in neuronal cells. Exp. Neurobiol. 2014, 23, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, K.; Fu, Z.; Yu, D.; Huang, H.; Zang, X.; Mo, X. Brain development and Akt signaling: The crossroads of signaling pathway and neurodevelopmental diseases. J. Mol. Neurosci. 2017, 61, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Higuchi, M.; Oishi, K.; Kishi, Y.; Okazaki, T.; Sakai, H.; Miyata, T.; Nakajima, K.; Gotoh, Y. PDK1–Akt pathway regulates radial neuronal migration and microtubules in the developing mouse neocortex. Proc. Natl. Acad. Sci. USA 2016, 113, E2955–E2964. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Gabriel, C.; Cacquevel, M.; Ali, C.; MacKenzie, E.T.; Buisson, A.; Vivien, D. Neurotrophin-3-induced PI-3 kinase/Akt signaling rescues cortical neurons from apoptosis. Exp. Neurol. 2004, 187, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Bassell, G.J. Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jiang, H.; Gray, V.; Dedhar, S.; Rao, Y. Role of the integrin-linked kinase (ILK) in determining neuronal polarity. Dev. Boil. 2007, 306, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Macklin, W.B. Integrin-linked kinase (ILK) deletion disrupts oligodendrocyte development by altering cell cycle. J. Neurosci. 2017, 37, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.A.; Benninger, Y.; Baumann, R.; Gonçalves, A.F.; Özçelik, M.; Thurnherr, T.; Tricaud, N.; Meijer, D.; Fässler, R.; Suter, U.; et al. Integrin-linked kinase is required for radial sorting of axons and schwann cell remyelination in the peripheral nervous system. J. Cell Boil. 2009, 185, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Soltani, M.H.; Pichardo, R.; Song, Z.; Sangha, N.; Camacho, F.; Satyamoorthy, K.; Sangueza, O.P.; Setaluri, V. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am. J. Pathol. 2005, 166, 1841–1850. [Google Scholar] [CrossRef]
- Johnson, G.V.; Jope, R.S. The role of microtubule-associated protein 2 (map-2) in neuronal growth, plasticity, and degeneration. J. Neurosci. Res. 1992, 33, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lu, N.; Ding, Y.; Wang, Y.; Chan, L.T.; Wang, X.; Gao, X.; Jiang, S.; Liu, K. Rapamycin-resistant mTOR activity is required for sensory axon regeneration induced by a conditioning lesion. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Driving neural regeneration through the mammalian target of rapamycin. Neural Regen. Res. 2014, 9, 1413. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Senile dementia and Alzheimer’s disease. Brain blood flow and metabolism. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 1986, 10, 447–478. [Google Scholar] [CrossRef]
- Misiak, B.; Leszek, J.; Kiejna, A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease—The emerging role of systemic low-grade inflammation and adiposity. Brain Res. Bull. 2012, 89, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.N.F. Inside the diabetic brain: Role of different players involved in cognitive decline. ACS Chem. Neurosci. 2015, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes—Evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Tong, M.; Bowling, N.; Moskal, P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: Relevance to fetal alcohol spectrum disorder. Mol. Brain 2011, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S. CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J. Alzheimer’s Dis. Park. 2013, 3, 128. [Google Scholar]
- Shuvaev, V.V.; Laffont, I.; Serot, J.-M.; Fujii, J.; Taniguchi, N.; Siest, G. Increased protein glycation in cerebrospinal fluid of Alzheimer’s disease2. Neurobiol. Aging 2001, 22, 397–402. [Google Scholar] [CrossRef]
- Benedict, C.; Frey II, W.H.; Schiöth, H.B.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp. Gerontol. 2011, 46, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.; Watson, G.; Frey Ii, W.; Baker, L.; Cholerton, B.; Keeling, M.; Belongia, D.; Fishel, M.; Plymate, S.; Schellenberg, G. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. Insulin signaling in health and disease. Science (New York) 2003, 302, 1710–1711. [Google Scholar] [CrossRef] [PubMed]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Klintworth, H.; Kavanagh, T.; Costa, L. Apoptosis induced by domoic acid in mouse cerebellar granule neurons involves activation of p38 and JNK MAP kinases. Neurochem. Int. 2008, 52, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Laratta, E.; Procopio, C.; Hribal, M.L.; Sciacqua, A.; Perticone, M.; Miele, C.; Perticone, F.; Sesti, G. Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol. Cell. Boil. 2007, 27, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Ashe, K.H.; Sweatt, J.D. Β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Unger, J.W.; Livingston, J.N.; Moss, A.M. Insulin receptors in the central nervous system: Localization, signalling mechanisms and functional aspects. Prog. Neurobiol. 1991, 36, 343–362. [Google Scholar] [CrossRef]
- Moroo, I.; Yamada, T.; Makino, H.; Tooyama, I.; McGeer, P.; McGeer, E.; Hirayama, K. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 1994, 87, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, T.; Tooyama, I.; Moroo, I.; Kimura, H.; Yamamoto, T.; Okada, H. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci. Lett. 1996, 204, 201–204. [Google Scholar] [CrossRef]
- Bowen, B.C.; Block, R.E.; Sanchez-Ramos, J.; Pattany, P.M.; Lampman, D.A.; Murdoch, J.B.; Quencer, R.M. Proton MR spectroscopy of the brain in 14 patients with Parkinson disease. Am. J. Neuroradiol. 1995, 16, 61–68. [Google Scholar] [PubMed]
- Hu, M.; Taylor-Robinson, S.D.; Chaudhuri, K.R.; Bell, J.D.; Labbe, C.; Cunningham, V.; Koepp, M.; Hammers, A.; Morris, R.; Turjanski, N. Cortical dysfunction in non-demented Parkinson’s disease patients: A combined 31P-MRS and 18FDG-PET study. Brain J. Neurol. 2000, 123, 340–352. [Google Scholar] [CrossRef]
- Lalić, N.M.; Marić, J.; Svetel, M.; Jotić, A.; Stefanova, E.; Lalić, K.; Dragašević, N.; Miličić, T.; Lukić, L.; Kostić, V.S. Glucose homeostasis in Huntington disease: Abnormalities in insulin sensitivity and early-phase insulin secretion. Arch. Neurol. 2008, 65, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Montojo, M.T.; Aganzo, M.; González, N. Huntington’s disease and diabetes: Chronological sequence of its association. J. Huntington’s Dis. 2017, 6, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Patrone, C.; Eriksson, O.; Lindholm, D. Diabetes drugs and neurological disorders: New views and therapeutic possibilities. Lancet Diabetes Endocrinol. 2014, 2, 256–262. [Google Scholar] [CrossRef]
- Bayliss, J.A.; Lemus, M.B.; Santos, V.V.; Deo, M.; Davies, J.S.; Kemp, B.E.; Elsworth, J.D.; Andrews, Z.B. Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS ONE 2016, 11, e0159381. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, A.A.; Espinosa-Oliva, A.M.; Santiago, M.; García-Quintanilla, A.; Oliva-Martín, M.J.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases MPTP-induced damage to the nigrostriatal dopaminergic system. Toxicol. Appl. Pharmacol. 2016, 298, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Khang, R.; Ham, S.; Jeong, G.R.; Kim, H.; Jo, M.; Lee, B.D.; Lee, Y.I.; Jo, A.; Park, C. Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 2017, 8, 48603. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Dong, R.R.; Zhong, K.L.; Ghosh, A.; Tang, S.S.; Long, Y.; Hu, M.; Miao, M.X.; Liao, J.M.; Sun, H.B. Antidiabetic drugs restore abnormal transport of amyloid-β across the blood–brain barrier and memory impairment in db/db mice. Neuropharmacology 2016, 101, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.L.; Lee, M.-S.; Hsu, C.-C.; Chuang, S.-Y.; Lee, J.-T.; Tsai, H.-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with type 2 diabetes in a Taiwanese population cohort. Park. Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Luo, Y.-J.; Xiao, J.; Yu, N.-W.; Yi, G. Impact of insulin sensitizers on the incidence of dementia: A meta-analysis. Dement. Geriatr. Cogn. Disord. 2016, 41, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the insulin sensitizer metformin in Alzheimer’s disease: Pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain J. Neurol. 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 2013, 521, 4124–4144. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain J. Neurol. 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Mesulam, M. A horseradish peroxidase method for the identification of the efferents of acetyl cholinesterase-containing neurons. J. Histochem. Cytochem. 1976, 24, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.A.; Leavitt, J. Human memory and the cholinergic system: A relationship to aging? Arch. Neurol. 1974, 30, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.K.; Majovski, L.V.; Marsh, G.M.; Tachiki, K.; Kling, A. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N. Engl. J. Med. 1986, 315, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C. Scopolamine induced learning failures in man. Psychopharmacology 1977, 52, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, I. Mechanism of action of scopolamine as an amnestic. Trends Pharmacol. Sci. 1989, 10, 175–177. [Google Scholar] [CrossRef]
- Lovestone, S.; Howard, R. Alzheimer’s disease: A treatment in sight? J. Neurol. Neurosurgery, Psychiatry 1995, 59, 566. [Google Scholar] [CrossRef]
- Massoud, F.; Gauthier, S. Update on the pharmacological treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2010, 8, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.A.; Gartlehner, G.; Webb, A.P.; Morgan, L.C.; Moore, C.G.; Jonas, D.E. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. Clin. Interv. Aging 2008, 3, 211. [Google Scholar] [PubMed]
- Richter, N.; Beckers, N.; Onur, O.A.; Dietlein, M.; Tittgemeyer, M.; Kracht, L.; Neumaier, B.; Fink, G.R.; Kukolja, J. Effect of cholinergic treatment depends on cholinergic integrity in early Alzheimer’s disease. Brain J. Neurol. 2018, 141, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.W.; Spreng, R.N.; Weiner, M.W.; Aisen, P.; Petersen, R.; Jack, C.R.; Jagust, W.; Trojanowki, J.Q.; Toga, A.W.; Beckett, L. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun. 2016, 7, 13249. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, E.M.; Permanne, B.; Soto, C.; Wisniewski, T.; Frangione, B. In vivo reversal of amyloid-β lesions in rat brain. J. Neuropathol. Exp. Neurol. 2000, 59, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.; Hardy, J.; Duff, K.; Jantzen, P.; DiCarlo, G.; Wilcock, D. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer’s Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Lacey, L.; Bobula, J.; Rüdell, K.; Alvir, J.; Leibman, C. Quality of life and utility measurement in a large clinical trial sample of patients with mild to moderate Alzheimer’s disease: Determinants and level of changes observed. Value Health 2015, 18, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.; Sundell, K.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.; Pontecorvo, M.; Dean, R.; Demattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimer’s Dement. 1983, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Benzinger, T.L.; Berry, S.; Clifford, D.B.; Duggan, C.; Fagan, A.M.; Fanning, K.; Farlow, M.R.; Hassenstab, J.; McDade, E.M. The DIAN-TU next generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 8–19. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Sims, J.R.; Lowe, S.L.; Nakano, M.; Hawdon, A.; Willis, B.A.; Gonzales, C.; Liu, P.; Fujimoto, S.; Dean, R.A. Safety, pharmacokinetics (PK), and florbetapir F-18 positron emission tomography (PET) after multiple dose administration of LY3002813, a β-amyloid plaque-specific antibody, in Alzheimer’s disease (AD). Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, P352–P353. [Google Scholar] [CrossRef]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimer’s Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Blaettler, T.; Smith, J.; Smith, J.; Paul, R.; Asnaghi, V.; Fuji, R.; Quartino, A.; Honigberg, L.; Rabbia, M.A.; Yule, S. Clinical trial design of cread: A randomized, double-blind, placebo-controlled, parallel-group phase 3 study to evaluate crenezumab treatment in patients with prodromal-to-mild Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, P609. [Google Scholar] [CrossRef]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K. An effector-reduced anti-β-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-arcswe mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401-a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimer’s Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Satlin, A.; Wang, J.; Logovinsky, V.; Berry, S.; Swanson, C.; Dhadda, S.; Berry, D.A. Design of a bayesian adaptive phase 2 proof-of-concept trial for BAN2401, a putative disease-modifying monoclonal antibody for the treatment of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kastanenka, K.V.; Bussiere, T.; Shakerdge, N.; Qian, F.; Weinreb, P.H.; Rhodes, K.; Bacskai, B.J. Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J. Neurosci. 2016, 36, 12549–12558. [Google Scholar] [CrossRef] [PubMed]



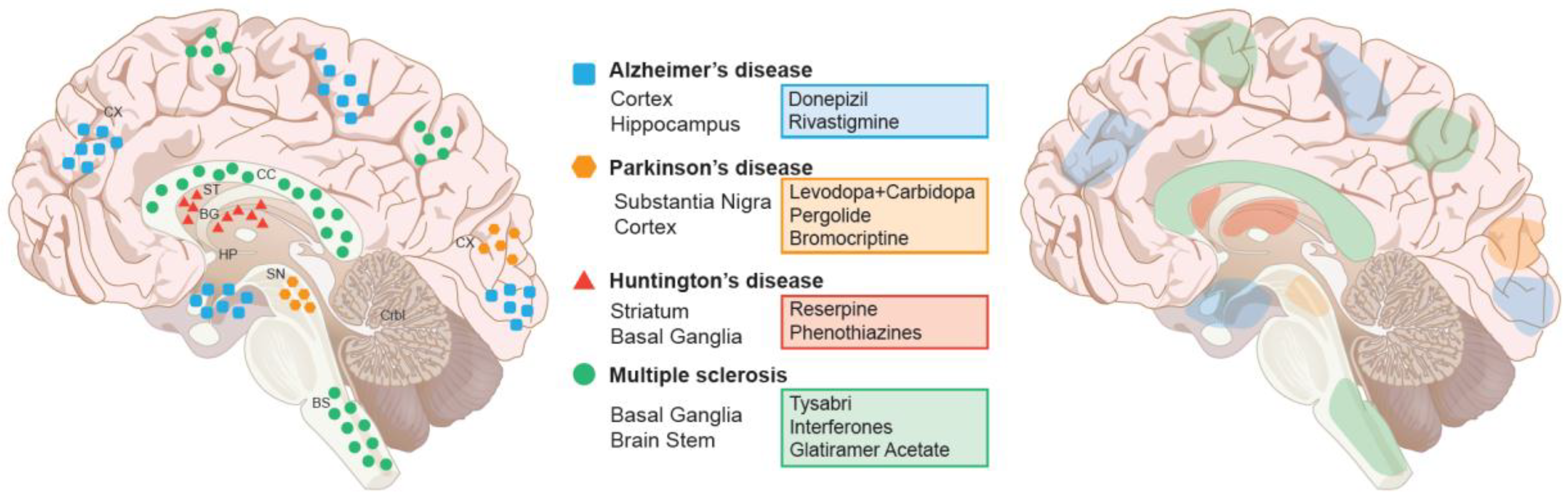{kind=link}
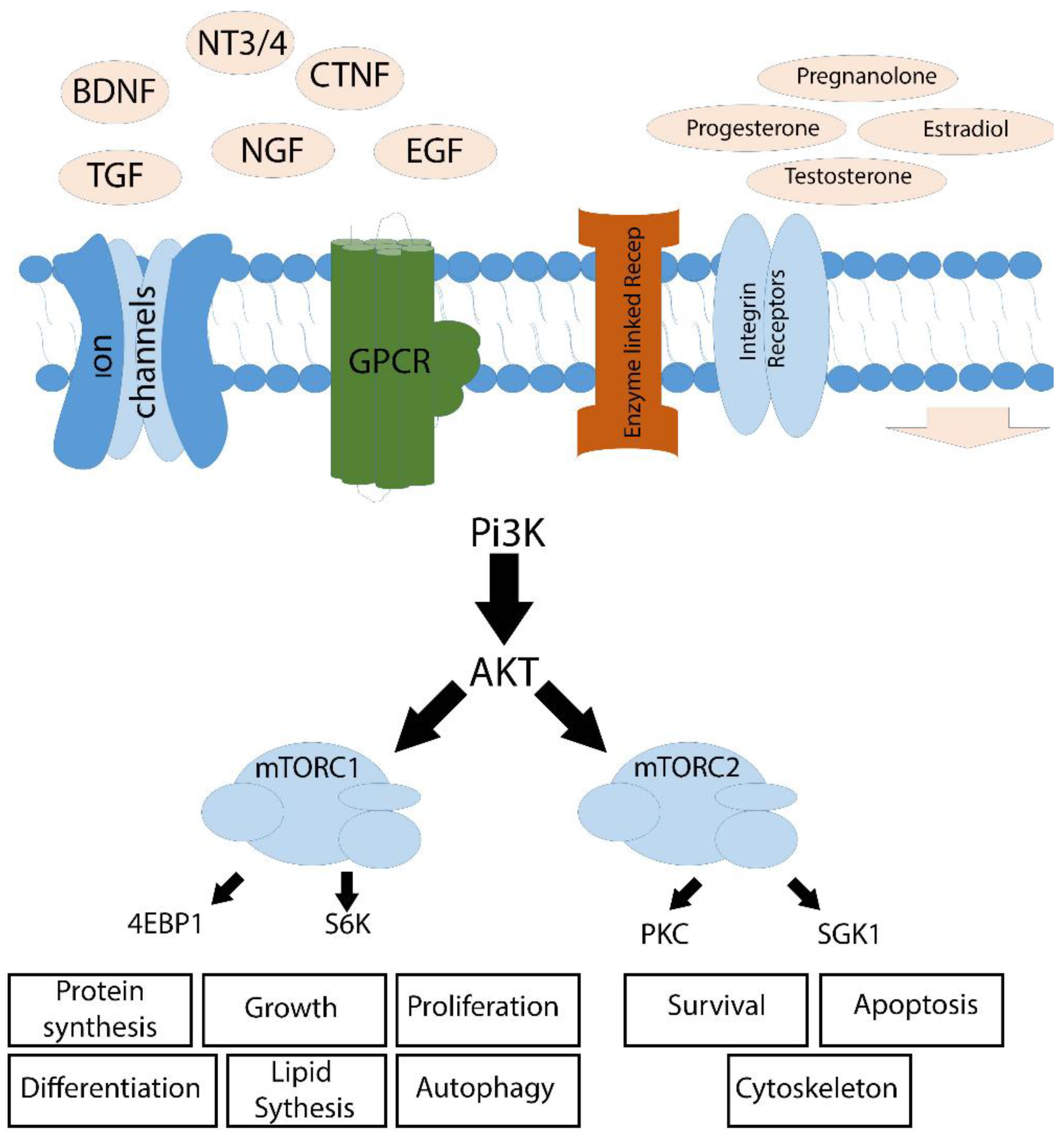{kind=link}
| Strategy | Model | Drugs | Dose | Results | Reference |
|---|---|---|---|---|---|
| Inhibiting protein aggregates | Rat | iAβ5 (Chaperon) | 200 nmol |
| [377] |
| Disaggregating misfolded proteins | Mouse | Hsp104 | - |
| [171] |
| Mouse | Geldanamycin | 1 or 10 µg/kg |
| [177] | |
| Immunomodulation | Mouse | Amyloid-β-Peptide | - |
| [378,379,380] |
| Mouse | Resveratrol | 50 mg/kg |
| [195] | |
| Mouse | Tanshinone IIA | 25 mg/kg |
| [197] | |
| Mouse | Tanshinone I | 10 mg/kg |
| [198] | |
| Mouse | 1H7, 5C1, 5D12 | - |
| [204] | |
| Mouse | mAb47 | - |
| [205] | |
| Induction of Autophagy | Drosophila | Rapamycin | 1 µM |
| [223] |
| Verapamil | Mice | 25 mg/kg |
| [225] | |
| Rapamycin ester (CCI-779) | Mice | 20 mg/kg |
| [102] | |
| Metformin | Mice | 2 mg/mL of drinking water |
| [229] | |
| Nilotinib | Mice | 10 mg/kg |
| [231] | |
| Nilotinib | Mice | - |
| [232] |
| Antibody | IgG Subtype | Specificity | Dose | Reference |
|---|---|---|---|---|
| Bapineuzumab | IgG1 AAB-001 (humanized mouse 3D6) | Aβ 1–5 (helical, N-terminal D sensitive) | Phase I: 12-month 0.5, 1.5, or 5 mg/kg Phase II: 18-month 0.15, 0.5, 1, or 2 mg/kg Phase III: 18-month 0.5 mg/kg 1.0 mg/kg | [381,382] |
| Solanezumab | IgG1 (humanized mouse [265]) | Aβ 16–26 accessible only on monomeric Aβ | 400 mg every week for 76 weeks Phase III A4 400–1600 mg every 4 weeks for 240 weeks | [383,384,385] |
| LY3002813 | IgG1 (humanized mouse mE8-IgG2a) | pE3-Aβ | 0.1 mg/kg to 10 mg/kg, infused monthly up to four times, and a single subcutaneous injection against placebo for safety | [386,387] |
| Gantenerumab | b (RG1450, RO4909832) IgG1 (full human) | Aβ 2–5 (−9) + 23–25 binds with subnanomolar affinity to a conformational epitope on Aβ fibrils. It binds both N-terminal and central amino acids of Aβ | Phase III 225 mg SC | [388] |
| Crenezumab | IgG4 (humanized mouse MABT5102) | Aβ 13–24 (conformational epitopes) Binds fibrillar, oligomeric, and monomeric Aβ | Phase III up to 60 mg/kg SC (every 2 weeks) for at least 260 weeks | [389,390] |
| BAN2401 | IgG1 (humanized mAb158) | recognizes Aβ protofibrils | Phase I: 2.5, 5 and 10 mg/kg Phase II: 2.5, 5 and 10 mg/kg | [391,392,393] |
| Aducanumab | b IgG1 (BIIB037/BART full human) | recognizes Aβ oligomer and fibrils | [394,395] |
| Drug Name | Type | Pathology | Model Type | Results | References |
|---|---|---|---|---|---|
| 1-methyl-4-phenylpyridinium | Dopaminergic neurotoxin | Culture models of Parkinson’s | Mouse | induce buildup of autophagic vacuoles | [218,219] |
| Rapamycin | selective inhibitor of TORC1 | Alzheimer’s Disease | Mouse | ameliorates Aβ and tau in AD mouse models | [226,228] |
| Latrepirdine | the stimulator of Atg5-dependent autophagy | Alzheimer’s Disease | Mouse | reduces Aβ in mouse models | [220] |
| Metformin | Protein phosphatase 2A agonist | Alzheimer’s Disease | Clinical Trials | Inhibits tau hyperphosphorylation in AD clinical trials | [220,230] |
| Lithium | UlK1 Kinase activator | Alzheimer’s Disease | Experimental/Clinical trials | AMPK activation and induces autophagic activation | [222] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. https://doi.org/10.3390/brainsci8090177
Hussain R, Zubair H, Pursell S, Shahab M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sciences. 2018; 8(9):177. https://doi.org/10.3390/brainsci8090177
Chicago/Turabian StyleHussain, Rashad, Hira Zubair, Sarah Pursell, and Muhammad Shahab. 2018. "Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches" Brain Sciences 8, no. 9: 177. https://doi.org/10.3390/brainsci8090177
APA StyleHussain, R., Zubair, H., Pursell, S., & Shahab, M. (2018). Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sciences, 8(9), 177. https://doi.org/10.3390/brainsci8090177